
Abstract
Introduction
The goal of this review is to cover recent discoveries related to the apoptosome, and more generally, the impact of reactive oxygen species (ROS) on the intrinsic pathway. However, as one might expect, ROS can potentially impact this pathway in many ways. For those familiar with pop culture, there is a trivia game known as the “Six Degrees of Kevin Bacon,” in which players attempt to connect any person in Hollywood with the actor, Kevin Bacon, in less than six steps. Similarly, there are numerous ways, both direct and indirect, to connect oxidative stress with the intrinsic pathway, far more than can be adequately addressed here. Thus, we shall focus primarily on those events that immediately impact cyt c (or its release), Apaf-1, or caspase-9—or to keep with the analogy, we will not stray more than two degrees from the apoptosome.
BCL-2 Family Members and MOMP
Arguably, the key step in the intrinsic pathway is permeabilization of the OMM, an event that is regulated by members of the BCL-2 family (Fig. 1). BCL-2 proteins are characterized by the presence of one to four BCL-2 homology (BH) domains and the ability to regulate apoptosis, at least in part, through the regulation of MOMP. The proapoptotic family members include a BAX-like subfamily (BAX, BAK, and BOK), as well as a “BH3-only” subfamily (BIM, BID, PUMA, BAD, NOXA, etc.). BAX and BAK proteins contain BH 1-3 domains, but lack a BH4 domain, and when activated, are thought to stimulate MOMP and the release of cyt c by forming pores in the OMM (Figs. 1 and 2). BH3-only proteins such as truncated BID (tBID), on the other hand, promote apoptosis by antagonizing antiapoptotic BCL-2 family members and/or by directly activating BAX-like family members (Figs. 1 and 2). Antiapoptotic family members (BCL-2, BCL-XL, MCL-1, BCL-W, and A1) contain all four BH domains (with the exception of MCL-1 and A1) and neutralize BAX-like and/or BH3-only family members, thereby preventing BAX/BAK oligomerization and pore formation (128, 142) (Figs. 1 and 2).

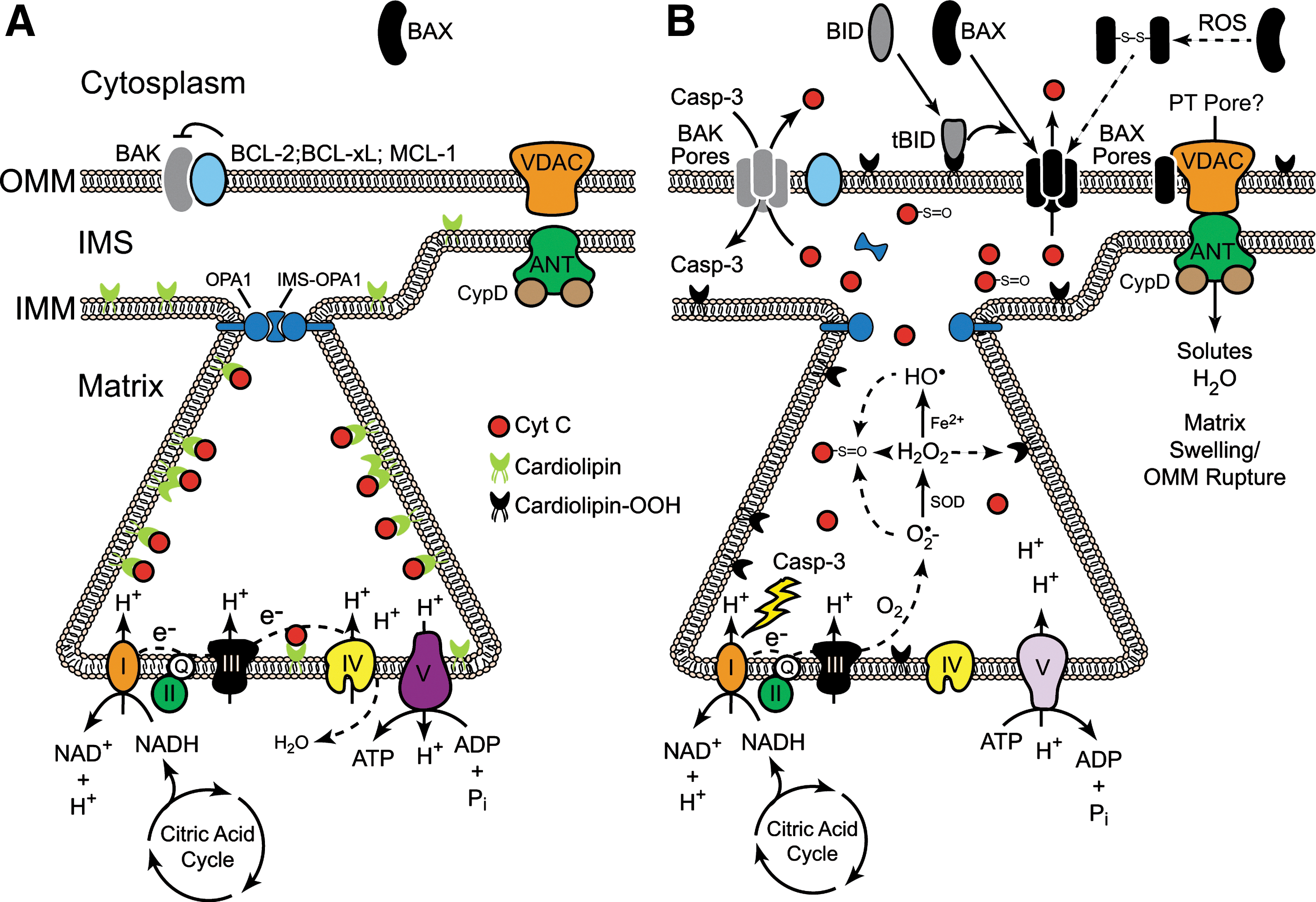
