
Abstract
Introduction
Increased availability of lipids and inflammatory processes contribute to the pathogenesis of obesity-related insulin resistance and T2DM (40). Moreover, proper mitochondrial functioning seems to contribute to the regulation of insulin sensitivity and secretion. Consequently, processes impairing mitochondrial function would lead to disturbed energy homeostasis with insulin resistance and deficiency (42). Indeed, impaired mitochondrial function has been associated with alterations of (i) glucose and fatty acid metabolism, (ii) production of electron transport system (ETS)-related reactive oxygen species (ROS), (iii) ATP-mediated insulin secretion from β-cells, (iv) synthesis of ATP for energy-consuming functions (i.e., insulin-stimulated glucose uptake), (v) exercise-induced production of ATP and aerobic capacity and, finally, (vi) calcium homeostasis which impacts on exercise-mediated glucose uptake (51).
Despite these associations supporting a critical role of mitochondria in glucose metabolism, the literature also provides controversial data, which could result from using different methods and terminologies (75). The present review summarizes the recent data on the associations between alterations in muscular and hepatic mitochondrial plasticity and insulin resistance in humans. In particular, some relevant endocrine and metabolic effects on mitochondrial plasticity will be reviewed, while the molecular regulation during aging and exercise training is beyond the scope of this review and will not be addressed.
Mitochondrial Plasticity
In animals, mitochondria are responsible for oxidative phosphorylation (OXPHOS), the main process of energy and fuel homeostasis regulation. Normally, mitochondria are “plastic” (i.e., they respond rapidly and adequately to metabolic alterations to meet the actual needs of the respective tissues). Recently, the term mitochondrial plasticity was introduced to define changes of mitochondrial activity or oxidative capacity in response to various metabolic conditions created by diet, exercise, insulin, and drugs (75). Of note, these changes could result from alterations of individual mitochondrial function and/or overall mitochondrial content.
Mitochondrial plasticity can be assessed by comparing the following three parameters under basal conditions and during stimulation (e.g., by exogenous lipids or exercise training) (75): (i) In vivo mitochondrial activity, assessed by 31P-magnetic resonance spectroscopy (MRS) (69), represents unidirectional flux through ATP synthase and thereby resting oxidative phosphorylation at low ADP concentrations, depending on substrate/oxygen availability and demand. (ii) In vivo submaximal ADP-stimulated oxidative phosphorylation, also assessed by 31P-MRS, represents the rate of phosphocreatine (PCr) re-synthesis (35) upon submaximal exercising and is also influenced by substrate/oxygen flux controlling processes. (iii) Ex vivo oxidative capacity, assessed from oxygen consumption rates in isolated mitochondria or permeabilized fibers, represents maximal ADP-stimulated oxidative phosphorylation reflecting maximal energy demand at unlimited substrate/oxygen supply (23).
Mitochondrial Plasticity in Insulin-Resistant States
As insulin resistance is predicting and preceding the onset of T2DM (78), it is of interest to compare mitochondrial function with insulin sensitivity in cohorts of variable insulin sensitivity and at increased risk of T2DM.
Mitochondrial plasticity during aging
The aging-related decline in insulin sensitivity is held responsible for the greater risk of T2DM in the elderly (52, 70). Impaired insulin sensitivity was found in some elderly cohorts (52, 76), who were matched for physical activity to younger controls, but physical activity was not assessed using the state-of-the-art method, maximal oxygen uptake (V
Mitochondrial plasticity in first-degree relatives of patients with T2DM
