
Abstract
Introduction
Hypofunction of the glutamatergic system, in particular of the N-methyl-
Investigations using positron-emission tomography (PET) have shown that ketamine administration acutely increased metabolism in the frontal regions of the brain, and that this increase was correlated with psychotic-like symptoms in healthy volunteers (231) as well as in rodents (43). Principal neurons in the prefrontal cortex (PFC) receive inputs from glutamatergic afferents projecting not only from intracortical structures but also from the thalamus, as well as they receive dopaminergic afferents from centers in the midbrain (73, 201); postsynaptically, principal neurons express dopamine receptors and NMDARs that make synapses bidirectionally (32, 61, 87, 130, 199), with relevant consequences for neuronal output (see 249). Dopaminergic neurons are in turn downstream to glutamatergic neurons, either directly or via GABAergic interneurons, mechanisms through which NMDAR antagonists can influence dopamine release (see 31). Thus, it has been suggested that elevated baseline levels of dopamine observed in schizophrenia may be secondary to hypoglutamatergia. In support of this hypothesis, NMDAR antagonists can enhance spontaneous and amphetamine-induced release of dopamine (159). In addition, acute application of NMDAR antagonists to nonhuman primates was shown to increase glutamate and dopamine release in PFC, leading to cortical disinhibition (220, 226). This is due to an enhanced sensitivity to antagonists of inhibitory GABAergic cells, specifically parvalbumin-positive (PV+) fast-spiking interneurons (85, 184). In sum, these facts support a multifactorial view of schizophrenia, involving interactions among the glutamatergic, GABAergic, and dopaminergic systems.
In the cerebral cortex, multiple types of GABAergic inhibitory interneurons are present that differ in their morphology, electrophysiological properties (e.g., fast-spiking and low-threshold spiking), synaptic connectivity, and expression of specific markers (e.g., parvalbumin [PV], somatostatin, and vasointestinal peptide) (103, 151). One type of GABAergic neurons, the PV-expressing fast-spiking interneurons, provides strong perisomatic inputs onto pyramidal neurons, and therefore can strongly suppress their activity (41, 158, 244). These cells are critical for the synchronization of the neural network through generation and synchronization of gamma-frequency oscillations (28, 210), which are critical for cognitive processes and are impaired in neuropsychiatric disorders (reviewed in 12, 149).
In rodents and nonhuman primates, prolonged treatment with NMDAR antagonists leads to significant alterations of the PV+ neuron phenotype, including decreased expression of PV and glutamic acid decarboxylase GAD67, the major gamma-aminobutyric acid (GABA)-synthesizing enzyme in the cortex (14, 15, 42, 107, 193). A similar loss of GAD67 expression in PV+ neurons has been consistently observed in the PFC of schizophrenic patients (222, 230), supporting the hypothesis that a dysfunction of the PV+ fast-spiking interneurons may be a core feature of the disease (135).
Thus, insights into the origins of schizophrenia could arise from understanding the mechanisms by which the exposure to NMDAR antagonists leads to prolonged alterations of PV+ neurons and their functions in cortical circuits. A key discovery was that ketamine exposures produce increased oxidative stress in the brain through activation of Nox2-dependent reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox2) (14, 212), which activates a cascade of events that ultimately leads to the sustained alteration of PV+ neuronal function. Oxidative stress has been also found to be a mediator of neurochemical and behavioral alterations in other rodent models of schizophrenia, such as social isolation from rearing (198), and in rodents with diminished glutathione (GSH) production (reviewed in 48).
In this review, we will first briefly summarize the literature describing the presence of Nox2 in neurons. We will then discuss the function of NMDARs, their expression and development in the brain, and the acute and chronic consequences of exposure to NMDA antagonists. Next, we will focus on the role of Nox2 in the propsychotic effects of NMDAR antagonists and redox dysregulation. Finally, we will explore possible consequences of oxidative dysregulation of the inhibitory system for the pathophysiology of schizophrenia.
Expression of Nox2-Dependent NADPH Oxidase in Neurons
The first NADPH oxidase described, gp91phox or Nox2-dependent NADPH oxidase (Nox2), was identified in phagocytic cells as the enzyme complex responsible for the respiratory burst, an essential host response to microbial invasion (reviewed in 125). Nox2 and other members of the family are expressed in numerous tissues and cells types, including neurons (211). At least six proteins form part of the active Nox2-complex: the membrane flavocytochrome b588 core complex consisting of gp91phox and p22phox subunits and four cytoplasmic components (p40phox, p47phox, p67phox, and Rac1/2) (Fig. 1). Nox2 is inactive in resting cells, but becomes activated to generate superoxide upon assembly of the core complex with the cytoplasmic components, which occurs upon phosphorylation of p47phox (125). All the subunits required for an active Nox2 complex are expressed in neurons (reviewed in 152, 211), where activation of this complex seems to follow similar mechanisms as those described in non-neuronal cells (221). Interestingly, a specific effect of interleukin-6 (IL-6) in the activation and induction of Nox2 expression in neurons was recently demonstrated (15).