In terms of the direct effects of ROS on BCL-2 family members, hydrogen peroxide (H2O2) reportedly induces BAX dimerization through direct formation of a Cys-62/Cys-126 disulfide bond that promotes its translocation from the cytoplasm (where it normally resides) to the OMM (30) (Fig. 2B). However, in most cases, oxidative stress alters the function of BCL-2 family members by regulating the kinases that phosphorylate them. Broadly speaking, ROS are capable of activating p38 mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and Akt pathways, depending upon the context (132). However, a detailed discussion of all the mechanisms whereby ROS alter kinase function, and consequently BCL-2 function, are beyond the scope of the current review. Suffice it to say, these kinases have established roles in the regulation of many pro- and antiapoptotic BCL-2 family members, often converging upon them and regulating their expression or function in highly complex ways, as in the case of MCL-1 regulation by ERKs, JNKs, and glycogen synthase kinase-3β (GSK-3β) (32, 53, 83, 86).
Cyt c and ROS
Oxidants are thought to play important roles in the release of cyt c from mitochondria following MOMP and in the ability of cyt c to stimulate formation of the apoptosome. However, cyt c release also disrupts the electron transport chain (ETC) and stimulates the production of ROS. In the next section, we discuss these mechanisms in greater detail.
Role of ROS in cyt c release
Cardiolipin is a unique phospholipid found in the inner mitochondrial membrane (IMM), where it associates with several enzymes required for oxidative phosphorylation, including cyt c (108, 109). Since mitochondria are the main intracellular source of ROS, especially at Complexes I and III, cardiolipin is directly exposed to relatively high concentrations of ROS in stressed mitochondria. Moreover, cardiolipin contains four typically unsaturated fatty acyl chains (compared with two in most phospholipids) and is therefore more vulnerable to oxidation. Cardiolipin-bound cyt c also serves as a cardiolipin-specific peroxidase that can oxidize cardiolipin in an H2O2-dependent manner (54). Once oxidized, cardiolipin dissociates from enzymes in the IMM, including cyt c, and becomes enriched in the OMM (91, 117). tBID appears to target the OMM, at least in part, due to its strong binding affinity for cardiolipin and then promotes BAX oligomerization, which ultimately leads to pore formation and cyt c release (43, 44, 66, 72, 73). Thus, in short, cyt c release is a cardiolipin-dependent process in which cardiolipin oxidation plays an important role (Fig. 2).