First-degree relatives (FDR) are frequently insulin resistant and at greater risk of T2DM. Young, lean, but severely insulin-resistant FDR have ∼30% lower in vivo flux through muscular ATP synthase than insulin-sensitive otherwise matched controls in line with impaired basal mitochondrial activity (53). FDR with marginally lower insulin sensitivity only show a nonsignificant tendency of lower in vivo submaximal oxidative phosphorylation and ex vivo mitochondrial oxidative capacity than age-matched controls (56). Likewise, we reported that insulin-sensitive FDR have similar in vivo mitochondrial activity rather than carefully matched controls (29). Of note, both studies found that associations of mitochondrial function with V
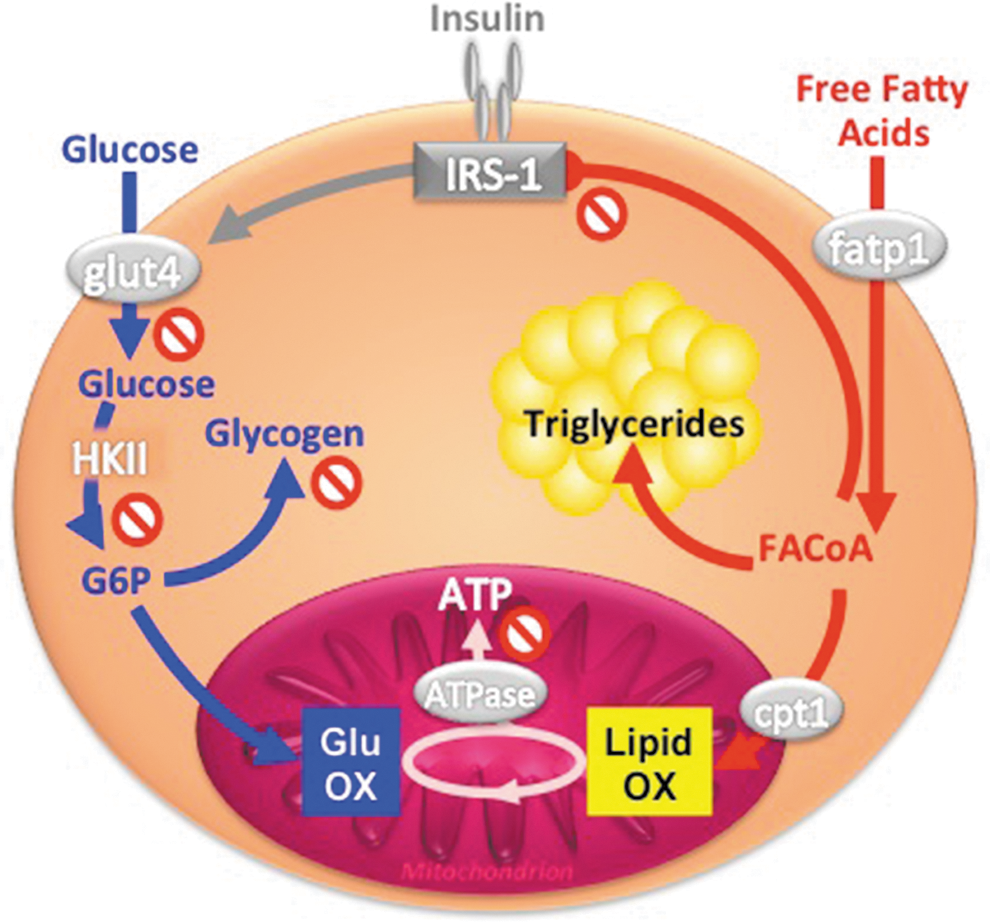
Insulin-resistant FDR also showed lower mitochondrial plasticity from diminished insulin-stimulated in vivo flux through ATP synthase, which further associated with lower insulin-stimulated rises of myocellular Pi but not PCr/ATP concentrations (54). This suggests that reduced Pi supply, possibly due to decreased phosphate transport, accounts for the compromised mitochondrial plasticity in insulin-resistant FDR. We further studied mitochondrial plasticity in a cohort of FDR with low degree of insulin resistance employing short- and long-term exercise training (29, 30). In vivo flux through ATP synthase increased with both training interventions, while insulin sensitivity improved only upon short-term exercise training. The beneficial effects on mitochondrial plasticity were present only in some FDR and did not depend on common single nucleotide polymorphisms of PGC1α but on the G/G gene polymorphism of the NADH dehydrogenase (ubiquinone) 1 ß subcomplex (NDUFB6) rs540467 gene, a component of complex I of the ETS. These data show that the role of mitochondrial plasticity for the adaptation of insulin sensitivity to environmental demand may depend on inherited factors. However, it remains unclear whether impaired plasticity results from primary inherited mitochondrial defects or from cellular accumulation of deleterious lipids (Fig. 8A).
Mitochondrial Plasticity in Overt T2DM
T2DM is a heterogeneous disease, which is currently defined by a certain degree of hyperglycemia and explained by insulin resistance and relative insulin deficiency. Here, we focus on mitochondrial function in liver and muscle (75).
Skeletal muscle
Already 40 years ago, evidence was provided for decreased activity of mitochondrial TCA enzymes in skeletal muscle of humans with T2DM (3). However, not all subsequent studies found compromised mitochondrial function in muscles of insulin-resistant patients with T2DM. While mitochondrial area and activity of ETS enzymes were decreased in T2DM, and the size of mitochondria positively correlated with insulin sensitivity (33), such correlation disappeared after matching subjects for their physical activity (13), known to stimulate mitochondrial density (79). Human muscle biopsies revealed that mRNA expression of genes encoding key enzymes of oxidative metabolism and mitochondrial function, such as PGC-1α, PGC-1β, and nuclear respiratory factor-1 (NRF-1)-dependent genes (50), as well as activity of NADH oxidase (63), are lower in patients with T2DM (Fig. 8A). Another modulator of mitochondrial activity is mammalian target of rapamycin complex 1 (mTORC1) pathway. Inhibition of mTORC1 resulted in decreased mRNA of PGC-1α and its target genes along with lower oxygen consumption in C2C12 myotubes (15). Patients with T2DM have increased muscle mTORC1 levels, again implying that mitochondrial function can dissociate from insulin sensitivity and/or is under control of other signaling pathways (39). Interestingly, fasting (22) or administration of resveratrol (59) induce PGC-1α, mitochondrial genes, and fatty acid oxidation in myotubes by activating AMP-activated protein kinase (AMPK) and sirtuins, a family of NAD+-dependent acyltransferases (Fig. 8A). Although resveratrol improves insulin sensitivity (7), the relationship between insulin resistance in human T2DM and sirtuins or AMPK is not yet clear.