NMDAR and Its Antagonists
Glutamate, the main excitatory neurotransmitter, activates three different classes of ion-gated channels: α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors, kainate receptors, and NMDARs, named after their preferred synthetic agonists (142). The NMDAR has unique features that distinguish it from other ion-gated glutamate receptors (as illustrated in Fig. 2): first, it requires the binding of a co-agonist (glycine or
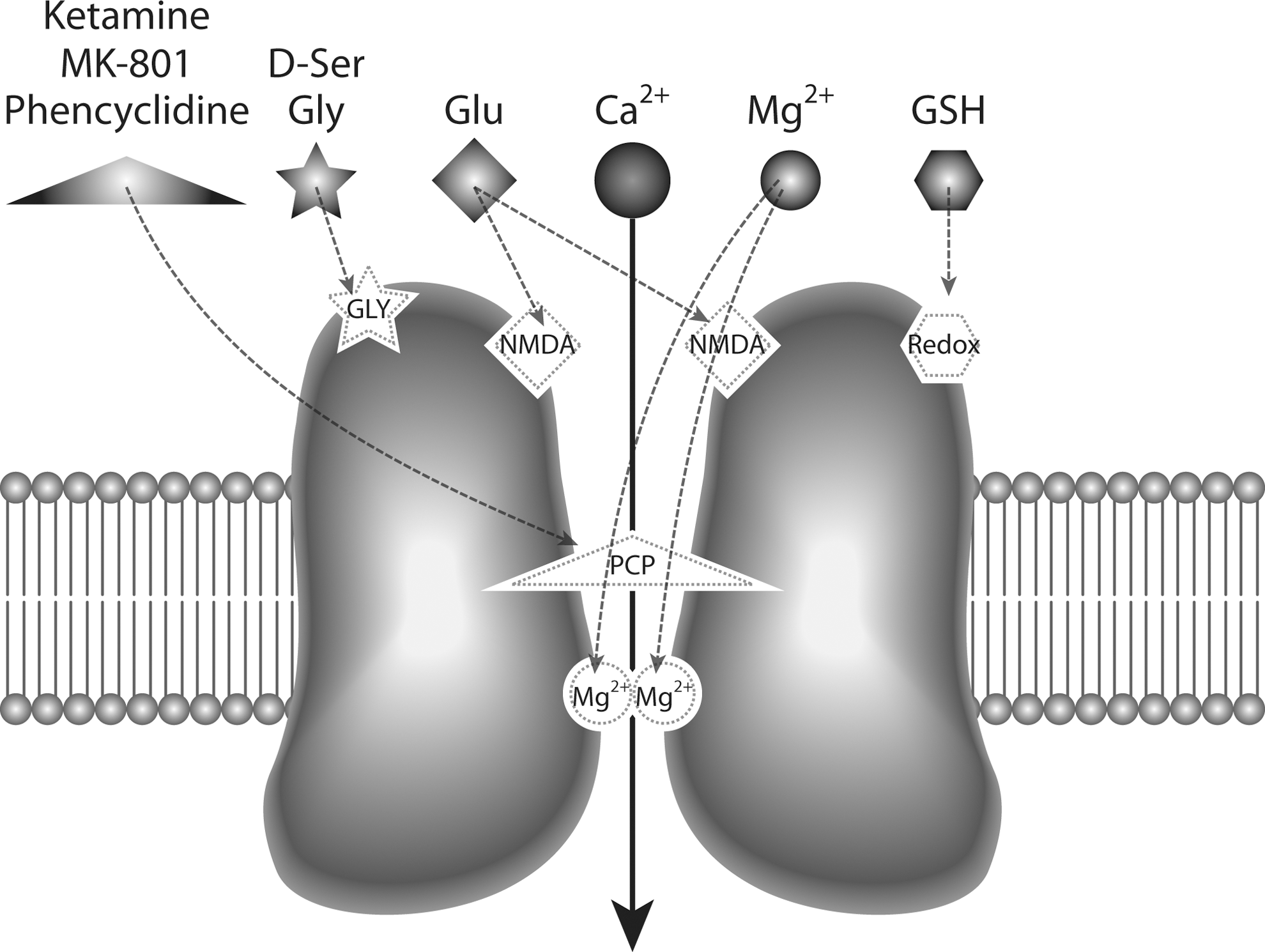
The NMDAR is composed of several subunits, namely the glycine-binding core NR1, the glutamate-binding NR2, including NR2A-D, and the recently discovered glycine-binding NR3, which includes NR3A and NR3B (35, 48, 56, 144). The presence of NR1 is required for forming functional NMDARs, and this subunit combines with several NR2 and/or NR3 subunits to form heteromeric receptors (2, 40, 218). The biophysical and pharmacological properties of NMDARs depend on the subunit composition, which varies across brain regions and is subject to change during development (164).
The NMDAR is highly sensitive to redox modulation through a redox-sensitive site (Fig. 2) (1, 38, 39, 112) and decreases in the main antioxidant in the brain, GSH, or reduced activity of GSH-peroxidase can lead to oxidized hypofunctional NMDARs (125, 211). Mutagenesis studies have revealed three distinct pairs of cysteine residues on NMDAR subunits—Cys79/Cys308 and Cys744/Cys798 on the NR1 subunit and Cys87/Cys320 on the NR2A subunit—which can be specifically oxidized or reduced by agents that affect the redox state of the NMDAR (38, 112). As a result, the formation and destruction of disulfide bonds have direct consequences upon NMDAR function: while reducing agents, such as dithiothreitol and GSH increase NMDA-evoked current, oxidizing agents that promote disulfide formation, such as 5,5′-dithio-bis(2-bisnitrobenzoic acid), attenuate it (112). The oxidation status of this redox site can affect the regulation of these receptors by spermine and protons, as well as the inhibition mediated by the high-affinity Zn2+ site (139). In particular, NMDARs composed of NR1/NR2A subunits were shown to undergo a rapid and highly reversible current potentiation by sulfhydryl redox agents, including GSH, acting on a specific redox site in NR2A (112). In contrast, the effects of reducing agents on NMDARs containing NR2B and NR2C are very slow and may not be reversible unless exposed to oxidizing agents (112).
Several selective and nonselective antagonists have been used to modify NMDAR function (Table 1). Some of these drugs act as competitive antagonists, competing with glutamate or glycine at the NMDAR agonist-binding sites, and therefore can be rapidly displaced from the receptors by high local concentrations of glutamate and glycine. On the other hand, noncompetitive antagonists, such as PCP, ketamine, and MK-801, directly block the NMDAR channel when it opens (Fig. 2), and thus can affect normal physiological functions when administered to animals or humans, regardless of local concentrations of glutamate and glycine, resulting in robust cognitive alterations and psychotic events.
Listed are some of the available compounds that have been used to modify NMDAR activity in vitro and in vivo [modified from (252)]. Relative affinities are specified in parenthesis.
NMDAR, N-methyl-
Expression and Function of NMDARs in Cortical Neurons
Excitatory synaptic transmission among neurons is achieved by release of glutamate from presynaptic neurons, which generates excitatory postsynaptic potentials (EPSPs) in postsynaptic neurons through activation of glutamate receptors. NMDARs are known to mediate EPSPs in multiple regions and cell types in the brain. Therefore, it is not surprising that NMDAR antagonists inhibit EPSPs in cortical pyramidal neurons in vitro and in anesthetized preparations (37, 84, 100). Interestingly, however, when administered in vivo, these drugs cause cortical activation instead of suppression in human subjects (122, 231) and rodents (92). Recent studies have suggested that these apparent contradictory effects may result from decreases in inhibitory neuron activity, due to a predominant effect of the antagonists on NMDARs present in cortical inhibitory neurons (85, 184). Disinhibition, in turn, leads to an increase in the firing rate of the majority of pyramidal neurons. The higher sensitivity of NMDARs expressed in interneurons to NMDAR antagonists may be due to a different composition in NR2 subunits (107, 246), a key factor for voltage-dependent Mg2+ blockade (90, 119, 161, 162). Although Mg2+ dependence of NR2 subunits is determined by a shared element located in the M2 domain of NR2 subunits (26, 163, 194), it has been shown that the M1–M4 segment contains subunit-specific determinants for Mg2+ block (117). This is possibly the reason why NR2A or NR2B is more sensitive to Mg2+ block as compared to NR2C- or NR2D-containing channels (161), and thus less prone to blockade by NMDAR antagonists. In the following sections, we summarize what is known about the subunit composition of NMDARs in the different neuronal types present in the cortex.