Notably, the vast majority of cyt c (>85%) is present within cristae—folded structures formed by the IMM—and it appears that crista junction opening (CJO) is required to liberate oxidized cyt c from the intra-cristae space into the IMS (115). Optic atrophy 1 (OPA1), a dynamin-related GTPase located on the IMM, regulates mitochondrial fusion and CJO. OPA1 is proteolytically cleaved, most likely by OMA1 and Yme1 (and possibly PARL), and the truncated form of OPA1 reportedly oligomerizes with noncleaved OPA1 to form a complex at the crista junction that regulates its “tightness” (29, 33, 36, 48, 97, 121). Remarkably, CJO and MOMP are separable but required events for cyt c release and apoptosis. Indeed, while tBID and other BH3-only proteins induce the disassembly of OPA1 complexes in a BAX/BAK-dependent manner, unlike MOMP, CJO does not require oligomerization of BAK. Moreover, cells expressing a disassembly-resistant OPA1 mutant (Q297V) undergo normal MOMP, but fail to release significant amounts of cyt c (140). Finally, it is worth noting that some antiapoptotic BCL-2 family members, once thought to reside only in the OMM, have recently been observed in the IMM and appear to regulate mitochondrial fission/fusion dynamics, cristae ultrastructure, membrane potential and/or F1F0-ATP synthase activity (3, 25, 90). Thus, collectively, it appears that cyt c release occurs through a two-step process: in the first step, CJO, along with cardiolipin oxidation, allows for cyt c to be released from the intra-cristae space into the IMS; and in the second step, cyt c is released into the cytoplasm through an oligomerized BAX/BAK pore in the OMM (88, 140) (Fig. 2B).
While formation of a BAX/BAK pore in the OMM is the most widely accepted model for MOMP, an alternative model involves BAX/BAK-dependent activation of a permeability transition pore (PT pore), comprised of the voltage-dependent anion channel (VDAC), the adenine nucleotide transporter (ANT), and cyclophilin D (Fig. 2). Unlike the OMM, the IMM is charged and impermeable to water and solutes, but in response to certain proapoptotic stimuli, the PT pore opens, resulting in catastrophic matrix swelling, rupture of the OMM, and ultimately cyt c release (40) (Fig. 2B). Importantly, most well-known inducers of the permeability transition involve oxidative stress and/or the influx of Ca2+ (70). While there is little debate regarding the permeability transition per se, there is significant disagreement as to the existence of the PT pore (as defined) and its role in cyt c release, as well as the mechanisms whereby pro- and antiapoptotic BCL-2 family members regulate the permeability transition. Indeed, many studies invoke a role for the PT pore in oxidative stress-induced apoptosis, and both BID and BAX reportedly interact with the PT pore, promote its formation, and/or regulate its opening (74, 82, 107, 118, 119, 145). However, other studies have demonstrated that cells deficient in all isoforms of VDAC, ANT or cyclophilin D are still capable of releasing cyt c and undergoing apoptosis in response to most stimuli, though some do show defects in the permeability transition (7, 65). Thus, the specific mechanisms underpinning the permeability transition and its role in MOMP remain unresolved.
Direct effects of ROS on cyt c
The notion that the redox state of cyt c affects apoptosis is also controversial. On the one hand, replacing iron in the heme group of cyt c with copper or zinc dramatically inhibits its capacity to transfer electrons, but only partially inhibits its ability to induce apoptosome formation in vitro (63). Cyt c is also rapidly reduced in cytosolic extracts and in the cytoplasm of stressed cells following MOMP (46, 104), implying that the redox state of cyt c has no major impact on apoptosome formation or apoptosis. Other studies, however, suggest that only oxidized cyt c (Fe3+) possesses pro-apoptotic activity (13, 89, 127). This controversy notwithstanding, it is clear that ROS can directly modify specific amino acids in cyt c (Fig. 3A). Superoxide anion (O2 •−), H2O2, hydroxyl radicals (HO•), and peroxynitrite (ONOO−) oxidize methionine residues, particularly Met-80 (26, 61, 87, 129) (Fig. 3B), and singlet oxygen (1O2) not only oxidizes the ferrous form of cyt c, but also modifies several residues including His-26, His-33, Met-65, Met-80, and Phe-82 (60) (Fig. 3A). Importantly, oxidative modification of Met-80 dissociates it from its heme group and inhibits cyt c-dependent apoptosome formation (42, 78, 79, 127). Cyt c is also susceptible to nitration by ONOO- at Tyr-67, Tyr-74, and Tyr-97 (Fig. 3A), resulting in disruption of the heme iron-Met-80 bond (2, 21, 42), and NO-dependent nitration of Tyr-46 (human), and Tyr-48 (Fig. 3A) reportedly causes cyt c to assemble into a nonfunctional apoptosome (39). Finally, in addition to generating ROS, some redox-cycling quinones have been shown to directly arylate key lysines in cyt c, including Lys-72, thereby disrupting its interaction with Apaf-1 and preventing formation of the apoptosome (34, 35, 47, 62).

