
Abstract
Introduction
The presence of the oxidative phosphorylation machinery in the inner membrane reflects the evolutionary origin of mitochondria. As detailed in the endosymbiotic theory, the bacterial ancestor of mitochondria joined an archaebacterial cell (58) and this laid the foundation of today's eukaryotes. Both the mitochondrial and the bacterial respiratory chains couple the stepwise reduction of terminal electron acceptors to the formation of an electrochemical gradient across the inner membrane. This gradient is then used to drive ATP synthesis or to fuel a number of transport processes. Thus the general catalytic principles of the mitochondrial respiratory chain complexes are similar to that of their bacterial counterparts.
However, while all subunits of bacterial respiratory chains are produced by one genetic system, the mitochondrial respiratory chain is a mosaic of components produced by two genetic systems (Fig. 1). In the course of mitochondrial evolution, most of the former bacterial genes were transferred to the nucleus. This movement of genetic information to the nucleus co-evolved with dedicated machineries allowing post-translational import of the nuclear gene products destined for mitochondria (30). Consequently, N-terminal extensions were added to the mitochondrial proteins, allowing their post-translational import (Fig. 1) into the organelle. Proteomic, as well as genetic studies, have revealed that mitochondria contain up to 1500 different proteins (56, 73). Most of these are the products of nuclear genes. Conversely, the amount of genes kept within the organelle was significantly reduced so that mitochondria of today typically synthesize a small number of different proteins. These mitochondrial-encoded proteins represent the essential reaction centers of the respiratory chain complexes and are therefore indispensable for oxidative phosphorylation. Thus, any mutation affecting the expression of the mitochondrial-encoded genes can severely compromise the oxidative phosphorylation system. In this review, we will summarize the current knowledge on how mitochondrial protein synthesis is regulated, and how mutations and environmental stress might influence the efficiency and accuracy of mitochondrial translation and therefore mitochondrial functionality. We focus on work employing yeast but also include results from the mammalian systems. Straight-forward, powerful genetics and the possibility for well-defined biochemistry have made Saccharomyces cerevisiae and Schizosaccharomyces pombe important tools to unravel basic principles of mitochondrial biogenesis and the factors controlling aging.
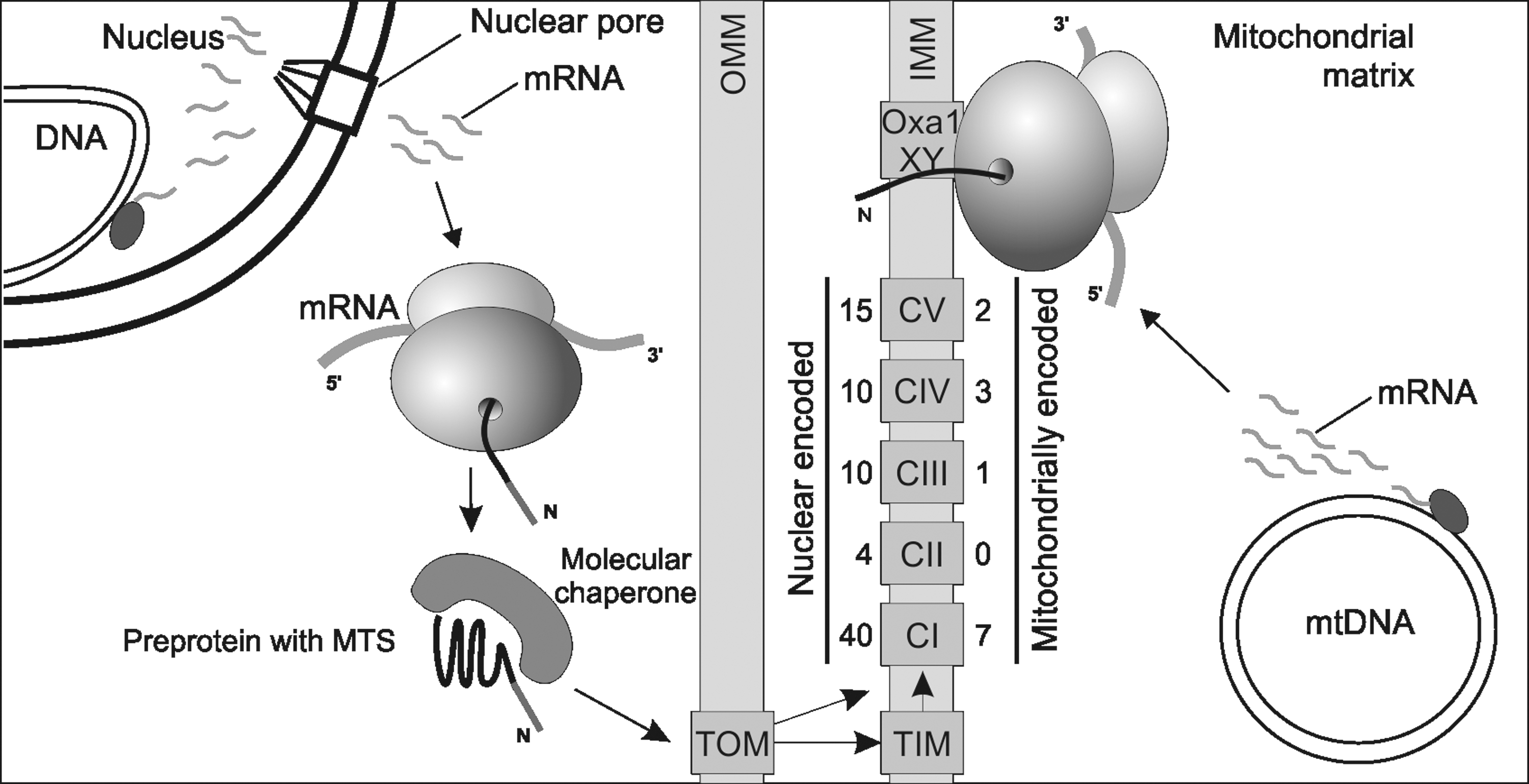
General Function of Mitochondrial-Encoded Proteins
The genes typically present in mitochondrial genomes of animals and fungi encode the ribosomal and transfer RNA (rRNA and tRNA) as well as the membrane-embedded reaction centers of the respiratory chain subunits (Fig. 2). In most eukaryotes, these proteins include ND1, ND2, ND3, ND4, ND4L, ND5, and ND6 of complex I, CYTb of complex III, COX1, COX2, and COX3 of complex IV, and ATP6 and ATP8 of ATP synthase (complex V). Mitochondria of the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe lack complex I and hence the mitochondrial genes encoding its subunits, but contain the genes coding for Atp9 of ATP synthase and for Var1/Rps3, a constituent of the small ribosomal subunit.