In addition to studies on enzyme activities and expression levels, ex vivo studies reported lower oxidative capacity in T2DM. Also, ADP-stimulated state 3 respiration was lower in permeabilized muscle fibers from patients with T2DM than in matched controls (56), but not after normalizing the rate of oxygen consumption to mitochondrial DNA copy number (5). But even mitochondria isolated from muscles of patients with T2DM had decreased pyruvate plus malate-driven state 3 mitochondrial oxidative capacity (44). While the majority of these data suggest impaired mitochondrial density and/or function in overt T2DM, ex vivo rates of ATP synthesis and OXPHOS capacity were similar in mitochondria isolated from muscles of healthy humans and Asian Indians with T2DM (48). Although the Asian Indians were more insulin resistant, their OXPHOS capacity was even greater compared with Northern European Americans, again indicating dissociation of mitochondrial function and insulin sensitivity.
Further studies measured features of myocellular mitochondria in vivo, which offer the possibility to examine mitochondrial function in their intact cellular and metabolic environment. Submaximal oxidative phosphorylation was also decreased in T2DM compared to age- and BMI-matched controls (56) and associated with insulin resistance, but not with intramyocellular lipids. PCr recovery kinetics did not differ between controls and patients at early and advanced stages of T2DM (16). Of note, submaximal oxidative phosphorylation positively correlated with insulin sensitivity across a larger cohort, but this relationship was lost within T2DM (2). Discrepancies between these studies may result from differences in exercise tolerance or load used to deplete PCr, variable degree of muscular acidosis, which may affect the rate of PCr re-synthesis. Of note, in vivo flux through ATP synthase at rest was lower in T2DM than in young, but not carefully matched humans without T2DM (76). Nevertheless, the observed heterogeneity could reflect the multifactorial pathogenesis of T2DM.
These studies did not clarify whether mitochondrial function per se or density might be impaired or compensatory altered in T2DM. But even normalization of mitochondrial function for mitochondrial content revealed functional impairment in some (44, 56, 63) but not all studies in T2DM (5, 33). These discrepancies might be due to the use of indirect markers of density such as mitochondrial DNA content or citrate synthase activity, which is affected by acute exercise (18) and hyperinsulinemia (73). Direct quantification of mitochondrial content in situ by transmission electron microscopy showed lower mitochondrial content in insulin-resistant humans with as well as without T2DM. This was explained by reduced intermyofibrillar mitochondrial subpopulations (13), in contrast to a study previous showing reduced subsarcolemmal mitochondrial content in T2DM (64). Again, the observed discrepancy could be the result of methodological differences (13).