Impact of cyt c release on ROS production
In general, when cyt c is released, electron transport is disrupted, and free electrons are donated to oxygen to produce O2 •− (18) (Fig. 2B). Studies have shown, however, that generation of O2 •− precedes the loss in IMM potential (ΔΨ m) (18), and that even following cyt c release, a fraction of cyt c can diffuse back across the permeabilized OMM, reengage the ETC, and maintain some level of ETC function (138). Mitochondria may also utilize ATP, generated through glycolysis, to help maintain a near normal ΔΨ m by allowing the F1F0-ATP synthase (complex V) to operate in reverse, hydrolyzing ATP to help generate a proton gradient (137) (Fig. 2B). Indeed, in many cellular systems, ΔΨ m can be maintained for many hours, so long as caspases are inhibited and not allowed to attack the ETC. If caspase-3 gains access to the ETC, it can cleave the p75 NDUFS1 subunit in complex I, which in turn disrupts electron transport and promotes ROS production (102) (Fig. 2B). Other more recent studies argue that caspases-9 and -3 may have opposite effects on the ETC, in that caspase-9 activity promotes ROS production, whereas caspase-3 activity inhibits it (22). These studies underscore an important question, as it relates to the intrinsic pathway: at what point can a cell no longer survive, or more succinctly, when is the point of no return? Is it MOMP, loss of ETC function, loss of ΔΨ m, apoptosome formation, or effector caspase activation? Antiapoptotic BCL-2 family members inhibit all of these events and promote clonal growth, implying that one of these downstream events is likely the final lynch pin to guarantee cell death.
The Apoptosome and ROS
In this last section, we will provide basic information on the structure and function of Apaf-1, caspase-9, and the apoptosome. Where applicable, we will also discuss how this complex and its individual components are regulated directly and/or indirectly by ROS.
Structure and function of Apaf-1
The adapter protein Apaf-1 possesses an N-terminal CARD domain, through which it binds to caspase-9; a nucleotide binding and oligomerization domain (NBD/NOD) that contains a AAA+ ATPase cassette, with Walker's A and B boxes that bind to (d)ATP and Mg2+, respectively; and a series of C-terminal WD40 repeats that form seven and eight-blade ß-propellers (101, 103, 149) (Fig. 4). In normal cells, monomeric Apaf-1 is likely present in an autoinhibited state, bound to (d)ATP (50, 101, 103). Cyt c, following its release from mitochondria, is thought to bind Apaf-1 between its ß-propellers and induce (d)ATP hydrolysis, providing the energy necessary to adopt a semi-open conformation (10, 49, 57). At this stage, in the absence of nucleotide exchange, cyt c-bound Apaf-1 is prone to aggregation that results in the formation of inactive apoptosome complexes (57) (Fig. 4). Thus, nucleotide exchange is critical, and a nucleotide exchange factor complex, composed of PHAPI, Hsp70, and cellular apoptosis susceptibility (CAS), has recently been identified for Apaf-1 (58). Following nucleotide exchange, the NBDs are exposed, and Apaf-1 undergoes proper oligomerization to form the functional complex (19, 111, 150). Based upon cryo-electron microscopy and structural modeling techniques, the functional complex appears to contain seven Apaf-1 molecules, arranged in a wheel-like structure with the NBDs forming a central hub and the CARDs forming a ring that is situated directly above the hub (143, 144). The WD-40 repeats then form spokes that radiate outward from the hub and end in a two-pronged fork that stabs cyt c (143, 144) (Fig. 4). Once formed, the Apaf-1 apoptosome then sequentially recruits and activates the initiator caspase-9 and the effector caspase-3 (17).