NMDARs in excitatory neurons
The NR1 subunit of NMDARs is widely expressed across all brain regions in rodents from birth (44, 161, 166), but expression of the NR2 subunit changes throughout postnatal life (127, 161, 203). In situ hybridization studies and protein analyses have shown that the newborn rodent cortex is enriched in NR2B and NR2D subunits, and that the expression of these two subunits, especially NR2D, progressively decreases throughout postnatal life. These studies have also shown that the expression of NR2A and NR2C increases during postnatal development. Although these results were obtained at the tissue level, the high proportion of excitatory neurons in the cortex (∼80%) suggests that they relate to the pattern of subunit expression in principal neurons.
The most visible consequence of this developmental modification is the progressive change from synaptic NMDARs containing predominantly NR1/NR2B/NR2D to those containing NR1/NR2A subunits (11, 243). Such changes in the subunit composition may crucially affect the permeability of NMDARs, since NR2A or NR2B subunits have larger conductance and higher sensitivity to blockade by Mg2+ than receptors containing NR2C or NR2D subunits (44, 48).
NMDARs in inhibitory neurons
Even within the same brain region, different cell types can express different combinations of NMDAR subunits, reflecting different roles in the neuronal network. Cortical inhibitory neurons expressing the neurotransmitter GABA comprise diverse subtypes that can be grouped according to the expression of calcium-binding proteins and specific peptides, as well as by their morphology and electrophysiological properties (103, 248). The expression of NMDARs is evident in several types of inhibitory neurons (72), where they control subthreshold calcium dynamics and participate in long-term synaptic plasticity (116). One particular anatomical subtype of cortical inhibitory neurons, those expressing PV, tightly regulates the activity of principal cells by providing them with strong perisomatic inhibition and can thus control the activity of neural networks physiologically, including the generation and synchrony of network rhythms in the gamma-frequency band (28, 210). Because physiological gamma-oscillations are correlated with cognitive mechanisms, including attention and working memory, it is currently thought that the perturbation of NMDAR function in PV+ neurons may be responsible for cognitive impairments associated with psychiatric disorders (185, 228).
PV+ neurons in the rodent PFC express high levels of functional NMDARs during the first 3–4 postnatal weeks, and the activity of these receptors is necessary for the normal development of their characteristic fast-spiking (107, 236, 246, 252). Interestingly, NMDARs in these neurons have a different subunit composition than that found in neighboring pyramidal neurons, with NR2A/NR2B ratios of 5:1 in PV+ neurons and 1:1 in pyramidal neurons (107, 246). PV+ neurons also express NR2C subunits, which differentiate them from principal neurons at the same developmental age (246).
In fully matured PFC, the number of NMDAR-expressing PV+ neurons is considerably reduced, due to a dramatic downregulation of functional NMDARs in these neurons during late adolescence (191, 236). Although this seems in contradiction to the findings that NMDAR antagonism in adults produced a disinhibited state with hypoactive PV+ neurons and hyperactive pyramidal neurons (85, 184), there is evidence suggesting that a residual ∼30% of the PV+ neuron population still expresses NMDARs in the PFC when animals reach adulthood (236). Thus, it is possible that this subpopulation of NMDAR-expressing PV+ neurons could be sufficient to lead to disinhibition upon NMDAR antagonism, as suggested by computational modeling of cortical networks (232).
These studies suggested that different subtypes of PV+ neurons might be present in the adult PFC (for example, subtypes of basket and/or chandelier neurons) as occurs in the entorhinal cortex, where PV+ neurons in layer 2 express NMDARs and respond to NMDAR antagonists with altered gamma-oscillatory activity in adults (45, 102). Thus, alterations in the normal maturation of PV+ neurons in different brain regions at different times could lead to the differential maturation of the neuronal circuits involved in working memory and selective attention.
Use of NMDAR Antagonists to Model Schizophrenia-Related Behavior
The hypoglutamatergic theory of schizophrenia is based on the effects of exposure to the NMDAR noncompetitive antagonists PCP, ketamine, and MK-801, which constitute the best pharmacological models of schizophrenia in rodents, nonhuman primates, and humans (96, 173, 176). Acute exposures to these substances produce a classic psychotic episode in humans and hyperdopaminergia/hyperglutamatergia in animals as well as cognitive deficits, reminiscent of both positive and negative symptoms of schizophrenia. However, these effects are reversible and short lasting. Chronic or developmental exposures, on the other hand, reproduce enduring cognitive, neurochemical, and physiological alterations, even in absence of psychosis, as observed in schizophrenia.
Acute exposures
The discovery that the dissociative anesthetics PCP and ketamine are noncompetitive antagonists of the NMDAR (8, 95) brought new insights into the study of schizophrenia (reviewed in 96). These two drugs were known to produce a psychotic state in humans emerging from anesthesia (reviewed in 52), and were later shown to exacerbate psychosis in schizophrenic patients (91, 145). The undesirable propsychotic side effects of PCP prevented its use as an anesthetic, and its abuse as a street drug led to its prohibition [see (52) for recent review of the clinical history of PCP]. The synthesis of ketamine, an analog of PCP, and the demonstration of its potency as an anesthetic and its more manageable side effects have led to its continued use in humans (52). Nonetheless, the propsychotic effects are still observed in healthy volunteers (115, 124), and ketamine exacerbates psychosis in schizophrenia patients (123, 148).
Subanesthetic concentrations of ketamine produce psychotic-like positive symptoms in humans, as well as reductions in working memory and sustained attention performance, similar to the cognitive deficits observed in schizophrenia patients (115, 124, 229). Acute ketamine exposure can impair performance on tasks testing executive function in humans (114) and nonhuman primates (217), resembling executive functioning deficits that are associated with treatment-refractory aspects of schizophrenia (105).
Similar to its effects in humans, systemic administration of NMDAR antagonists in rodents produces deficits in spatial working memory, in reversal learning, and in sustained attention (99, 213). Exposure to PCP, ketamine, and MK-801 is therefore widely used in adult animals as acute pharmacological models to study behavioral and neurochemical disruptions relevant to schizophrenia (reviewed in 169). At subanesthetic doses, ketamine also induces a wide range of behavioral alterations in rodents, including disruption of prepulse inhibition of the startle reflex (150), working memory deficits (33, 182), reversal learning deficits (64), and alterations in social behavior (205) that further resemble aspects of the cognitive and behavioral alterations observed in schizophrenia patients.
Chronic exposures
Although acute exposures to NMDAR antagonists can produce some symptoms of schizophrenia in healthy subjects, these are transient and disappear after drug washout. On the other hand, chronic exposure, as it occurs in drug addiction, can produce, in some cases, a syndrome that is indistinguishable from schizophrenia (171), and substance abuse of NMDAR antagonists can, in fact, hinder its diagnosis (192).
Repetitive NMDAR antagonist treatment in animals produces more persistent effects on stereotypy, locomotor activity, and social withdrawal (169), as well as enduring cognitive deficits and neurochemical changes that resemble more accurately the alterations observed in schizophrenia (99, 167, 169).
