Abstract
Introduction
Free radical theory of aging
The “free radical theory of aging,” as formulated by Denham Harman proposes that reactive oxygen species (ROS), generated due to oxygen metabolism, are the driving force behind aging and age-related diseases (68). ROS include the free radicals oxygen singlet (1O2), superoxide anion (O2 •−), and hydroxyl radical (OH•–) as well as the nonradical H2O2. ROS are normal by-products of metabolic processes, such as oxidative phosphorylation in mitochondria, or they are generated by growth factors and cytokines through the action of NADPH oxidases [reviewed in ref. (43)]. Cellular homeostasis of ROS is maintained by the function of certain detoxification systems, namely, the superoxide dismutases (SOD), catalase, the glutathione/glutathione peroxidase, and the thioredoxin/peroxiredoxin system. However, intracellular free radicals increase with age due to accumulation of dysfunctional mitochondria and functional decline of the cellular antioxidant defenses. Eventually, ROS react with cellular constituents and cause oxidative damage; it is proposed that this accumulation of ROS-induced damage over time drives the aging process (Fig. 1).
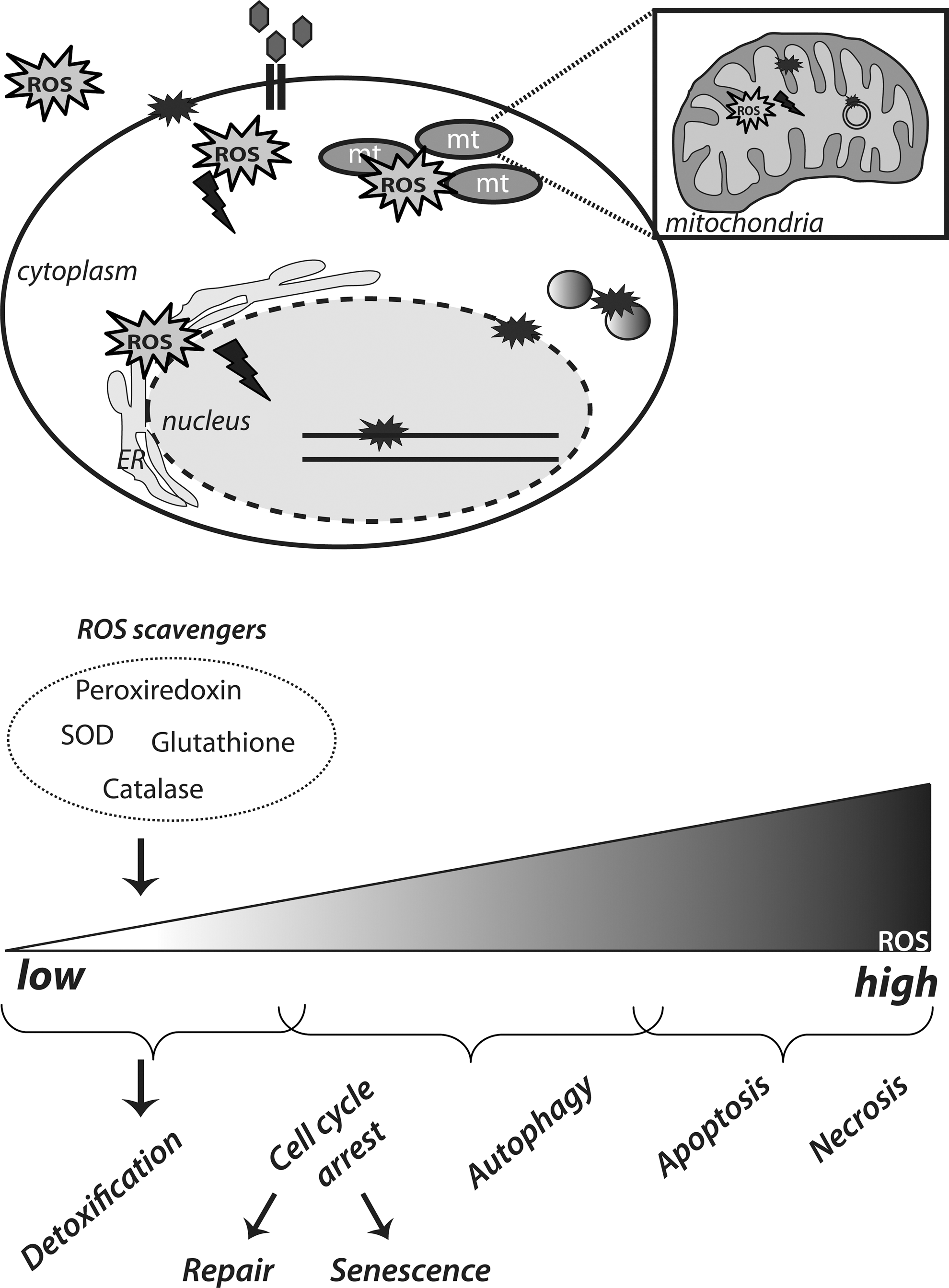
In support of the “free radical theory of aging,” lifespan in most animals inversely correlates with their metabolic rates and, therefore, the rates of ROS generation (78). Moreover, mouse models with impaired antioxidant defenses and, thereby, increased intracellular ROS, show increased prevalence of age-related diseases. For example, SOD1 null mice exhibit a shortened lifespan and age-related degeneration (44, 127). However, the reverse, a mere increase in expression of ROS detoxifying enzymes does not always result in increased longevity. Mice engineered to overexpress catalase targeted to the nucleus or the peroxisomes do not show any change in their median or maximal lifespan. Rather, targeted overexpression of catalase in the mitochondria resulted in increased lifespan of these mice compared to their wild-type littermates, accompanied by attenuation of aging associated pathologies (147). Interestingly, SOD2−/− mice are short lived (101, 109), but this is in apparent contrast with recent studies in Caenorhabditis elegans that show SOD2 lacking worms to have extended lifespan, despite increased oxidative damage (181). At the same time, overexpression of SOD1 in C. elegans increases the lifespan, but this is not attributable to improved ROS scavenging (20). Moreover, depletion of all nematode SODs does not appear to affect the lifespan (182). Overall, despite the fact that data from studies in C. elegans appear to challenge the free radical theory of aging, one could argue that they actually reflect