The hydrophobicity of the translation products is considered to be a major reason why mitochondria still synthesize proteins. Because these proteins are so hydrophobic, they tend to aggregate in a soluble compartment such as the cytoplasm, which would render their transport to mitochondria via the standard route almost impossible. Synthesis of these proteins within mitochondria allows their co-translational insertion into the inner membrane, thus circumventing the problem of transport (55) and allowing a coordinated synthesis and assembly of respiratory complex subunits (see section Differences: Translation Initiation and Post-Transcriptional Control of Gene Expression). In addition, mitochondrial genomes encode all the rRNAs, and transport of such large and negatively charged polymers would be a major challenge for the cell, as for the hydrophobic membrane proteins. Thus, it appears likely that keeping these genes encoded within the organelle circumvents the potential problems of such complex transport pathways and facilitates ribosome assembly in mitochondria.
Similarities and Differences to Bacterial Protein Synthesis
Similarities: Antibiotic sensitivity, conserved proteins and factors
To date, no robust in vitro translation system could be established to study protein synthesis by mitochondrial ribosomes. Hence, much of our current understanding on mitochondrial translation stems from the characterization of protein synthesis in prokaryotes, the system most closely related to mitochondria. Because of the evolutionary relationship between bacteria and mitochondria, a series of in vitro complementation experiments with mammalian translation factors could be performed, allowing to shed light on conserved and diverged aspects of mitochondrial translation (14, 21, 61, 75).
All ribosomes consist of small and large subunits that are composed of protein and RNA elements. Three essential binding sites for tRNAs are located at the surface of the small subunit that contacts the large subunit: the aminoacyl-tRNA site (A-site), the peptidyl-tRNA site (P-site), and the exit site (E-site). These serve as docking sites for the tRNAs and are implicated in the repeated decoding of the mRNA in a highly accurate manner. During elongation of the polypeptide, alternate rounds of decoding, peptide bond formation and translocation of the mRNA proceed until the stop codon signals termination of translation. These reactions are controlled and mediated by a number of translation factors. These factors, together with the catalytically active rRNA domains, are well conserved and found also in mitochondria (1, 75). Depending on the model organism, small changes in the set of translation factors occur (Table 1); their functions have been best understood for the mammalian system (14). While the general scheme of translation initiation has not been established for mitochondrial protein synthesis (see also later in this review), it is assumed that elongation of the peptide chain occurs by almost identical mechanisms while termination of translation and recycling are showing stunning deviations from the bacterial pathway (15).
The rRNAs of the small and large subunit play at least two main roles in translation, namely by proofreading the codon–anticodon interaction and catalyzing peptide bond formation, respectively. In bacteria, the molecular mechanisms determining fidelity and efficiency of translation are very well understood (85). In contrast, hardly anything is known about this in mitochondria. Many antibiotics that directly interact with the rRNAs of bacterial ribosomes decrease accuracy and efficiency of translation. Because of their evolutionary origin from bacterial ribosomes, mitochondrial ribosomes show a pronounced sensitivity to the same antibiotics, therefore limiting their relevance for medical treatment (11, 80). The aminoglycoside antibiotics are specifically important in this context since they decrease fidelity of translation in bacteria and presumably in mitochondria (87). Some mutations of the mitochondrial 12S rRNA cause a hypersensitivity to aminoglycosides that can lead to the loss of cochlear neurons and consequently to deafness (37).