Developmental exposures
Alteration of glutamatergic transmission by blockade of NMDAR during the postnatal period leads to a range of behavioral abnormalities in adults that are relevant to schizophrenia, from enhancement of exploration and psychomotor agitation, to impaired working memory (169). Prenatal or perinatal NMDAR antagonist exposures also lead to impairments in sensorimotor gating, spatial memory, social interaction behavior, and cognitive flexibility in adulthood (21, 23, 24, 169, 198, 234). In addition to cognitive deficits typical of schizophrenia, rodents treated during the perinatal period with NMDAR antagonists show alterations in conditioned fear responses (89, 238).
Functional Consequences of NMDAR Blockade
Neurochemistry
Acute exposure to NMDAR antagonists in adults is believed to result in acute disinhibition (85, 140, 157, 177), as measured by increased excitatory activity in the frontal and anterior cingulate cortex (220). PET studies have shown that hypermetabolic responses after ketamine administration were more severe in schizophrenic patients as compared to control subjects, even under resting conditions (68).
In rodents, the initial hypermetabolism observed after acute NMDAR antagonist exposure shifts to a decreased metabolic activity in several brain regions if repetitive exposures occur (43). While acute exposures to PCP or ketamine increase dopamine and glutamate release in the frontal cortex in rodents and primates (4, 98, 212), repetitive exposures decrease dopamine release and leads to a hypofunctional state (99). This repetitive regimen also elicits alterations in N-acetyl-aspartate and N-acetyl-aspartylglutamate in the temporal cortex and hippocampus as assessed by high-performance liquid chromatography (189), and decreases 5HT2A receptor binding in PFC (216), resembling schizophrenia pathology (126, 174).
Although the behavioral and neurochemical effects of acute exposures to NMDAR do not lead to enduring changes in PV+ neurons observed in schizophrenia (15, 135, 212), repetitive exposures to NMDAR antagonists produce a lasting reduction in GAD67 and PV expression in PV+ neurons of rodents (15) as well as decreased expression of PV in rodents and nonhuman primates (42, 104, 168, 193). Unlike the acute effects of NMDAR antagonists, which include hyperglutamatergia, these neurochemical changes eventually lead to a hypofunctional state through a homeostatic pathway (as discussed in the following sections), and thus reflect more faithfully the alterations observed in postmortem samples collected from patients with schizophrenia (reviewed in 135, 183).
Early postnatal NMDAR blockade produces a decrease in the number of PV+-labeled neurons and principal neuron spine density in the frontal cortex, nucleus accumbens, and hippocampus in rodents when these are analyzed in adulthood (21, 170, 235). Direct confirmation of the role of NMDAR function during postnatal development in the expression of schizophrenia-like behaviors comes from results showing that genetic ablation of these receptors from PV+ neurons decreases the expression of PV, produces disinhibition of pyramidal neurons, and leads to schizophrenia-related behaviors when mice reach adulthood (18, 29, 113). Interestingly, a more profound effect in behavior was observed in animals in which the disruption of NMDAR function in PV+ neurons occurred earlier in life—around the second postnatal week (18). This period coincides with the initiation of the maturational program of the cortical PV+ network (51, 71, 175, 195), which suggests that the normal development of PV+ neurons may depend critically on a well-preserved NMDAR function.
Electrophysiology
Dynamical activities of neural circuits, especially oscillations in the gamma-frequency range, accompany many important executive functions of the brain (66, 206), and thus constitute a crucial link between the neurochemical alterations and behavioral deficits in studies of schizophrenia (for review see 67, 219, 228). The hypo-NMDA hypothesis of schizophrenia (70, 94, 177) predicted electrophysiological consequences of acute and chronic noncompetitive NMDAR blockade, providing insights into distinct circuit mechanisms that contribute to the schizophrenic brain.
Acute application of subanesthetic concentrations of NMDAR antagonists, ketamine, MK-801, or PCP, resulted in an increase, instead of a decrease, of baseline power of gamma-frequency activity in electroencephalogram (EEG) and local field potential recorded from various cerebral cortical, hippocampal, and basal brain structures in awake, behaving rodents (Fig. 3) (55, 77, 129, 133, 146, 147, 180, 204). Later experimental work using in vivo single-unit recording suggested that the paradoxical increase of gamma-activity in response to NMDAR antagonists is due to a differential effect of these drugs on PV+ inhibitory neurons versus on pyramidal (excitatory) neurons, leading to disinhibition of excitatory activity (85, 184). Sensory-evoked activities, for example, measured auditory event-related potentials (ERPs) (Fig. 3), were observed to vary in peak amplitude and latency in the time domain (3, 153) and to have stronger gamma-frequency components in the frequency domain (55, 129, 204).
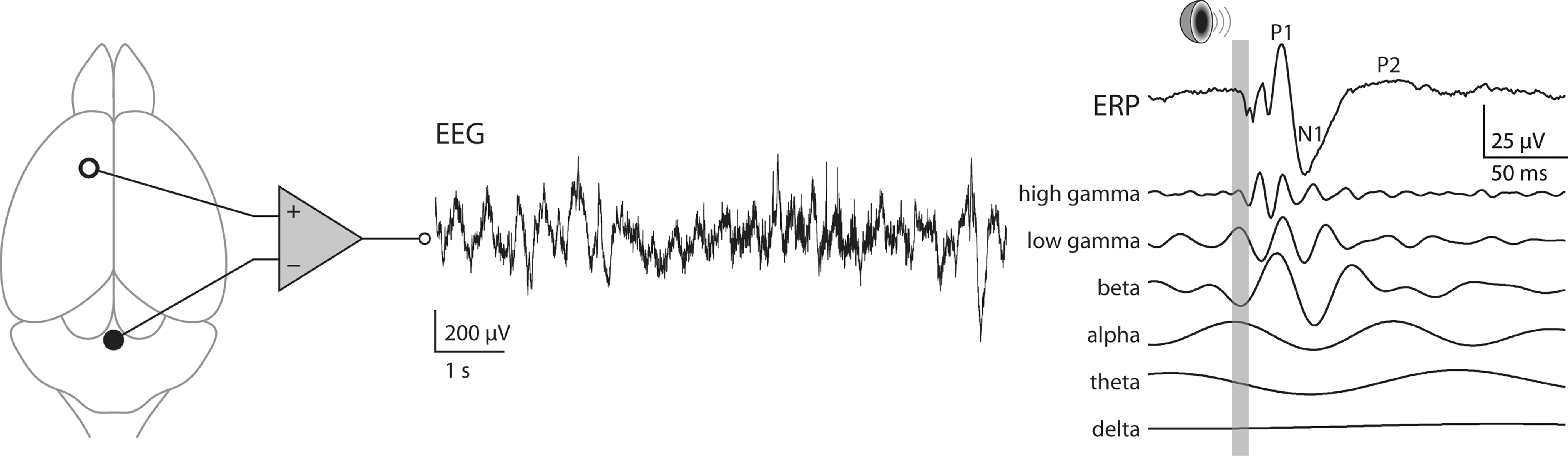
Consistent with the observations in rodent models, a rise in baseline gamma-power induced by acute NMDAR blockade was also found in healthy human subjects (86). Antipsychotic-treated schizophrenic patients usually have a lower level of gamma-activity in EEG, which correlates with their negative and cognitive symptoms (120, 136) and are better modeled by the enduring alterations caused by chronic or developmental NMDAR blockade, though a transient increase in gamma-power was associated with positive symptoms in psychosis (131, 136).