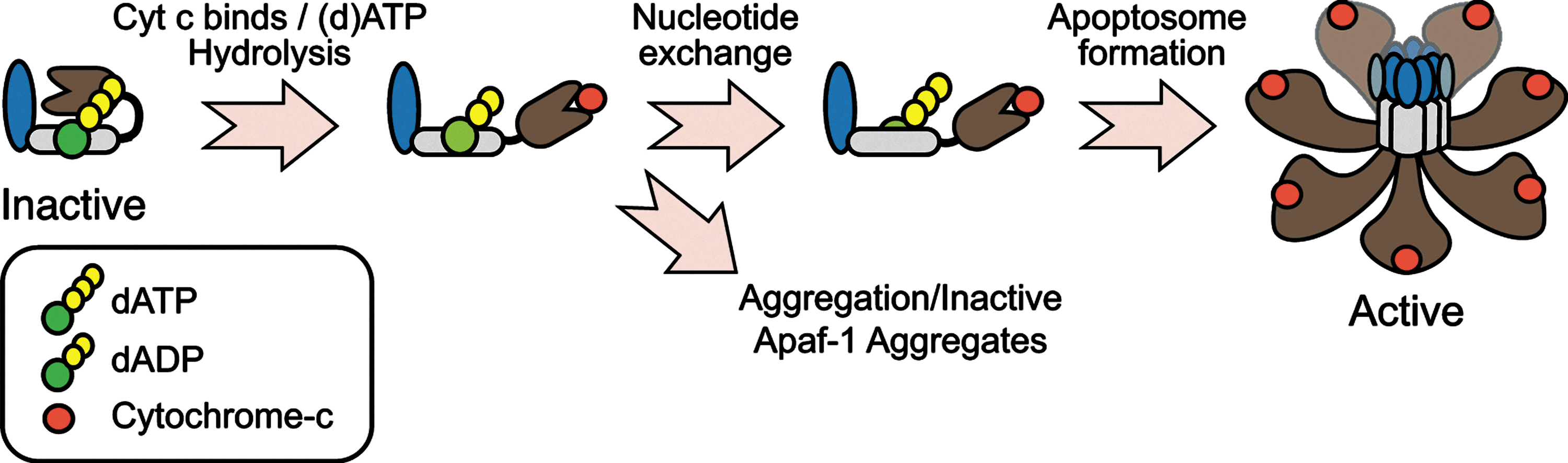
Direct and indirect effects of ROS on Apaf-1
How or if ROS directly regulate Apaf-1 remains unclear. However, in one study, ROS were found to be required for Fas-mediated apoptosome formation. Using in vitro translated Apaf-1 and caspase-9 (and purified cyt c), the authors reported that apoptosome formation was inhibited by the addition of antioxidants or reducing agents, and that oxidation of Apaf-1 in particular was essential for the activation of caspase-9 in their reconstituted system (112). In complete contrast, others report that ROS can indirectly inhibit apoptosome function through the activation of certain kinases. For example, one study found that JNKs, activated in response to ROS, bound to Apaf-1 and cyt c in a catalytically inactive ∼1.4–2.0 MDa complex, which the authors referred to as a “preapoptosome complex” (133). Interestingly, we previously reported the formation of active ∼700 kDa and inactive ∼1.4 MDa apoptosome complexes following cyt c/dATP activation of lysates (19). Given that similar inactive complexes are formed as a result of inadequate nucleotide exchange (57), it will be interesting to determine if JNK binding (or perhaps JNK-dependent phosphorylation of Apaf-1) interferes with nucleotide exchange. More recently, 90-kDa ribosomal S6 kinase (Rsk) has been reported to phosphorylate Apaf-1 at Ser-268 and Ser-357 (59). Phosphorylation at these sites results in Apaf-1 sequestration by 14-3-3ɛ and decreases cellular responsiveness to cyt c. Though not examined in the study, H2O2 reportedly activates Rsk (1), raising the possibility that ROS might suppress apoptosis in certain contexts via RSK-mediated phosphorylation of Apaf-1.
Structure and function of caspase-9
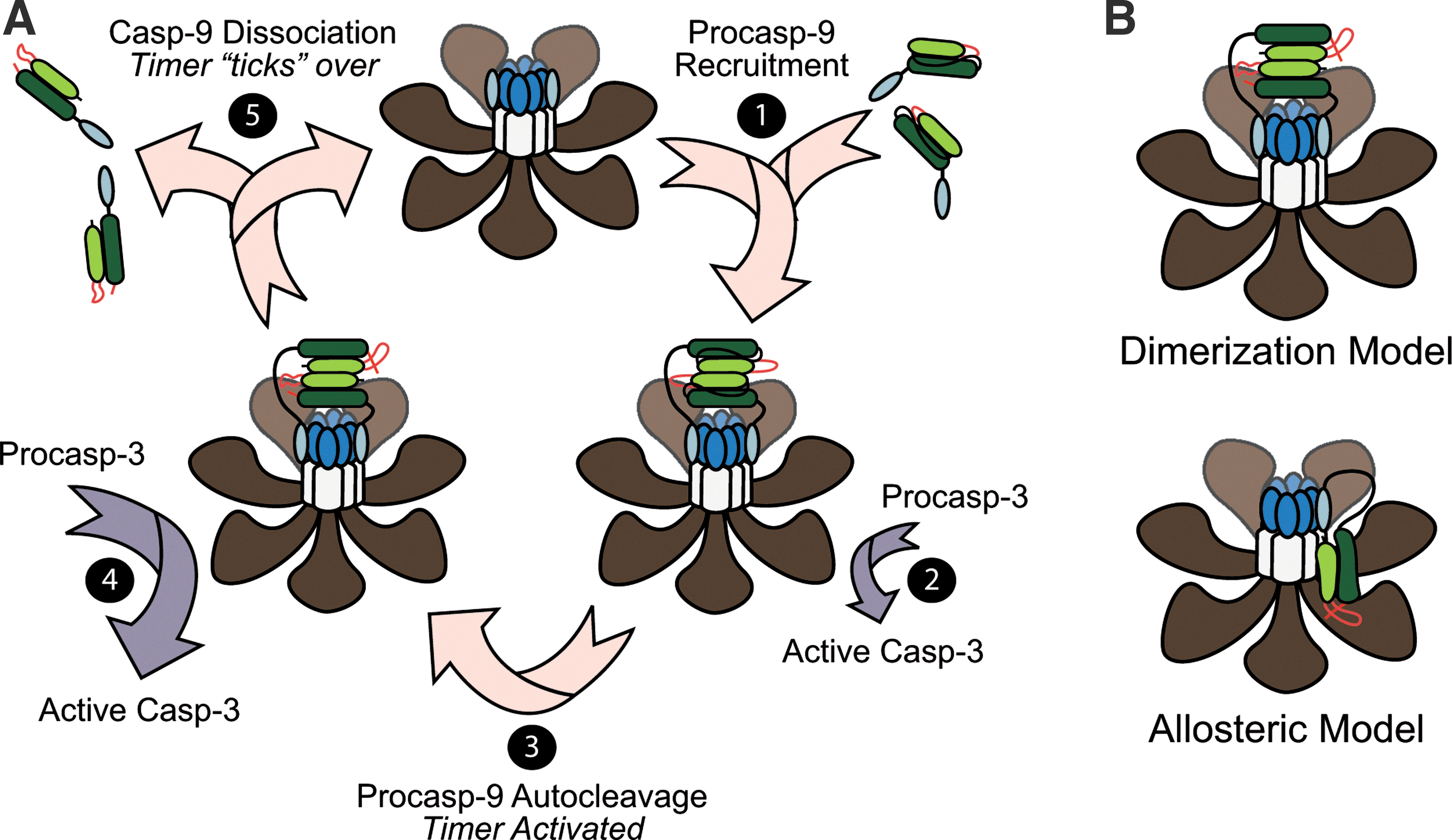