Differences: Translation initiation and post-transcriptional control of gene expression
While many aspects of protein synthesis in mitochondria are similar to those found in bacteria, a number of mechanisms diverged significantly during organellar evolution. Mitochondrial mRNAs differ from both bacterial and cytoplasmic mRNAs because they do not contain Shine-Dalgarno sequences or CAP structures (Table 1). The consequence is that initiation of translation in mitochondria must be mediated by other signals and/or factors. These factors have been most fully studied in the yeast S. cerevisiae, where the versatile genetic system combined with the possibility of manipulating the mitochondrial DNA have facilitated the identification of cis-signals, as well as trans-factors, involved in mitochondrial translation (8). These analyses revealed that, at least in yeast, translation of the different mRNAs requires the presence of one or more transcript-specific translational activator.
In this respect, translation of the COX2 mRNA has been particularly well investigated, possibly because its 5’-untranslated region (UTR) is relatively short. The study of respiratory mutants lacking the Cox2 protein has revealed that COX2 translation is under the control of the nuclear gene-product Pet111 (59), recently classified as an RNA binding protein of the penta-tricopeptide repeat (PPR) family (42). Pet111 acts on the 5’-UTR of COX2, as shown by the isolation of suppressors containing rearranged mitochondrial genomes (59). Computational and mutational analyses of this 5’-UTR have revealed that a stem-loop structure is especially important for Pet111 activity and thus could be the binding site of the translation activator (17). Like most of the other translational activators in mitochondria, Pet111 is associated with the inner membrane. This might both ensure a tight co-translational insertion of the newly synthesized proteins into the inner membrane and organize translation on the surface of the inner membrane (64). Such a function has been suggested by the observation that the translational activators controlling the synthesis of cytochrome c oxidase subunits can directly interact with each other (50), and therefore allow the synthesis of subunits destined for the same complex in a close proximity to facilitate assembly. Finally, the amounts of translational activators are strictly controlled, and overproduction experiments (e.g., for Pet111) have shown that the balance in the synthesis of the different subunits of a respiratory complex is important for efficient biogenesis and assembly (19). Thus, translational activators play a role both in the organization and regulation of organellar protein synthesis.
Whether translational activators can also participate in choosing the initiation codon is unclear. There clearly is a stringent START site selection in mitochondria since mutagenesis of the initiation codon of the COX2 mRNA in S. cerevisiae does not allow translation initiation at the nearby downstream AUG (7). A repeated 11-nucleotide sequence surrounding the AUG of the COX2 mRNA could possibly play a role in the recognition of the initiation codon (6).
In addition, three S. cerevisiae mRNAs (one for each of respiratory chain complexes of dual genetic origin: III, IV, and V) are subject to an assembly feedback control where efficiency to assemble the complex modulates synthesis of the mitochondrial-encoded subunits. The best described case is that of the COX1 mRNA whose translation is under the control of the activators Pet309 and Mss51 (48). Pet309 and Mss51 both activate COX1 translation, probably by interacting with the COX1 5’-UTR. In addition, Mss51 together with other factors can directly bind the newly synthesized Cox1 protein until it is assembled into complex IV. Thus, unassembled Cox1 sequesters its own translational activator and prevents further synthesis, despite the presence of Pet309. Upon assembly of the Cox1 protein into complex IV, Mss51 is released and can again activate translation. This allows a tight coupling of synthesis and assembly. A similar way to regulate synthesis of a mitochondrial-encoded protein according to efficiency of assembly has been demonstrated for the ATP6/8 co-transcript (60) and for COB mRNA (24, 26), respectively. However the mechanisms of these regulations have to be further documented.
Mitochondrial translational initiation and regulation in S. pombe and humans is much less well understood. The S. pombe and human mitochondrial genomes are rather compact and expressed by the production of large polycistronic transcripts that are processed to give individual mRNAs by excision of tRNAs that are located between the mRNAs. The formed UTR regions in mRNAs are rather short in S. pombe (70) while they are mostly absent in humans (79) (Table 1). Nevertheless, initiation complex formation on mammalian mitochondrial ribosomes requires mRNAs that contain a properly located START AUG codon (13). Because of the absence of long 5′-UTRs in mammalian mitochondria or their reduction in S. pombe, it appears likely that mitochondrial translation in these organisms is regulated in a different way than in S. cerevisiae.