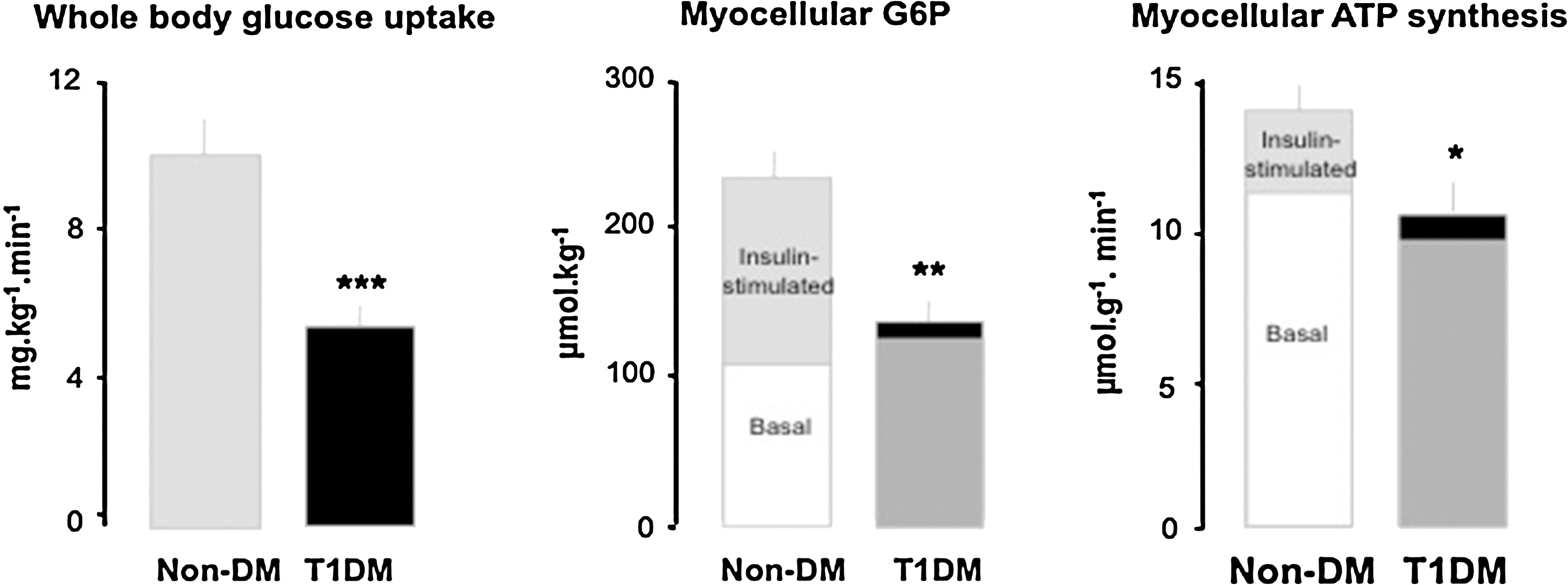
Only a few studies specifically address mitochondrial plasticity (e.g., by measuring the effects of insulin on myocellular mitochondrial function) in T2DM. Mitochondria isolated from skeletal muscle of young, lean, healthy subjects respond to high physiologic insulin concentrations with increases in ATP synthesis and expression of several ETS enzymes, both of which were absent in mitochondria obtained from T2DM patients (73). Similarly, we observed that insulin does not stimulate unidirectional flux through ATP synthase in vivo in insulin-resistant patients with T2DM compared to both body mass- and physical activity-matched controls at comparable or younger age (76). Myocellular ATP synthetic rate did not rise during hyperinsulinemic-hyperglycemic clamp conditions, which doubled rates of whole-body glucose uptake and myocellular glucose-6-phosphate (G6P) levels (Fig. 2A and 2B). Thus, impaired stimulation of ATP turnover did not simply result from lower substrate availability but rather from diminished mitochondrial plasticity in T2DM (Fig. 2C) (76).

Liver
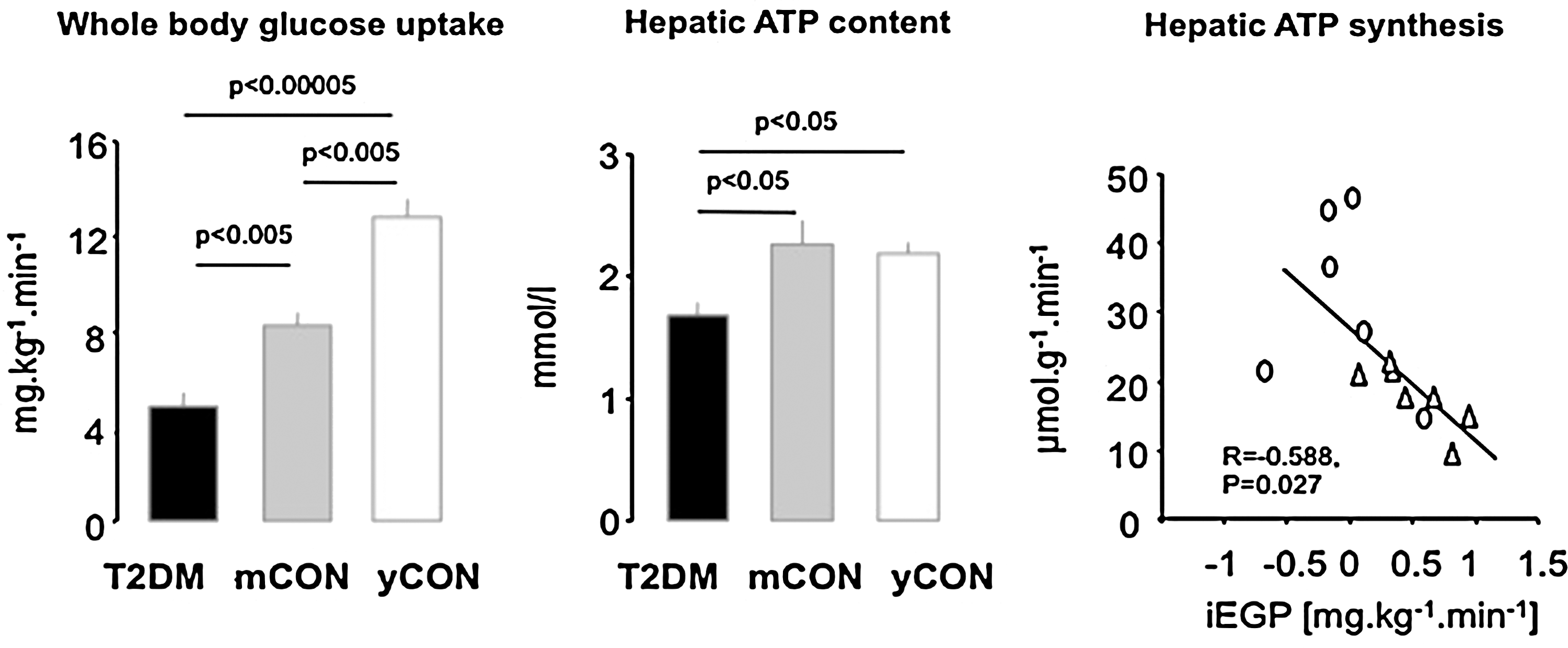
Proteomic analysis revealed distinct tissue-dependent variability in mitochondrial protein composition, suggesting that mitochondria may also differ functionally (21). Indeed, both ex vivo studies in isolated mitochondria and in vivo 31P-MRS showed that the OXPHOS pathway is thermodynamically more efficient (10) with 3-fold greater ATP turnover in liver than in skeletal muscle (68). Deterioration of mitochondrial OXPHOS capacity could lower lipid oxidation and raise lipid accumulation, thereby causing or contributing to hepatic insulin resistance and steatosis. Using a noninvasive 31P/1H-MRS technique, we found that hepatic energy metabolism is impaired in longstanding T2DM (74). Impaired whole-body insulin sensitivity in these subjects was associated with decreased liver ATP content compared to young, lean as well as age- and BMI-matched controls (Fig. 3A and 3B). Furthermore, basal ATP synthesis was also lower in T2DM (69) and negatively correlated with hepatic insulin resistance (Fig. 3C). Obese patients with T2DM also have lower hepatic expression of OXPHOS genes and exhibit a negative correlation of γ-subunit of ATP synthase with hepatic fat content (57). In other obese subjects, insulin resistance not only correlated with hepatic diacylglycerol content but also with markers of endoplasmic reticulum stress (38), which can impair mitochondrial function (41). However, less obese patients with T2DM can have greater hepatic OXPHOS expression than glucose-tolerant humans (43). Although this does not necessarily reflect greater hepatic mitochondrial function, more studies are needed to study the dynamic role of hepatic