These results led to the hypothesis that NMDARs in PV+ neurons may serve as sensors of activity in a homeostatic control loop that goes awry in response to NMDAR antagonists (140). In contrast to transiently enhanced glutamate transmission and gamma-oscillations associated with reversible disinhibition caused by acute NMDAR blockade, lasting increase in extracellular glutamate release is a consequence of activated Nox2-dependent NADPH oxidase triggered by prolonged or repetitive NMDAR antagonism (212). This is more than likely to alter neural circuit dynamics further, which suggests that an oxidative stress mechanism is involved in the alterations in gamma-oscillatory activity observed after repetitive NMDAR antagonist exposures (183).
In contrast to in vivo investigations, in vitro studies using slice preparations yielded diverse results in terms of pharmacologically or electrically induced gamma-oscillations after acute NMDAR blockade. Amplitude of the gamma-rhythm was reported to increase in certain cortical regions, for example, the primary auditory cortex, as observed universally in vivo, but to significantly decrease in others, for example, the entorhinal cortex (45, 157, 190). Although NMDAR blockade causes enhanced gamma-activity globally in the brain, local circuit mechanisms are possibly distinct to specific regions (240, 241).
Recent studies using genetically engineered mice have revealed the importance of PV+ neurons in generating the electrophysiological phenotypes of NMDAR hypofunction. Global NR1 hypomorphic mice did not completely reproduce the alterations of auditory ERPs in schizophrenia, though they exhibited a number of behavioral deficits relevant to the disease (22). Interestingly, however, mice lacking NMDARs specifically in PV+ neurons showed electrophysiological phenotypes consistent with schizophrenia (18, 29, 113), which essentially included a raised baseline gamma-power and reduced gamma-activity evoked or induced by sensory inputs. Pharmacological studies further suggested that such aberrant gamma-oscillations were primarily due to the block of the NR2A subunit (111). Interestingly, blockade of NR2A containing NMDARs was also responsible for the activation of Nox2-mediated superoxide production after ketamine exposure (183). It is also worth noting that the manifestations of these neural circuit and behavioral abnormalities are determined within a specific developmental time window (18) that coincides with the delayed maturation of PV+ neurons (51, 71, 175). During this time window, NMDAR-blockade-triggered oxidative stress is sufficient to cause loss of function in PV+ neurons (14, 183).
In sum, important electrophysiological endophenotypes of schizophrenia, especially baseline and sensory-driven gamma-oscillations, can be recapitulated in rodent models of noncompetitive NMDAR blockade using ketamine, MK-801, or PCP. These antagonists specifically target PV+ neurons, either through acute suppression of their activity or induction of lasting functional alteration by redox regulation of the NR2A subunit, leading to disruption of neural circuits that generate aberrant levels of functional gamma-oscillation as observed in schizophrenia. Recent computational studies that incorporate the neurochemical changes observed after NMDAR antagonist exposures corroborate these alterations in gamma-oscillatory activity (232).
The Role of Nox2 in the Propsychotic Effects of NMDAR Antagonists
Each of the steps involved in neurotransmission, including receptor binding, opening of ion-channel neurotransmitter receptors, membrane fusion, presynaptic neurotransmitter uptake, and synaptic vesicle refilling, are influenced by the redox state of regulatory sulfhydryl groups on cysteine residues in the proteins involved in each of those steps. Alterations in the neuronal redox milieu would be thus expected to have an important impact on neurotransmission. Several lines of evidence also suggest that reactive oxygen species (ROS), specifically superoxide and hydrogen peroxide, which are normally involved in age-dependent neurodegenerative processes, may be involved in learning and memory in normal brain function (reviewed in 152) (summarized in Fig. 4).
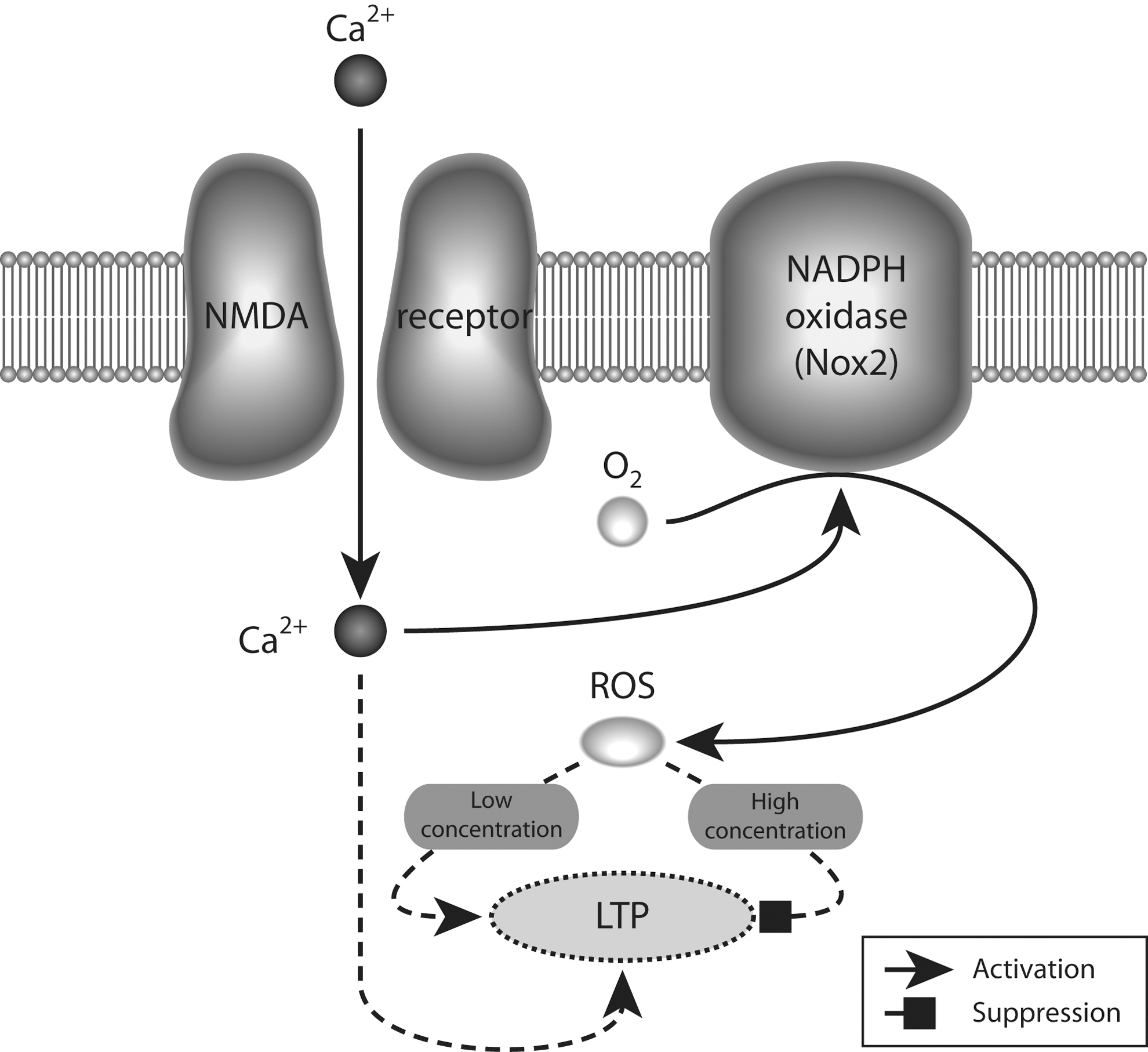
Mitochondria had been traditionally regarded as the site of formation of ROS (78). However, recent data show expression of the different subunits forming the NADPH (Nox2)-oxidase complex in the cell membrane of neurons (14, 106, 200, 221), suggesting that this enzyme plays an important role in normal brain physiology. Indeed, there is a requirement for Nox2-dependent superoxide production in NMDAR-mediated long-term signaling mechanisms, such as activation of the ERK-kinase system (109). Furthermore, a similar signaling role for Nox2-produced superoxide is suggested by experiments showing decreased long-term potentiation (LTP) and altered memory in animals lacking one of the subunits that forms the Nox2 oxidase (108). In addition, several studies have shown that blockade of NMDARs also produces an increase in superoxide production, which leads to detrimental effects in neuronal network performance (14, 212, 254). The superoxide produced by Nox2 oxidase is short lived, being rapidly converted to hydrogen peroxide by the action of superoxide dismutases. Then, most actions of Nox2 oxidase activation should be mediated by hydrogen peroxide. Indeed, several lines of evidence have suggested a dual role for hydrogen peroxide levels in learning and memory processes, where low-level increases are necessary for the functional changes associated with LTP, but higher doses are detrimental for its induction (152) (Fig. 4).