In accordance, only a few homologs of the S. cerevisiae mRNA translational activators described above have been detected by sequence comparison in S. pombe and humans, and their functions are not always conserved. For example, Mss51 has a clear sequence homolog in S. pombe, which is also important for the Cox1 protein biogenesis, but only at a post-translational level; this situation is similar for Cbp3 and Cbp6 (39). A human zinc finger protein ZMNYD-17 has been proposed to be the Mss51 structural homolog (48); however, a role in mitochondrial biogenesis has not yet been documented. Pet309 is also conserved in sequence in S. pombe (Ppr4 and Ppr5) and human (LRPPRC, involved in the French-Canadian Leigh syndrome (86). Ppr4 behaves as a bona fide cox1 translational activator, whereas Ppr5 acts as a general translational repressor (40). Likewise, LRPPRC is a factor with multiple effects on stability, polyadenylation, and translation of all mRNAs, and COX1 and COX3 mRNA are the most sensitive targets (63, 67). However, a bona fide cox1 translational activator has been identified in humans, TACO1, mutations in the gene coding this factor cause a late-onset Leigh syndrome (83). Thus, although sequence conservation is not a straight-forward way to identify mitochondrial translational activators, Cox1 synthesis appears as a universal point of control for translation, since activators of this mRNA can be found in both yeasts and humans. So far, the target sequence of these factors is unknown in S. pombe and humans, whereas Ppr4 could well bind the 220 bp S. pombe cox1 5’-UTR, the 5’-UTR of COX1 in humans is only 3 nucleotides long. Thus, the target of TACO1 probably lies within the cox1 coding region, as shown for LRPPRC (86). Whether feedback loops exist in S. pombe and humans is unclear, but if they do, they possibly could involve two components (e.g., Ppr4/Mss51 or TACO1/ZMNYD-17), rather than a factor with a dual function such as Mss51 in S. cerevisiae.
Significantly remodeled structures of mitochondrial ribosomes
Bacterial ribosomes consist of small and large subunits that sediment as 30S and 50S particles, respectively, and contain a well-defined set of rRNA components and proteins (Fig. 3). In contrast, mitochondrial ribosomes differ significantly from the bacterial ancestor, as well as between species. The most obvious alteration in comparison to the bacterial particles is that mitochondrial ribosomes contain much more protein and typically less rRNA. Importantly, mitochondrial ribosomes contain many components that are clearly conserved and found at the structurally and functionally important sites (Fig. 4). What could generally be the function of the novel, mitochondria-specific proteins that were recruited to the ribosome? A possible explanation is that these proteins compensate for the loss of structural RNA which is evident especially in mammalian mitochondria that contain very short RNAs in comparison to the prokaryotic ribosomes (36, 77). Such an interpretation is supported by the finding that around 20% of the bacterial-type RNA components that are missing in mitochondrial ribosomes are replaced by mitochondrial ribosomal proteins (71). The remaining mitochondria-specific proteins are found at the surface of the ribosomal subunits, thus shielding the rRNA components. In addition, these proteins appear to be important for establishing intersubunit bridges between the small and the large subunit (71), a feature that is different from cytoplasmic ribosomes that typically contain RNA–RNA intersubunit connections.
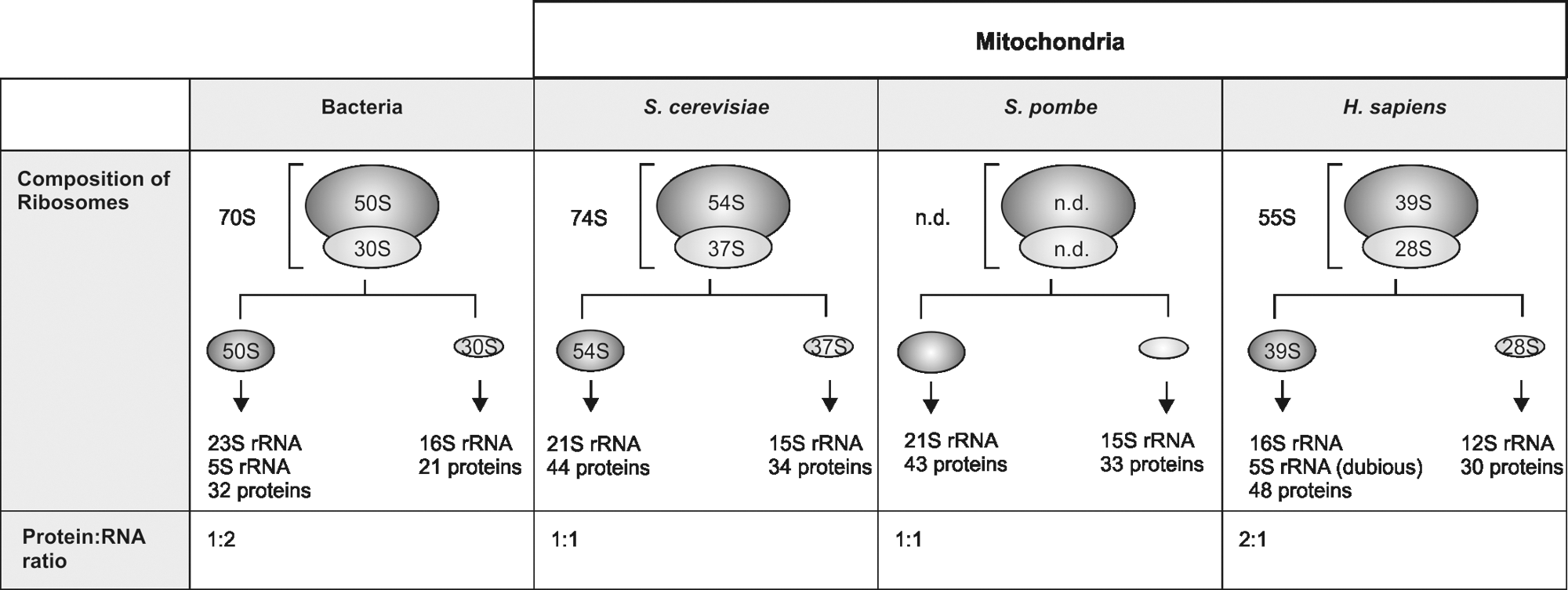