mitochondrial function during the development of obesity and T2DM. Of note, animal studies provide evidence for various modulators of hepatic mitochondrial function; for example, impaired insulin signaling in the liver-specific IRS-1/IRS-2 knock-down mice dysregulates mitochondrial biogenesis, morphology, and function by inhibition of forkhead box O1 (Foxo1) (12) and high-fat diet (HFD)-induced steatosis associates with decreased activity of sirtuins and components of ETS (36) (Fig. 8B).
Independent of differences resulting from measurement, genes, or aging, the metabolic environment could contribute to alterations of mitochondrial function in insulin-resistant states. In detail, in vivo insulin-stimulated myocellular ATP synthesis correlates inversely with both degree of hyperglycemia as assessed from hemoglobin A1c levels (Fig. 4A) and of hyperlipidemia, as assessed from postabsorptive plasma FFA concentrations (Fig. 4B).

Glucose-Induced Changes in Mitochondrial Plasticity
Chronic hyperglycemia is currently held responsible for the vast majority of microvascular and, to a minor degree, for macrovascular consequences of diabetes, but also for promoting insulin resistance and ß-cell dysfunction (67). Uncontrolled (excessively hyperglycemic) patients with type 1 diabetes mellitus (T1DM) display lower ex vivo rates of ATP production and altered mitochondrial gene expression profile in skeletal muscle (31). Of note, ex vivo mitochondrial respiration negatively correlates with fasting plasma glucose levels and near-normalization of glycemia by insulin improved respiration in T2DM patients with severe hyperglycemia (>15 mmol/l) (61). Recently, we showed that even near-normoglycemic patients with longstanding T1DM show substantial insulin resistance with impaired insulin-stimulated myocellular G6P concentrations, as well as compromised mitochondrial plasticity (28) (Fig. 5A and 5B). Moreover, in vivo myocellular ATP synthesis correlated positively with insulin sensitivity and negatively with glycemic control, as assessed from hemoglobin A1c levels (Fig. 4A). Thus, chronic hyperglycemia per se may deteriorate mitochondrial plasticity and thereby contribute to insulin resistance.
Lipid- and Obesity-Induced Changes in Mitochondrial Plasticity
Hypercaloric nutrition and obesity are frequently associated with hyperlipidemia (i.e., elevation of plasma triglycerides and FFA) and insulin resistance, and may lead to T2DM (65). Specifically, circulating FFA and/or their intracellular metabolites inhibit insulin signaling (Fig. 1) by several mechanisms including lipotoxic and inflammatory pathways (65, 66). Because of their critical role for fat oxidation, mitochondria may also be involved in lipid-induced insulin resistance, which occurs rapidly in the presence of high FFA (acute hyperlipidemia) or may evolve slowly during the development of obesity (chronic hyperlipidemia).