Procaspase-9 is generated as a single-chain 46 kDa protein with an N-terminal prodomain (CARD), followed by large (∼20 kDa) and small (∼12 kDa) subunits, connected via an intersubunit linker (16) (Figs. 5 and 6). Following apoptosome formation, procaspase-9 is recruited to the complex through CARD–CARD interactions with Apaf-1 (95). Procaspase-9 then undergoes autocatalytic processing within its intersubunit linker (Asp-315) to generate a two-chain (p35/p12) enzyme (122) (Fig. 5A). Importantly, unlike other initiator caspases, the prodomain in caspase-9 is generally not removed at any step in its activation. Thus, caspase-9 is unique in that it must remain bound to its caspase-activating complex, the apoptosome, in order to sustain significant catalytic activity (17, 105, 125). Once formed, the Apaf-1-caspase-9 complex activates the downstream effector procaspases-3 and -7 (17, 105, 141). In some cases, active caspase-3 can then feedback on and cleave unbound procaspase-9 or p35/p12 caspase-9 at Asp-330 to generate p37/12 or p35/p10 caspase-9 proteins, respectively (15, 123, 124, 151).

Apoptosome-bound caspase-9: an active monomer or a dimer?
One of the more highly debated topics in caspase biology has been the question of how caspase-9 is activated following its interaction with the apoptosome? Two models have been proposed: in the currently more well-accepted dimerization model, the apoptosome serves merely as a structural platform to recruit caspase-9 and increase its local concentration, thereby promoting its dimerization and subsequent activation (12, 92) (Fig. 5B). Indeed, unlike effector caspases, which are constitutive dimers, caspase-9 possesses a relatively low affinity for itself and is normally present as a monomer in solution. However, crystal structures of caspase-9 strongly suggest that it is a dimer, and mutations along the proposed dimerization interface kill the enzyme's activity (100) (Fig. 6). In addition, caspase-9 can be activated by artificially enforcing its dimerization (12, 92), and if one swaps the prodomain of caspase-8 for the CARD in caspase-9, the apoptosome then activates caspase-8, arguing that there is nothing special about the apoptosome, other than its ability to concentrate caspases (12, 92).