All ribosomes share the catalytic center that forms the peptide bonds. In cytoplasmic ribosomes, the secondary structure of the RNA involves base pairings and the strongest interactions are observed between cytosine and guanine. Interestingly, mitochondrial rRNAs are, in contrast to bacterial rRNAs, guanine-poor and adenine-rich (46). Because the cytosine content of mitochondrial rRNA is almost identical to that of bacterial ribosomes, the fraction of cytosine nucleotides found in base-pairing drops from ∼60% in prokaryotes to ∼45% in mammalian mitochondria (46). This suggests that mitochondrial rRNAs cannot adopt similarly tight folds compared to rRNAs from cytoplasmic ribosomes. Therefore, it is plausible that many mitochondria-specific ribosomal proteins were especially recruited to stabilize the rRNA. The significance of mitochondria-specific proteins for stabilization of the rRNA is reinforced by the finding that absence of most of these proteins (and also of many of the C-terminal extensions, our unpublished results) in yeast destabilizes the mitochondrial DNA. Because mitochondrial translation is a prerequisite to maintain a functional mitochondrial genome (49), destabilized ribosomes caused by the absence of a stabilizing ribosomal protein will eventually lose their ability to synthesize proteins and thereby provoke the loss of mitochondrial DNA. In addition to this stabilization of ribosomal structure, at least some mitochondria-specific ribosomal proteins could also introduce novel functions or organizational features to the mitochondrial translation system, as illustrated by a number of mitochondria-specific proteins found at the tunnel exit (23, 25) or involved in translational regulation (27, 84).
Susceptibility to Oxidative Stress and Influence on Aging
The respiratory chain of mitochondria is a primary source of reactive oxygen species (ROS) in the cell where as much as 1%–2% of the oxygen consumed by mitochondria is already converted to ROS under physiological conditions (54). Hence, mitochondrial components are primary targets of ROS. Mutations caused by oxidative modification of mitochondrial DNA (guanosine might be a primary target here, see also decrease in general guanosine content in mitochondrial genomes, previous section) can affect rRNAs and tRNAs that in turn can compromise the integrity/function of the mitochondrial translation apparatus (62, 72). Apart from the RNA components, proteins of the mitochondrial ribosomes can also be modified by ROS. Recently, it has been reported that the mitoribosomal protein Mrpl32 exhibits a sensing mechanism for ROS that allows the attenuation of mitochondrial translation under oxidative stress to limit further ROS production by the respiratory chain (5). Mrpl32 contains a conserved CxxC-X9-CxxC motif that binds zinc and thus is sensitive to oxidation by ROS. Oxidation and loss of the zinc ion destabilize Mrpl32. Since other ribosomal proteins were not affected upon exposure to H2O2, it was concluded that the susceptibility of mitochondrial translation to oxidative stress is at least partially caused by degradation of Mrpl32. This mechanism could be a conserved feature because certain bacterial proteins that bind zinc can act as redox switches sensing high levels of ROS (44).
The accumulation of oxidation-damaged mitochondrial components can cause respiratory deficiencies and thus might directly influence the aging process, during the course of which a decline of mitochondrial function is typically observed. It is well documented that ROS and mitochondrial dysfunction do play a role in the aging process (reviews in the forum issue from Nils and Alberto); however, it is currently unclear whether they are the cause or a consequence of cellular aging. The “mitochondrial free radical theory of aging” favors a strict causative role of mitochondrial ROS for mitochondrial aging (28). This hypothesis is supported by observations that lifespan of several organisms can be prolonged by increasing antioxidants (53, 69) and that oxidized proteins accumulate in aged cells. In its most simplified form, the theory postulates that oxidative injury accumulates over time and that the damaged cellular components give rise for further ROS, thus amplifying the initial insult in a feed-forward reaction. This theory, although very popular, has been questioned by a number of recent publications (22, 65, 66). However, alternative hypotheses exist that link primary causes of aging to other mechanisms. Among those alternative hypotheses is Orgels “error catastrophe theory” (52). This theory proposes that the increasing inaccuracy of the gene expression machinery during life might account for the observed age-related dysfunctions (Fig. 5).