Effect of acute hyperlipidemia
In vitro, primary human myocytes respond to physiologically increased palmitate concentrations with decreased ATP synthesis, oxygen consumption, mitochondrial complex activities, and membrane potential (1). Likewise, palmitate-overloading impaired β-oxidation and promoted intramyocellular lipid accumulation in myotubes from patients with T2DM compared with controls (37). In order to examine the time course of FFA-dependent changes in insulin sensitivity and mitochondrial plasticity, we monitored whole body glucose uptake (Fig. 6A), in vivo G6P levels and rates of ATP synthesis (Fig. 6B) in skeletal muscle during 6-hour lipid infusions in healthy humans. Within 3 hours, plasma FFA elevation impaired insulin sensitivity by inhibiting insulin-stimulated glucose uptake and myocellular G6P but did not affect ATP synthesis (9). Only at 6 hours, insulin-stimulated ATP synthesis was ∼24% lower, indicating that impaired mitochondrial plasticity is a consequence of insulin resistance during acute hyperlipidemia (8). Furthermore, 48 hours of lipid infusion decreased the expression of PGC-1 and nuclear encoded mitochondrial genes, along with decreased glucose uptake (62) and 3-day HFD downregulated muscular OXPHOS genes, ETS components, mitochondrial carrier proteins, and stimulators of mitochondrial biogenesis, even in lean humans (72). Intermediate duration of HFD revealed inconsistent data, showing greater muscular expression of genes regulating lipid oxidation after 5 days (6) and no effect on in vivo submaximal ADP-stimulated oxidative phosphorylation after 7 days (17). However, these dietary interventions also failed to show effects on insulin sensitivity. Thus, the available evidence indicates that acute hyperlipidemia first causes insulin resistance followed by impaired mitochondrial plasticity in skeletal muscle of lean humans.

Effect of chronic hyperlipidemia associated with obesity
Unlike lean, obese people are metabolic inflexible in that they do not respond appropriately to metabolic stimuli such as dietary fat intake with greater fat oxidation (34). Thus, HFD neither increases whole-body lipid oxidation nor expression of genes of lipid oxidation (6) in obese humans. Similar to some T2DM cohorts, obese individuals display decreased size (33) and content of muscular mitochondria (13, 64) without evidence for changes in one specific subpopulation of mitochondria. Transcription levels of genes encoding components of OXPHOS were also lower in adipose tissue of obese than in their non-obese monozygotic twins (47), pointing to the predominant role of acquired factors driving abnormalities in energy metabolism. Reduced mitochondrial function was confirmed in obese humans ex vivo in some studies on oxidative capacity (11) but not by bioluminescence assays (32). Interestingly, already obese children may have reduced in vivo submaximal ADP-stimulated oxidative phosphorylation, which associates with insulin resistance but not with obesity per se (20).
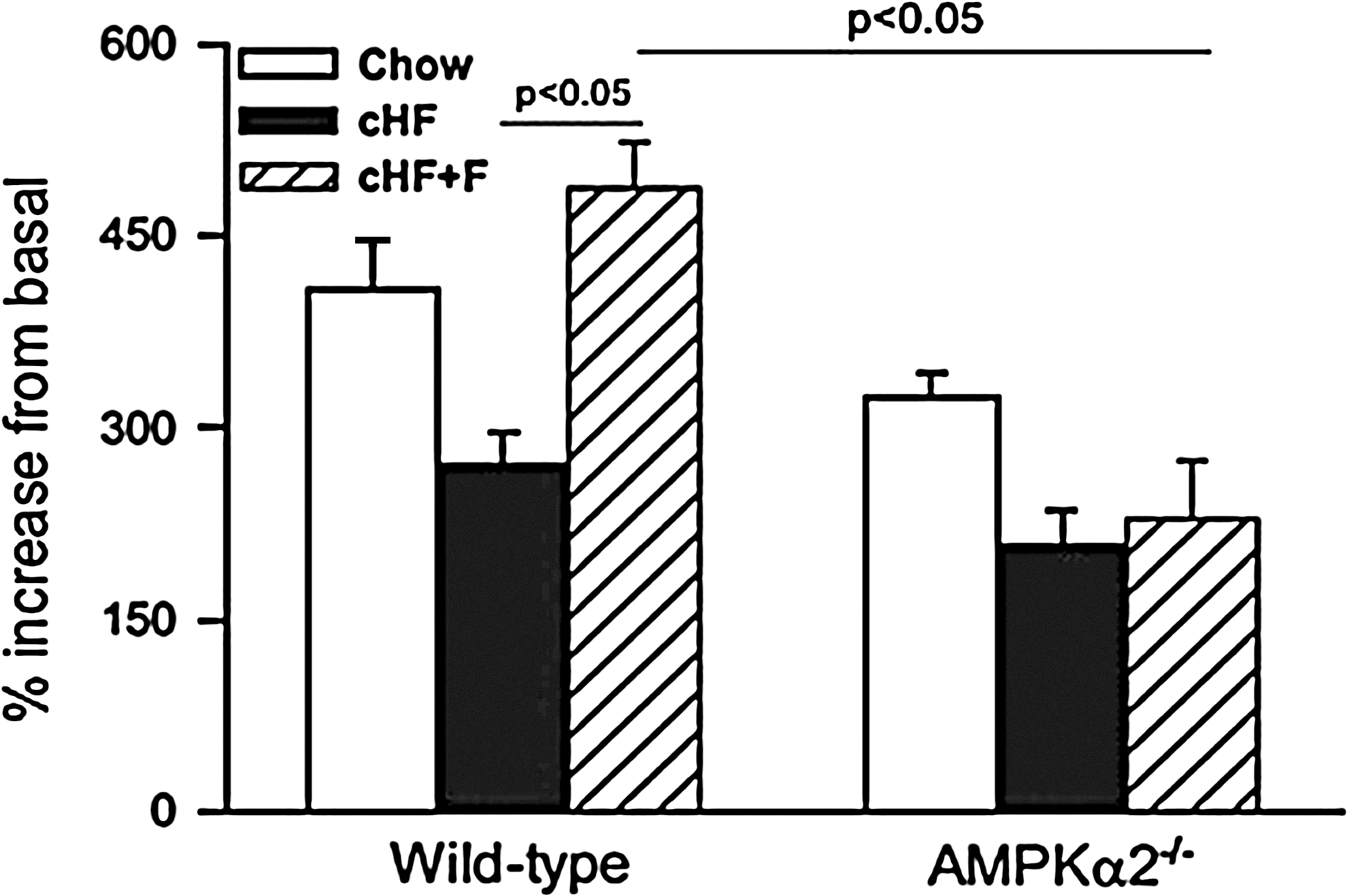