Proponents of the competing holoenzyme model argue instead that the apoptosome functions as a positive allosteric regulator of caspase-9, inducing conformational changes in the enzyme that are necessary for its activation (24, 105) (Fig. 5B). They insist that the seven-fold (rather than eight-fold) symmetry of the apoptosome is inconsistent with the idea that caspase-9 would form dimers and point out that caspase-9 has never been shown to dimerize in a wild-type Apaf-1-caspase-9 apoptosome complex. Moreover, while enforced dimerization of truncated caspase-9 (artificially lacking its prodomain) activates the enzyme, the observed activity appears to be significantly lower than that observed in a complex reconstituted with full-length Apaf-1 and caspase-9 (24). It is also notable that swapping the putative dimerization domain in caspase-9, with that found in caspase-3, produces a constitutive caspase-9 dimer, which is structurally indistinguishable from the wild-type enzyme. However, this caspase-9 dimer still exhibits significantly less activity than apoptosome-bound caspase-9 (24). Finally, a very recent cryo-electron microscopy structure of the apoptosome suggests that a single catalytic subunit of caspase-9 is bound near the hub of the apoptosome (143) (Fig. 5B). Thus, future studies are needed to assess not only the stoichiometry and dimerization status of caspase-9 within the apoptosome, but their requirements for activity.
The apoptosome as a molecular timer for caspase activation
We have recently demonstrated that procaspase-9 possesses higher affinity for the apoptosome and can displace processed p35/p12 caspase-9 from the complex, thereby facilitating a continuous cycle of procaspase-9 recruitment/ activation, processing, and release from the complex (75) (Fig. 5A). Importantly, we found that procaspase-9 undergoes autoprocessing so rapidly upon its recruitment to the complex that the proform of the enzyme contributes little to the activation of procaspase-3. Consequently, our results led us to the conclusion that the apoptosome functions as a proteolytic-based “molecular timer” (Fig. 5A). In our proposed model, the intracellular concentration of procaspase-9 sets the overall duration of the timer, the autoprocessing of procaspase-9 activates the timer, and the rate at which p35/p12 caspase-9 dissociates from the complex (and thus loses its capacity to activate procaspase-3) dictates how fast the timer “ticks” over (75). Our in vitro studies suggest that noncleavable caspase-9 should disengage the timer and result in an apoptosome complex that, once formed, cannot be “turned off” (75). However, in cellular systems it is important to note that processing of procaspase-9 to p35/p12 caspase-9 also produces a form of the enzyme that can be inhibited by X-linked IAP (XIAP), a potent endogenous inhibitor of caspases-9, -3, and -7 (15, 124). Indeed, XIAP specifically binds to a neo-epitope on the N-terminus of the small p12 subunit of processed caspase-9. Thus, in future studies, it will be important to determine the interplay between XIAP and the molecular timer, as well as the relative importance of these two mechanisms for regulating apoptosome function in vivo.
Direct and indirect effects of ROS on caspase-9
Regarding the direct effects of oxidative stress on caspases, several studies have shown that caspase-3 activation and/or activity can be negatively regulated by oxidants. In fact, several cysteines in caspase-3, including Cys-73, Cys-220, and its catalytic cysteine, Cys-163, undergo oxidation, nitrosylation, and/or glutathionylation (11, 51, 69, 76, 84, 106, 147). The reported effects of ROS on caspase-9, however, are more inconsistent. One study found that exposure of caspase-9 to H2O2 promoted its interaction with Apaf-1, through a disulfide bond involving Cys-403 (152). Another study reported the presence of active homodimerized caspase-9 within H2O2-treated mitochondria and suggested that multiple cysteines (including Cys-403) were sites of disulfide bond formation (55) (Fig. 6). While these studies are intriguing and warrant further investigation, some aspects are surprising. For example, a few studies have reported the presence of caspase-9 in mitochondria (67, 126, 148), but procaspase-9 does not possess a typical mitochondrial targeting sequence and others find that most of the caspase-9 that localizes to mitochondria does so following MOMP (23). The purpose of mitochondrial caspase-9 is also unclear, as it would presumably be released from the IMS along with cyt c during apoptosis. Given the consensus that far more procaspase-9 exists in the cytoplasm than in mitochondria, one might expect that apoptosome-driven activation of cytoplasmic caspase-9 would be far more active and plentiful. Finally, Cys-403 is one of the five amino acids that constitute the dimerization domain in caspase-9 (Fig. 6), and mutations along this interface often kill enzyme activity (100, 120). By contrast, other studies suggest that ROS inhibit caspase-9 activation. H2O2 reportedly blocks caspase-9 activation through oxidation of cysteine residues, including its catalytic cysteine (Cys-287) (Fig. 6), and this process appears to be iron-dependent, implying that Fenton chemistry and the production of HO• is involved (8). Similar to caspase-3, the caspase-9 active site cysteine is also susceptible to S-nitrosylation (131), and nitric oxide donors have been shown to block formation of the Apaf-1-caspase-9 complex by disrupting normal CARD-CARD interactions (146).