Several studies have reported an altered oxidative state in schizophrenia patients (reviewed in (49). GSH, responsible for detoxification of ROS and other radical species, is consistently decreased in the cerebrospinal fluid of drug-naïve schizophrenia patients (25, 50, 138, 185), as well as in postmortem tissue (250). Polymorphisms in genes coding for enzymes that participate in GSH synthesis have been linked to the risk of schizophrenia (76, 223), and recent results show that genetically compromised GSH synthesis affects the morphological and functional integrity of hippocampal PV+ neurons (215). Furthermore, results showing that treatment with N-acetylcysteine, a precursor of GSH, improves negative symptoms and corrects mismatch negativity in schizophrenic patients provide strong support for a redox imbalance in schizophrenia (19, 128).
A role for oxidative stress mechanisms in the propsychotic effects of NMDAR antagonists was recently suggested by studies showing that there is a rapid increase in brain ROS and reactive nitrogen species as a consequence of the exposure of adult animals to those drugs (14, 15, 60, 212, 254). It was also found that the acute exposure to ketamine increased extracellular glutamate and dopamine in the prelimbic region of wild-type mice, but failed to do so in Nox2-deficient animals (212). The specific role of Nox2 was confirmed in this study by a normal response to amphetamine in Nox2 animals. These results suggest the interesting possibility that activation of superoxide production by Nox2 is required for the acute disinhibition of pyramidal neurons (17). However, such a direct role for Nox2 in glutamatergic transmission has not been clearly elucidated. It is possible that the acute oxidative stress produced by activation of Nox2 is sufficient to produce the functional inactivation of PV+ neurons, and that this is the mechanism by which disinhibition occurs. Indeed, the fast-spiking characteristic of PV+ neurons entails a high metabolic cost (79), which could render these neurons highly sensitive to oxidative stress.
Elevated synaptic activity through NMDAR activation enhances thioredoxin activity, facilitates the reduction of hyperoxidized peroxiredoxins, and promotes resistance to oxidative stress (178). However, overactivation of NMDARs, such as it occurs in excitotoxic insults, can lead to overproduction of ROS and oxidative damage (54, 121, 134, 188). Thus, a tight control of antioxidant systems must exist in neurons, and an imbalance in redox regulatory systems would be expected to alter neuronal function.
Repetitive exposures of adult mice to NMDAR antagonists showed that injections of ketamine on two consecutive days induced an increased oxidative state in the brain, which was sufficient to produce the loss of GAD67 and PV expression in PV+ neurons (14), as well as an enduring inhibitory dysfunction in the rat prelimbic region (251). This altered oxidative state produced by ketamine was due to a sustained increase in the proinflammatory cytokine IL-6 and activation of the superoxide-producing enzyme NADPH oxidase-2 (Nox2). Such effects of ketamine were absent in Nox2-deficient and IL-6-deficient animals, confirming the role of the IL-6/Nox2 pathway as mediator of ketamine effects (15) (Fig. 5).

A similar increase in superoxide production as that produced by ketamine was observed using an NR2A-preferring antagonist (NVP-AAM007), at concentrations known to preferentially affect NR2A-containing receptors (57, 183). These results give support to a specific role for NR2A-containing NMDARs in mediating the increased oxidative stress in the adult brain, as was suggested by earlier results showing that NR2A-containing NMDARs are involved in the maintenance of the inhibitory phenotype of PV+ neurons in primary cultures (107).
Oxidative Dysregulation of the Inhibitory System and Its Possible Causative Relation to the Pathophysiology of Schizophrenia
There is increasing evidence that schizophrenia, with symptoms typically appearing during late adolescence or early adulthood, is a consequence of alterations in early brain development (58, 186). Several animal models have been used to understand neurobiological processes relevant to the developmental hypothesis of schizophrenia (58, 138, 155). These models have provided insight into the vulnerability of the developing embryo and the importance of the late-prenatal/early-postnatal environment for normal maturation of brain function.
Developmental models specific to schizophrenia have focused on epidemiological and pharmacological risk factors that include, but are not limited to, prenatal/postnatal infections, NMDAR antagonist administration, and neonatal ventral hippocampal lesions (183). In all of these models, the development of behavioral and anatomical alterations resembling those found in schizophrenia has been analyzed when animals reach adulthood. Common features include alterations in gating response, decreased working memory, alterations in dopaminergic responses, and diminished inhibitory activity, specifically of PV+ neurons (183).