Aside from longstanding obesity, the large variety of diets due to unlimited combinations of dietary FFA species will have various effects on lipid bioavailability and distribution with complex consequences for cellular energy metabolism. There is growing evidence from mouse studies that lipid class, saturation index, and/or chain length of FFA differently affects insulin sensitivity and mitochondrial plasticity. For example, treatment of rat muscle cells with fatty acids of varying degree of saturation revealed that only saturated fatty acids, such as palmitate, impair both insulin response and ATP synthesis (26). The mono-unsaturated fatty acid oleate dose-dependently protected from palmitate-induced inhibition of insulin signaling and induction of ROS production in rat hepatocytes (49). Combining HFD enriched in n-3 polyunsaturated fatty acids (n-3 PUFA) and caloric restriction resulted in synergistic increase in mitochondrial oxidative capacity and lipid catabolism in adipocytes of mice (19). Eicosapentaenoic fatty acid rescued impaired mitochondrial oxidative capacity in LPL-deficient myotubes, probably via PPAR-δ-mediated activation of mitochondrial biogenesis (46). We showed that raising the ratio of dietary n-3 PUFA prevents HFD-induced reduction of palmitate oxidation in mouse hepatocytes (Fig. 7), which was dependent on (AMPK) and associated with improvements of insulin sensitivity and liver lipid content (27).
In summary, short-and long-term hyperlipidemia can impair muscular mitochondrial function and plasticity. But these changes do not seem to cause lipid-mediated insulin resistance but rather arise as a consequence of the altered cellular metabolism.
Conclusions and Future Directions
Some nondiabetic but insulin-resistant cohorts such as severely insulin-resistant FDR show reduced myocellular mitochondrial density, function, and plasticity (4, 45, 53, 54, 56). In less insulin-resistant FDR (29, 56) or women with a history of gestational diabetes, pGDM (60) do not show abnormal basal mitochondrial function. Thus, abnormalities of mitochondrial function do not necessarily precede insulin resistance or T2DM, which is supported by findings that muscle- and liver-specific deficiency of OXPHOS does not cause insulin resistance, at least in mice (58, 80). However, it is uncertain whether insulin-sensitive FDR and pGDM also show altered mitochondrial plasticity or will develop T2DM later in their lives. At least, in some certain variants of mitochondrial diabetes, the inherited mtDNA mutation seems to cause both impaired myocellular mitochondrial plasticity and insulin resistance (77). Of note, patients with congenital mutations of insulin receptor and severe insulin resistance can secondarily develop impaired in vivo mitochondrial function (71). Similarly, mice with liver-specific downregulation of IRS-1/IRS-2 are insulin resistant and show dysregulated mitochondrial biogenesis and function (12), supporting the hypothesis that insulin resistance leads to impaired mitochondrial function (Fig. 8B).