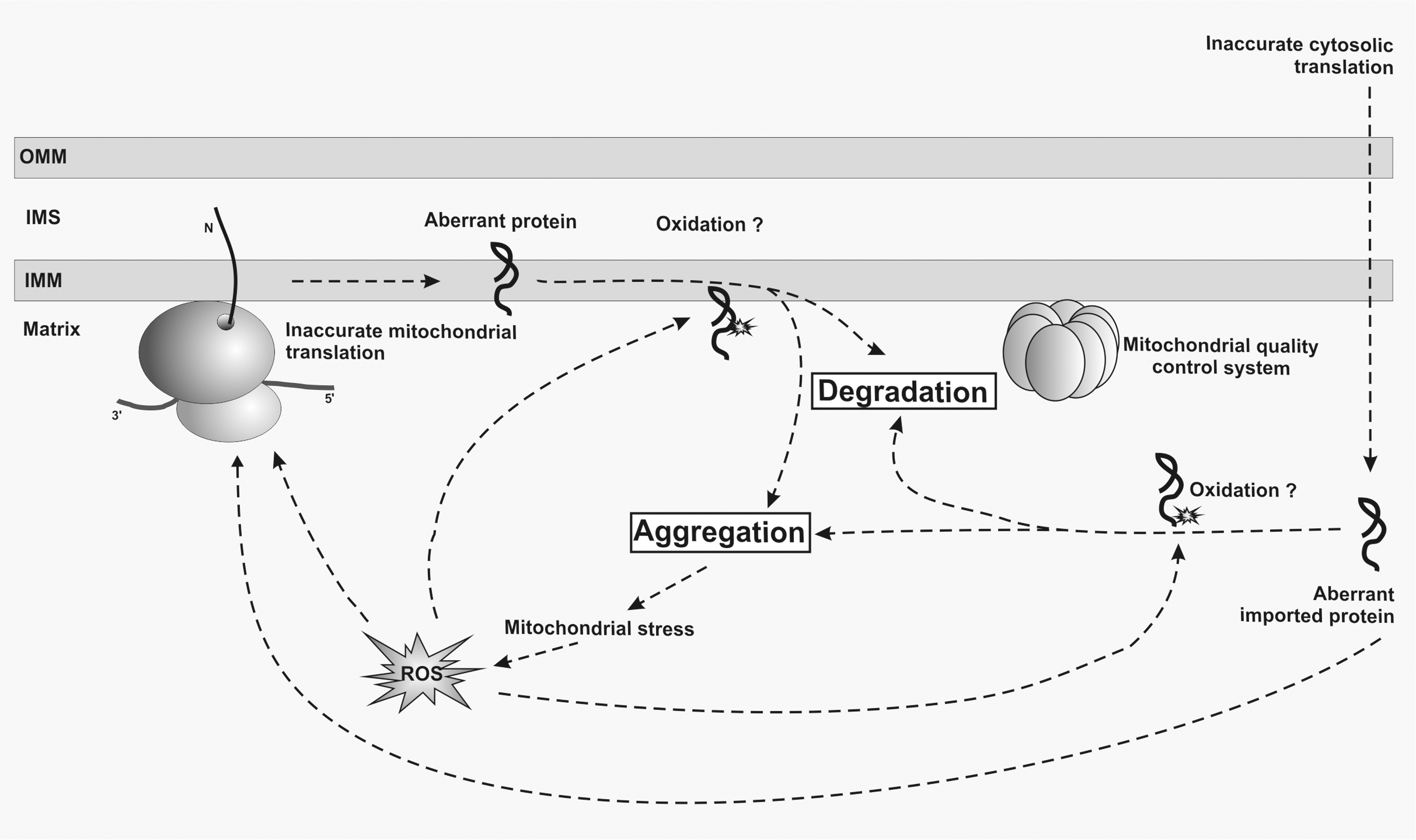
Orgels hypothesis is supported by a number of observations. Work in the filamentous fungus Podospora anserina has revealed that enhanced fidelity of cytosolic protein synthesis increases lifespan (69). Experiments by Thomas Nyström and co-workers in bacteria have shown that oxidatively damaged proteins accumulate during starvation and that higher levels of these proteins decrease viability (3). Importantly, increasing the quality of newly synthesized proteins reduces oxidative damage to proteins, while lowering the fidelity of translation has the opposite effect (3, 16). A molecular explanation for this increased sensitivity for oxidative modification might be that the aberrant translation products fail to fold properly and thus expose more oxidation-sensitive moieties (3). As mutants with error-prone or hyper-accurate translation have similar respiratory activities and SOD activities compared to wild-type cells, the authors concluded that the accumulation of oxidized proteins in growth-arrested cells is not the result of increased oxidative stress. Therefore, the observed age-related increase in oxidized proteins might be a secondary effect of oxidative metabolism and not a primary cause of increased oxidative stress. Importantly, aberrant and oxidized proteins are rapidly eliminated by the cellular quality control system (16, 20). This mechanism would ensure that the functionality of the translation machinery is not jeopardized by aberrant proteins which could cause an exponential increase of inaccurate translation (3). This interpretation is in contrast to one of the initial postulates of the error catastrophe theory that suggested a feedback loop amplifying inaccuracy of gene expression during aging. These observations in bacteria nevertheless demonstrate that a decreased accuracy of translation has a direct influence on viability of aged cells.