the complicated balance of SOD function(s) in maintaining optimal cellular redox. This is, particularly, important in the light of the essential and nondamaging roles of ROS in normal signaling. In addition, when comparing these apparent discrepancies in lifespan determination between nematodes and mice, it should be taken into account that there are crucial differences in the physiology of animals having mainly postmitotic somatic cells (C. elegans) versus animals in which somatic cells do still divide.
Overall, even though the ‘free-radical theory’ provides a framework for understanding lifespan and aging, the biology of the aging process appears to be more complicated.
ROS and DNA damage
ROS can induce a variety of DNA lesions, including base modifications and DNA breaks (150). Base modifications (for example, 8-oxo-guanine, 8-oxo-G) have a high mutagenic potential by generating mismatches, whereas DNA breaks are predominantly cytostatic/cytotoxic lesions, inducing growth arrest and apoptosis (72) (Fig. 2). To avoid the accumulation of ROS-induced DNA mutations, cells are equipped with systems dedicated to the detection and clearance of modified nucleotides, referred to as base excision repair (BER). After the induction of DNA breaks, specialized signaling cascades are initiated (30); recognition of DNA breaks activates the phosphoinositide-3 kinase (PI3K)-related kinases family (PIKKs), that subsequently orchestrate the cellular response for DNA repair. DNA breaks are repaired by two main pathways, namely, nonhomologous end-joining (NHEJ) and homologous recombination (HR) (30). NHEJ occurs throughout the cell cycle and is a very rapid mechanism for DNA repair, as it is based on bridging together, the broken DNA ends. This mechanism of DNA repair, however, is error-prone, as it may lead to the loss or gain of bases as well as to chromosomal translocations and eventually genomic instability. In contrast to NHEJ, HR utilizes the genetic information of the intact sister chromatid for the faithful repair of the lesion and, thus, is considered an error-free mechanism of repair; HR occurs exclusively in the late S and G2 phase of the cell cycle, when the sister chromatid is available.
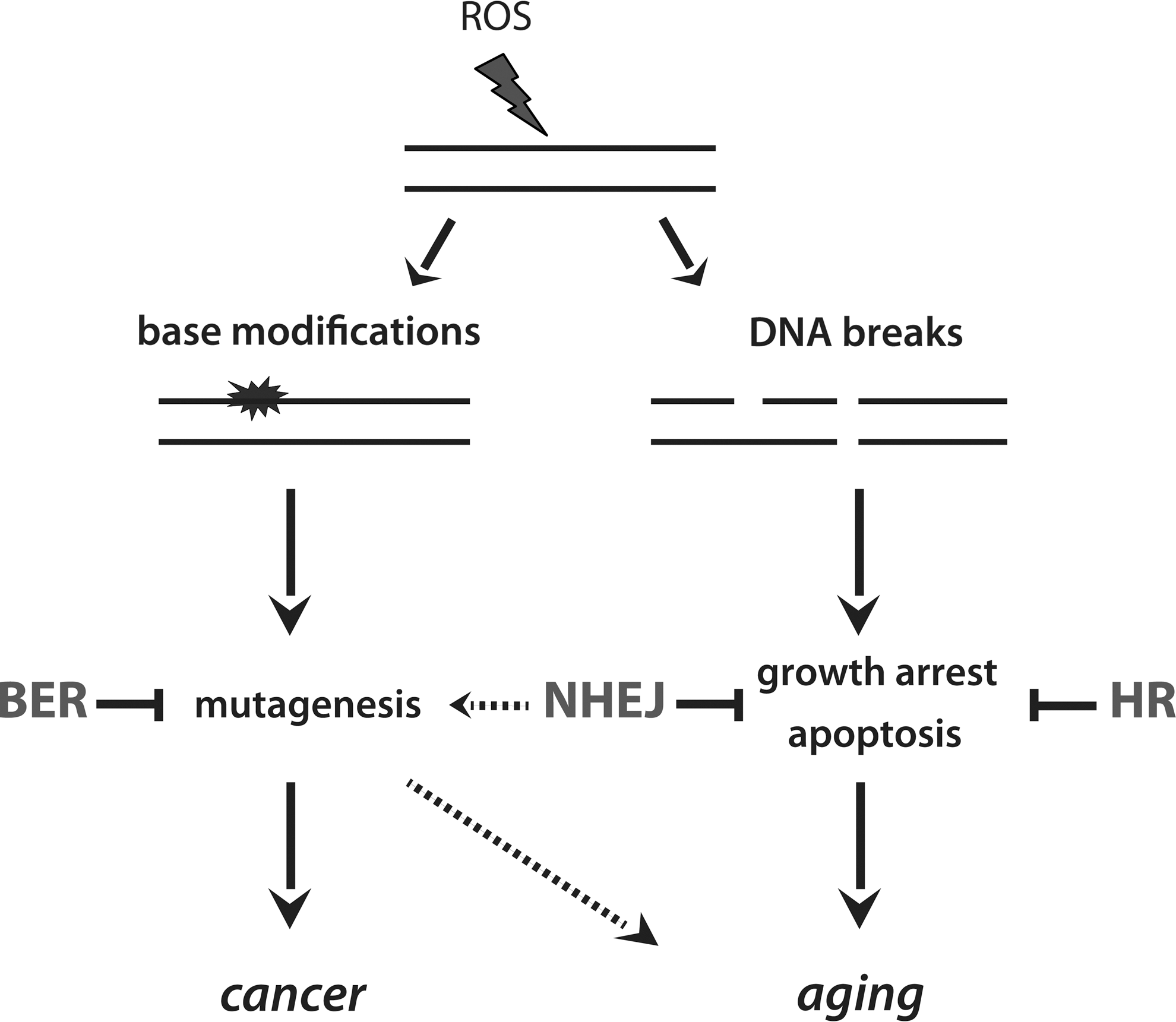
When damage is too severe or not properly repaired, other protection mechanisms are activated. Cell cycle arrest, which is normally induced by the DNA damage response signaling to facilitate DNA repair, can become irreversible, a phenomenon termed senescence. Moreover, cells experiencing severe DNA damage can commit to programmed cell death (apoptosis).
The DNA damage theory of aging