In addition to the direct effects of oxidative stress on caspase-9, there are also a few potential indirect effects, many of which involve the activation of kinases. Over the last decade, there have been several reports that phosphorylation of caspase-9 regulates its activity. Most prominently, human caspase-9 is phosphorylated at Thr-125 by ERK2, DYRK1A, and CDK1/cyclin B1 kinases during survival signaling and mitosis, and phosphorylation at this site directly inhibits the activity of procaspase-9 following its recruitment to the apoptosome (4, 5, 80, 81, 116). PKCζ and Akt also phosphorylate human caspase-9 at Ser-144 and Ser-196, respectively, leading to its inhibition (14, 20), but interestingly, neither of these sites is conserved in the mouse. Finally, and perhaps most surprising, c-Abl appears to activate caspase-9 through phosphorylation at Tyr-153 (98). It remains unclear how any of the aforementioned phosphorylation sites regulate caspase-9 activity, since none are located in the active site or putative dimerization domain. Thus, in the coming years, it will be exciting to determine, mechanistically, how phosphorylation impacts caspase-9 structure and function, as well as to test the importance of phosphorylation in vivo. For additional insight into ROS-independent regulation of the Apaf-1-caspase-9 apoptosome, the reader is directed to the following reviews (16, 113).
Other effects of ROS on apoptosis in vitro and in vivo
As previously noted, in light of the pleiotropic effects of ROS on apoptosis, we have chosen to focus the majority of this review on the more immediate effects of ROS on the intrinsic pathway. However, we wish to emphasize that oxidative modification of DNA, proteins, and lipids can impact a variety of signaling pathways, both upstream and downstream of the intrinsic pathway. For example, ROS can activate the kinase ATM, either through direct modification or as a consequence of DNA damage, resulting in the activation of p53, a transcription factor that drives expression of proapoptotic BCL-2 family members, including NOXA, PUMA, and BAX (27, 45, 68, 85, 136). ROS also regulate the upstream production of sphingolipids through direct oxidative modification of the acidic sphingomyelinase at Cys-629, or through oxidation of glutathione (GSH), which normally serves as an endogenous inhibitor of neutral sphingomyelinases (71, 96). In an elegant biochemical study, Green and colleagues have recently shown that two sphingolipid metabolites, sphingosine-1-phosphate and hexadecenal, directly promote BAK and BAX-induced MOMP, respectively (28). ROS may also promote apoptosis via the extrinsic pathway through upregulation of death receptors or by serving as intermediates in the activation of kinases, such as JNKs, which promote MOMP through direct phosphorylation of BCL-2 family members (52, 53, 56, 93, 110, 130, 139).
How ROS mediate apoptosis in vivo has been more challenging, in part due to difficulties in monitoring cell and tissue-specific generation of ROS, as well as isolating oxidatively-modified macromolecules, which can be short-lived or artificially introduced from the isolation procedure. There is little doubt, however, that ROS play important roles in apoptosis in vivo, as transgenic and knockout mice for various antioxidant genes, including superoxide dismutases (SOD1 and SOD2), catalase, and glutathione peroxidases, exhibit the expected decreases or increases in apoptosis in response to ischemic injury and other pro-death stimuli (37, 38, 41, 64, 94, 99, 134, 135). Moreover, the recent development of various genetically-encoded fluorescent probes and imaging techniques can now be used to monitor the production of ROS in a spatiotemporal manner (6, 9, 31, 77). Thus, it should be possible to establish the relative importance of ROS-induced apoptosis versus inflammation, mutagenesis, etc. in the development of disease and to determine the relative importance of a given oxidative modification.
Concluding Remarks
After many years of research, the intrinsic pathway remains an important area of investigation. Each step in the pathway is regulated by various modulators and posttranslational modifications. However, while the importance of many of the genes in this pathway (e.g., BCL-2, cyt c, Apaf-1, caspase-9, and XIAP) have been verified in transgenic and knockout animal models, particularly during development, many of these gene products and their post-translational modifications, including oxidative modifications, have not been thoroughly vetted in animal models of disease. This is certainly true in cancer where most studies have been limited to hematopoietic malignancies. Thus, in the future, it will be critical to fully characterize in detail not only how the apoptosome is formed and regulated at the biochemical level, but to assess its importance in vivo.
Footnotes
Acknowledgments
This work was supported in part by NIH Grants R01CA129521 and R01GM096101.
Author Disclosure Statement
No competing financial interests exist.