Currently, it is not known whether the error catastrophe theory also applies to eukaryotic aging at the level of mitochondria. It is firmly established that the intactness of the mitochondrial genetic system determines aging (18, 81), but how accuracy or efficiency of mitochondrial protein synthesis modulates aging is just beginning to be explored. Currently, there is one study showing that mitochondrial translation accuracy might influence longevity. Treating cells with erythromycin, which presumably also increases translational accuracy of mitochondrial ribosomes, can extend replicative life-span by about 27% in comparison to untreated yeast cells. Because this effect was not observed in cells lacking mitochondrial DNA, it was suggested that the fidelity of the mitochondrial ribosome contributes to lifespan (31). Mitochondria have a very efficient quality control system that removes aberrant proteins employing various proteases, among them Lon protease that plays a dedicated role in removal of oxidized proteins (78). There are several studies demonstrating a decline of Lon activity with age (2, 9). Deletion of Lon in yeast and mammals results in respiratory deficiency and DNA deletions (10, 76). Conversely, overexpression of Lon in Podospora anserina results in lifespan extension and resistance to oxidative stress (43), supporting the idea that accumulation of oxidatively modified aberrant proteins might contribute to aging at the level of mitochondria (Fig. 5).
Mitochondria are deeply embedded in cellular physiology and, consequently, mitochondrial dysfunction triggers many cellular responses that allow the cell to survive under suboptimal conditions (Fig. 6). A prime example is the so-called “retrograde response” that is activated by the loss of the mitochondrial membrane potential and causes an extension of replicative lifespan (32, 47). Thus, the retrograde response can compensate for the gradual increase of mitochondrial dysfunction during aging. The molecular nature of the trigger of the retrograde response is not known; the signal arises from the loss of the mitochondrial membrane potential rather than from increase in ROS or oxidized proteins. Consequently, treatment of cells with a ROS scavenger does not change the retrograde response or affect lifespan in this setting (47).
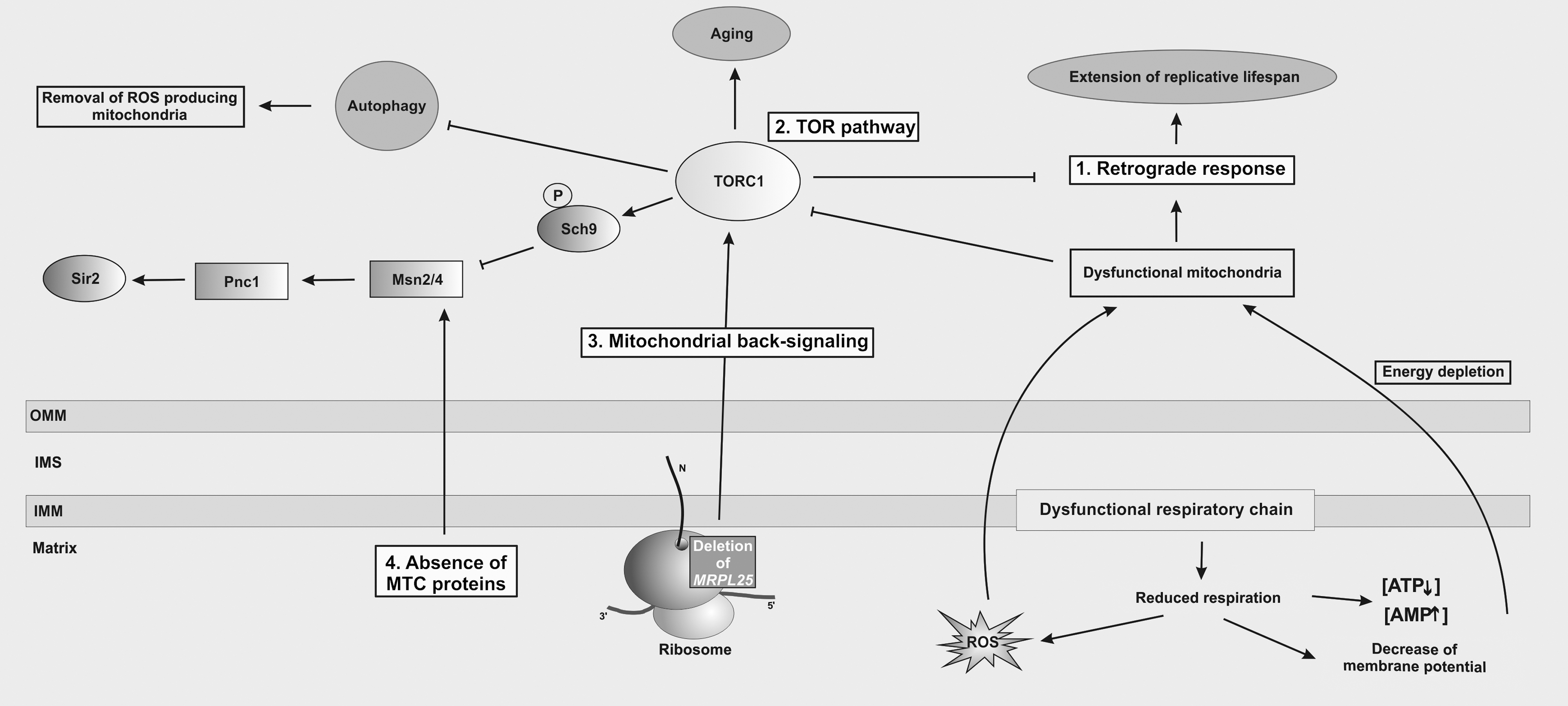
The retrograde response is interconnected with several other signaling pathways that are stimulated by internal and external signals, such as the Target of Rapamycin (TOR) signaling pathway (Fig. 6). The TOR pathway employs two structurally and functionally distinct multiprotein complexes (TORC1, TORC2) and has a key role in development and aging by regulation of cell growth and metabolism in response to availability of nutrients, cellular energy, growth factors, and stress. TORC1 is a negative regulator of the retrograde response (38) and might ensure that the retrograde response is shut off when the cell is well supplied with nutrients. Moreover, TORC1 has a key function in quality control of mitochondria, as it actively inhibits mitophagy (33) under normal conditions. In turn, dysfunctional mitochondria provoke energy depletion and ROS generation and both stimuli activate a signaling cascade that eventually inhibits TORC1 (88), thus promoting removal of damaged mitochondria to protect the cell from further stress. Inhibition of TORC1 directly inactivates the kinase Sch9 (35, 82) that plays a major role in modulation of the stress responses controlled by the Msn2/Msn4 transcription factor (45). This transcription factor controls genes that protect the cell against reductive and oxidative stress such as SOD2, TRX2, or TRX3 (18, 45).