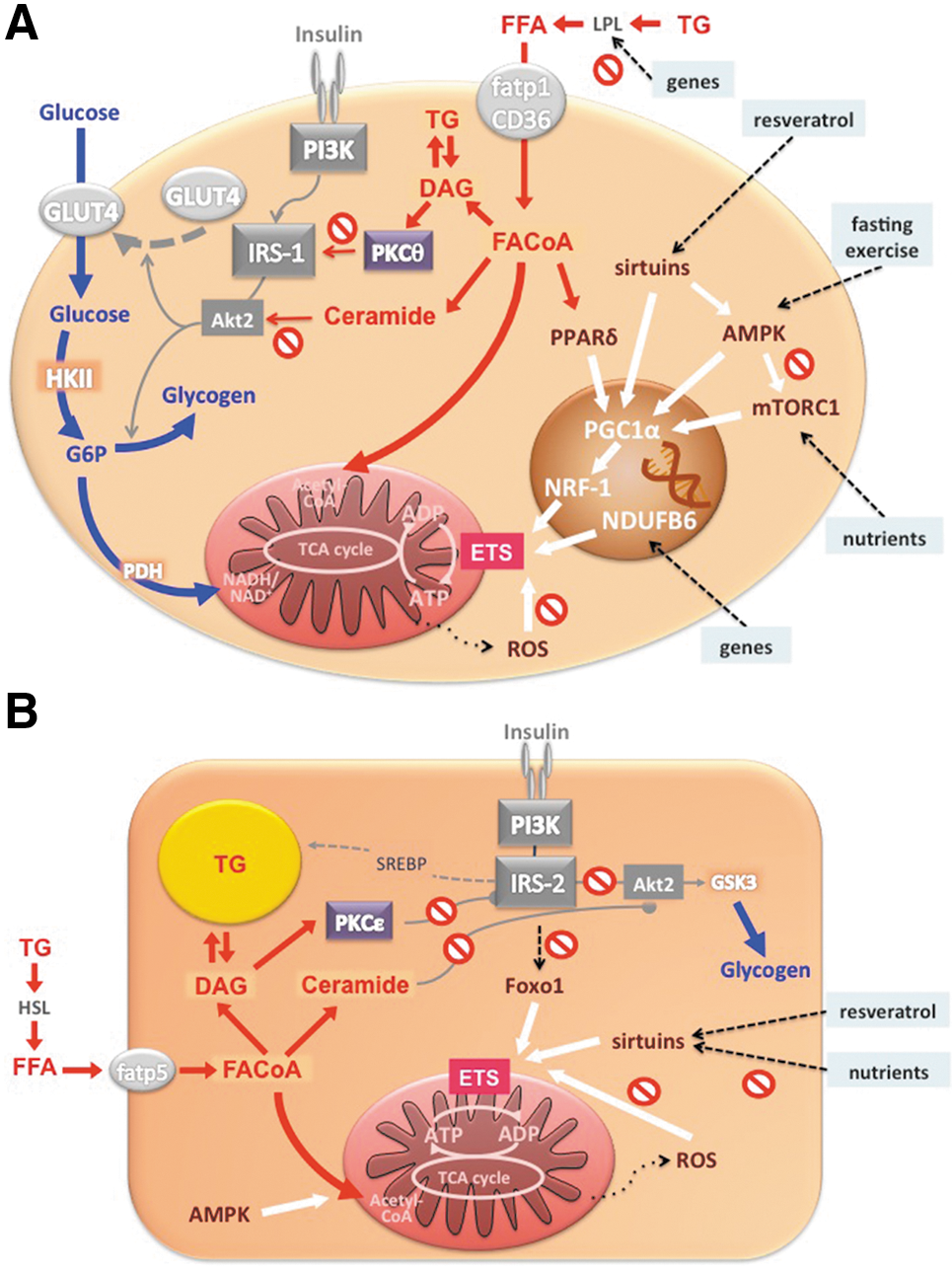
Acutely, excessive lipid availability induces insulin resistance, which can be followed by lower mitochondrial plasticity (Fig. 6) (8, 9). Chronic elevation of lipid availability during the development of obesity seems to induce an adaptation of mitochondrial oxidative capacity in that it increases proportionally with the degree of obesity in insulin-sensitive humans but falls in the most obese at the onset of insulin resistance (11). The concept of such adaptation has been supported by findings in animal models on HFD and in human athletes in whom myocellular lipid accumulation is associated with high oxidative capacity and insulin sensitivity. HFD- and obesity-induced activity of mTORC1, a positive regulator of mitochondrial biogenesis in muscle (15), could be a link between nutrient overload and increased mitochondrial function (39) (Fig. 8A). However, all these models do not mirror human hyperlipidemia and obesity, which may exhibit differences in amount and composition of cellular lipids inhibiting insulin signaling (e.g., diacylglycerols or ceramides), and in production of ROS and lipid peroxides. For example, ex vivo treatment of skeletal muscle cells with physiological concentrations of palmitate suppressed, while lower palmitate levels improved ATP production (1).
Finally, the contributions of various insulin-sensitive tissues to the pathogenesis of T2DM may vary. In glucose-tolerant women with pGDM, liver lipid storage rather than muscular mitochondrial activity correlated with insulin sensitivity (60), indicating that abnormal hepatic rather than muscular energy metabolism might be present in certain groups at increased risk of T2DM, as suggested for overt T2DM (69, 74).
In conclusion, insulin resistance can impair mitochondrial function or vice versa, but both abnormalities are not always causally related. The different findings cannot be exclusively attributed to differences in the employed methods or protocols, but also result from the multifactorial alterations underlying the heterogeneous condition, currently defined as T2DM. Alterations of the intracellular lipid profile, maybe resulting from inadequate fatty acid oxidation, can impair insulin signaling and thereby serve as a causal link between abnormal mitochondrial function and insulin resistance.
Footnotes
Acknowledgments
The studies by the authors were supported in part by a DAAD-Leibniz scholarship and by a grant from the Czech Science Foundation (P301/10/1420; Grant recipient: Martin Rossmeisl) to TJ, and by grants from the European Foundation for the Study of Diabetes (EFSD), Juvenile Diabetes Foundation (JDRF), Schmutzler-Stiftung, Skröder-Stiftung, German Research Foundation (DFG; SFB 512) to MR, and from the German Federal Ministry of Education and Research (BMBF) to the German Center for Diabetes Research (DZD e.V.).
Author Disclosure Statement
No competing financial interests exist.