Starting at the end of the first postnatal week in rodents, inhibitory neuronal networks are critically involved in experience-dependent refinement of neural networks. During this period, cortical inhibition is fundamental to the formation of critical periods for sensory plasticity (81). The levels of the inhibitory neurotransmitter GABA profoundly influence inhibitory neuron axon growth and synapse formation during brain postnatal development, and alterations in GAD67, the main enzyme producing GABA, can have profound effects on the proper development of inhibitory circuits (reviewed in 88). Among all inhibitory neurons in the cortex, and across species, the last to mature are the PV+ inhibitory neurons (74, 82, 196). In rodents, the maturational process of these neurons starts at the end of the first postnatal week (47, 51, 175). Throughout the next 3 weeks, they slowly mature into fast-spiking inhibitory neurons (51, 71, 175, 195). This period of maturation occurs concomitantly with a striking transcriptional change (71, 175) in which most of the genes characteristically expressed in mature PV+ interneurons are turned on in an orchestrated fashion between the second and fourth postnatal weeks, coinciding with their electrophysiological maturation (51, 71, 175).
Among all interneuron subtypes, PV+ interneurons receive the highest number of glutamatergic synapses from thalamic afferents and intracortical networks in the adult rodent brain (75). Glutamatergic neurotransmission preferentially activates Ca2+-permeable AMPA receptors on mature PV+ interneurons, and these receptors appear slowly during postnatal development (72, 237). The expression and function of NMDARs in PV+ neurons change during postnatal development, with high levels being expressed early during postnatal development and profound functional changes occurring during adolescence (21, 107, 236, 252).
The high NMDA/AMPA receptor ratio during postnatal development might make PV+ neurons extremely sensitive to alterations in glutamatergic transmission, specifically on NMDAR function. Such effect could be detrimental for the transcriptional program that controls the maturation of the PV+ neuronal system, leading to structural alterations of the cortical network. Accumulating evidence shows that embryonic and perinatal NMDAR antagonist exposures, contrary to the reversible effects observed in adults (15), can induce a loss of PV+ neurons in several regions, and produce persistent behavioral and neurochemical deficits when animals reach adulthood (7, 21, 53, 170, 207, 214, 235, 242). High doses of NMDA antagonists during the perinatal period were previously shown to lead to diminished numbers of PV-expressing neurons when animals reached adulthood. Since exposure to these same antagonists was shown to trigger cell death, through apoptotic mechanisms in several brain regions (6, 20, 208, 233), it was believed that the reduced number of PV+ neurons observed was due to their death (177, 234, 235). However, the lack of expression of a cell marker such as PV does not necessarily incur cell death. Using a mouse line expressing GFP exclusively in PV+ neurons in the cortex (G42 line, 34), we recently demonstrated that perinatal exposures to low doses of ketamine do not lead to the death of PV+ neurons, but to the absence of PV expression in around 35% of the neurons (183). These results suggest that while the cells are still alive, their developmental maturation may be affected. Similar results were recently obtained using a mouse line expressing GFP in all GABAergic neurons (252). In this study, using an NR2A-preferring antagonist, the authors showed that prolonged blockade of NR2A-containing receptors in vivo during the critical period of plasticity in the barrel cortex produced a decrease in PV expression and an alteration of fast-spiking-mediated inhibitory postsynaptic currents onto principal neurons.
Alterations upon brain development during specific periods of pre- or postnatal life produce a decrease in the expression of PV in several brain regions (13, 65, 97, 135, 137, 143, 156, 179, 187, 209, 222, 225). Interestingly, reverse translational models using schizophrenia risk genes such as DISC1, NRG1/ErbB4, and Reelin show similar alterations of PV expression (5, 10, 59, 62, 83, 172, 202, 239).
Several animal models with disruptions in genes involved in the development and maturation of PV+ neurons are now available. From these studies, we have learned the key roles played by schizophrenia risk gene products such as TrkB, ErbB4, (receptors for brain-derived neurotrophic factor and neuregulin, respectively), dysbindin, and cannabinoid receptor 1 in the maturation process of PV+ neurons (30, 36, 63, 239, 253). Interestingly, it was recently shown that deletion of selenoprotein expression or of the Se-dependent glutathione peroxidase 4 severely affects the maturation of PV+ neurons (245). Furthermore, specific deletion of the catalytic subunit of glutathione synthase from PV+ neurons leads to a cell autonomous increased oxidative stress and pronounced decrease in PV+ synaptic contacts (165). These results suggest that PV+ neurons show a heightened sensitivity to oxidative stress conditions that could be due, in part, to their high metabolic demand (80).
Mechanisms involving oxidative stress during development are hypothesized to underlie the origin of schizophrenia pathophysiology. Decreases in antioxidant capacity during early postnatal periods in rodents, as well as genetic deficiencies in GSH, produce a loss in PV+ neurons and induce cognitive derangements relevant to the disease (27, 215). Acute GSH depletion potentiates the release of dopamine produced by amphetamine in the striatum and enhances the behavioral effects of NMDAR antagonists, as a well as those of amphetamine (93).
Antioxidants can prevent the appearance of behavioral disruptions in adult animals that were treated with PCP during the perinatal period, suggesting that oxidative mechanisms in the perinatal NMDAR antagonist model were involved (234). Exposures to PCP and to more selective NMDAR antagonists, such as MK-801 and CPP, were shown to produce a rapid increase in ROS and reactive nitrogen species in vitro (247) and in vivo (60, 254), and repetitive exposures in vivo led to a substantial elevation of baseline levels of free radicals, suggesting that this treatment results in a persistent change in the oxidative state of the cortex (254). The lack of effects of ketamine on Nox2-deficient animals supports the role of Nox2 in the loss of PV+ interneurons in the perinatal ketamine model (183). Furthermore, recent studies have implicated Nox2-dependent oxidative mechanisms in the loss of PV+ neurons and development of schizophrenia-like behavior in the isolation- rearing model (79, 198), a model that was positively associated with oxidative stress as measured by increased superoxide dismutase, decrease oxidized/reduced GSH ratio, and increased concentrations of malondialdehyde (160).
So far, the targets of oxidative stress in PV+ neurons that lead to their enduring dysfunction in neurodevelopmental models of schizophrenia are unknown. One possible hypothesis is that glutamatergic synapses onto these neurons might be particularly sensitive to oxidative stress (17). Such increased sensitivity could be due to the fundamental role that glutamatergic transmission has on the maturation of these interneurons, where both NMDA and AMPA receptors slowly appear during the second postnatal week, and show a developmental switch in subunit expression during the next weeks of development (51, 71, 175, 236, 252). Oxidative modification of neurotransmitter transporters (such as glutamate transporters) and ligand-gated ion-channels (such as GABAA and NMDARs) and consequent decrease of their activity might be another target by which oxidative stress alters the development of PV+ neurons. In neurons, cysteine, the limiting substrate in the synthesis of GSH, the antioxidant that neutralizes ROS, is taken up from the extracellular space by EAAC1, the main neuronal glutamate transporter (9). EAAC1 as well as EAAC2 have been shown to be highly sensitive to oxidative conditions, where reducing agents activate, and oxidation inactivates glutamate transport (9, 224). Regulatory redox sites have also been found in proteins that are key to glutamatergic neurotransmission through NMDARs, including the enzyme serine racemase, which is responsible for the synthesis of glycine co-agonist at NMDARs (169a), and glutamine synthase, which is responsible for glutamate synthesis (181). As discussed above, the NMDAR itself is highly sensitive to redox modulation through redox-sensitive sites (Fig. 2).