Two recent publications unraveled two additional signaling cascades that are involved in sensing intactness of the mitochondrial genetic system to modulate replicative life span in yeast. Deletion of MRPL25/AFO1 coding for a mitochondrial-specific protein of the large ribosomal subunit (74) leads to a 60% increase in lifespan compared to control cells (29). Additionally, the absence of Mrpl25 causes resistance to oxidative stress as well as a reduced internal ROS production. Since lifetime extension was only observed in cells lacking Mrpl25 but not in cells lacking two other proteins of the mitoribosome (Mrp17, Ppe1; proteins of the small ribosomal subunit), it was suggested that the Mrpl25 protein itself might establish a connection between mitochondria, cellular metabolism, and aging. As the observed increase in lifespan was also induced in cells grown on glucose that suppresses the retrograde response, the authors concluded that the lifetime extension in the absence of Mrpl25 must be different from this pathway. The authors proposed to call this new signaling pathway “mitochondrial back-signaling” to distinguish it from the retrograde response (Fig. 6). However, whether a cross-talk connects both pathways is currently unknown. Interestingly, it has been shown that Mrpl25 is associated with a ribosome-assembly subcomplex containing several proteins of the ribosomal tunnel-exit indicating that Mrpl25 might be present at this site (34). It would be interesting to test whether mitochondrial back-signaling is mediated by a block in ribosome assembly of the large ribosomal subunit and whether absence of other proteins of this subcomplex might provoke a similar phenotype as deletion of MRPL25.
Similarly, a recent report indicated that the absence of some MTC module (mitochondrial translation complex) proteins prolongs replicative lifespan in yeast (12). The class of MTC modules comprises 15 proteins implicated in translation of mtDNA-encoded genes, such as translational activators and mRNA metabolism enzymes (57). Deletion of at least nine MTC proteins increases replicative lifespan independently of mitochondrial respiration, ROS formation, mtDNA, or mitochondrial translation. The observed lifespan extension is, similar to the MRPL25 deletion, independent of the retrograde response but requires the cAMP/PKA-dependent activation of the Msn2/Msn4 transcription factor that mediates induction of the stress response (12), indicating that this pathway is related to the Tor1 pathway (Fig. 6). Furthermore, lifespan extension of at least SOV1 and CBS1 deletion mutants requires histone deacetylase Sir2, leading to elevate rDNA silencing in the nucleus (12). The molecular identity of the signal activating this pathway is not known.
These results indicate that there has to be an extensive communication between mitochondria and the nucleus that signals the quality of mitochondria and allows the adaptation of mitochondrial function in respect to the cellular conditions to assure survival of the cell. This communication likely depends on a complex crosstalk between the mentioned pathways. Furthermore, there are several indications that mitochondrial translation efficiency might affect aging. However, for future research, numerous exciting and challenging experiments will be needed to determine how accurate mitochondrial translation is, whether accuracy changes during life, and how these changes affect aging. To achieve these goals, many important steps of mitochondrial gene expression still need to be better understood.
Footnotes
Acknowledgments
We thank all members of our group for stimulating discussions. Work in our laboratories is supported by grants from the Center for Biomembrane Research (CBR) at Stockholm University, Swedish Science Foundation (VR), German Science Foundation (DFG), Jaenssons Stiftelse, Sweden, and German academic exchange service (DAAD) to MO and from the Association Française contre les Myopathies (AFM) to NB. Kirsten Kehrein was a recipient of a pre-doctoral fellowship from the Carl-Zeiss Foundation, Germany.
Author Disclosure Statement
No competing financial interests exist.