Redox-mediated changes in transcriptional activity might affect the maturational process of PV+ neurons. Under physiological conditions, nuclear antioxidants are critical for maintaining the environment needed for proper gene transcription. A number of transcription factors contain redox-sensitive residues, and in most cases, oxidation inhibits their deoxyribonucleic acid (DNA)-binding activities (227). The transcriptional activation responsible for PV+ neuronal maturation occurring during the perinatal period could thus be the target of oxidative stress that results in enduring disruption of PV+ neuronal function later in life (Fig. 6). Indeed, recent data show alterations in several frontal cortex messenger ribonucleic acid (mRNAs) after perinatal exposure to PCP (141). Whether any or all of these mRNA changes occur in PV+ neurons remains to be elucidated.

Conclusions
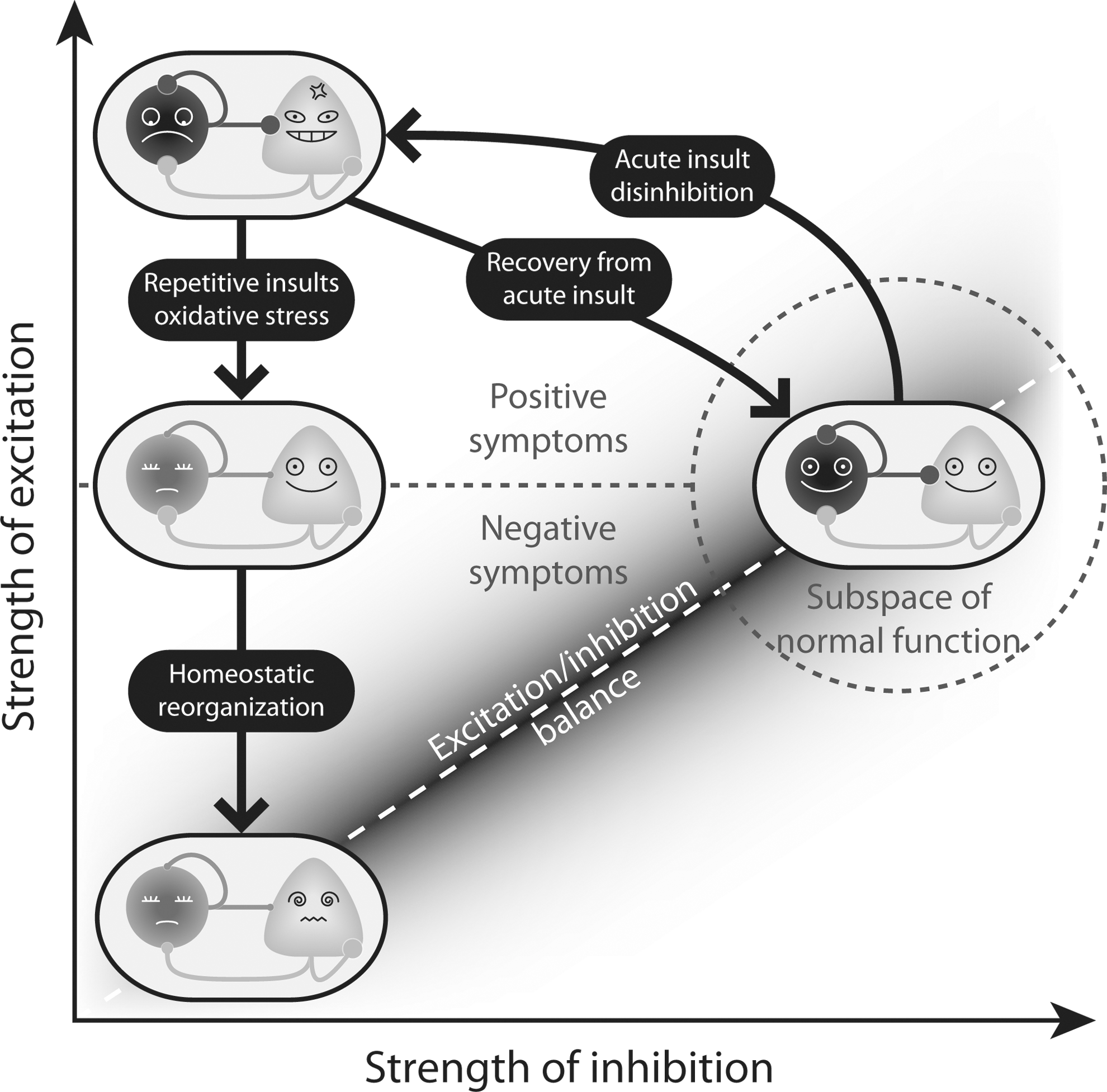
The possibility that PV+ inhibitory neurons are not just one of the many types of neurons that are affected in schizophrenia, but are at the nexus of the disease, with alterations starting early in development, is supported by mounting experimental evidence summarized in this review. In particular, the evidence points toward the early involvement of oxidative stress mechanisms and proinflammatory cytokines in derailing the normal development of PV+ neurons. Disruption of one of the main inhibitory circuits in the cortex would affect the way excitatory neurons develop throughout the same developmental period, through a homeostatic process that restores the excitatory/inhibitory balance. Although compensatory mechanisms may restore this balance, the altered state of the cortex could remain vulnerable to further episodes of oxidative stress (Fig. 7).
The ultimate mechanism by which oxidative stress produces long-term effects in the maturation of cortical circuits could be related to epigenetic alterations during sensitive periods of neurodevelopment, as suggested by recent results from our group showing that ketamine effects on PV+ neurons are prevented by inhibition of DNA-methylation processes (16). These new findings are exciting because they suggest that some aspects of schizophrenia may be treated by reversing the dysregulated genes in the PV+ cells, thus restoring the cortical circuits to a normal state. The next steps are underway to test this hypothesis.
Footnotes
Acknowledgments
The authors were supported by the grants from NARSAD (Gwill Newman Investigator, MMB), NIMH (MH091407, MMB), and Howard Hughes Medical Institute (TJS). APD is a recipient of a Calouste Gulbenkian Foundation Fellowship, and XW is a recipient of a Life Sciences Research Foundation Pfizer Fellowship.