The aging process and aging related pathologies have been linked to the accrual of DNA damage (11, 65, 150). This accrual may derive from the increased ROS load in aged tissues, as well as the functional decline of DNA repair mechanisms. Ultimately, accumulated DNA lesions are associated with increased prevalence of mutations and blockage of physiological processes, including replication and transcription. Responses that are activated eventually result in the induction of senescence and apoptosis, processes that are both linked to the functional decline observed in aging. Furthermore, it is important to consider that, whereas other damaged cellular macromolecules (lipids, proteins) can be efficiently removed and replaced by new undamaged molecules, this is not the case for the genetic material. Damaged DNA persists for the cell's lifetime, while carrying the information for the generation of all other cellular components. Therefore, accumulation of genomic instability over time results, among others, in the production of defective cellular components and is, thereby, connected to the cellular deterioration associated with aging [DNA damage theory of aging, reviewed in ref. (48)].
In agreement with the above, human genetic disorders with disruptions in certain DNA repair pathways are characterized by early onset of aging associated diseases (Table 1). Some typical examples are the Werner syndrome and ataxia telangiectasia (A-T), both with mutations in proteins associated with double-strand breaks (DSB) repair, as well as syndromes associated with defects in transcription-coupled repair. Interestingly, most of these genetic disorders are also characterized by increased intracellular ROS (Table 1). These observations beg the question what is the cause and consequence with respect to the aging phenotype and whether impaired DNA repair or the increased ROS load, is the cause of premature aging observed in these disorders.
DSB, double-strand breaks; ND, not determined; ROS, reactive oxygen species; UV, ultraviolet.
Nonetheless, whether the “free radical” and “DNA damage theory” are complementary or not is still under debate. Indeed, mice engineered with proofreading deficient mitochondrial DNA (mtDNA) polymerase exhibit reduced lifespan and early onset of age-related disorders (169). However, no evidence for increased oxidative mtDNA damage was observed in this model. Moreover, as mentioned, targeted overexpression of catalase in the nucleus does not have an impact on lifespan, suggesting that nuclear oxidative damage may not be a cause of aging. Regarding the “DNA damage theory,” the main criticism is raised on the grounds that, whereas reduced DNA repair is associated to premature aging, the opposite, an increase in lifespan due to enhanced efficiency of DNA repair has not been observed (38).
Aging and Cellular Homeostasis
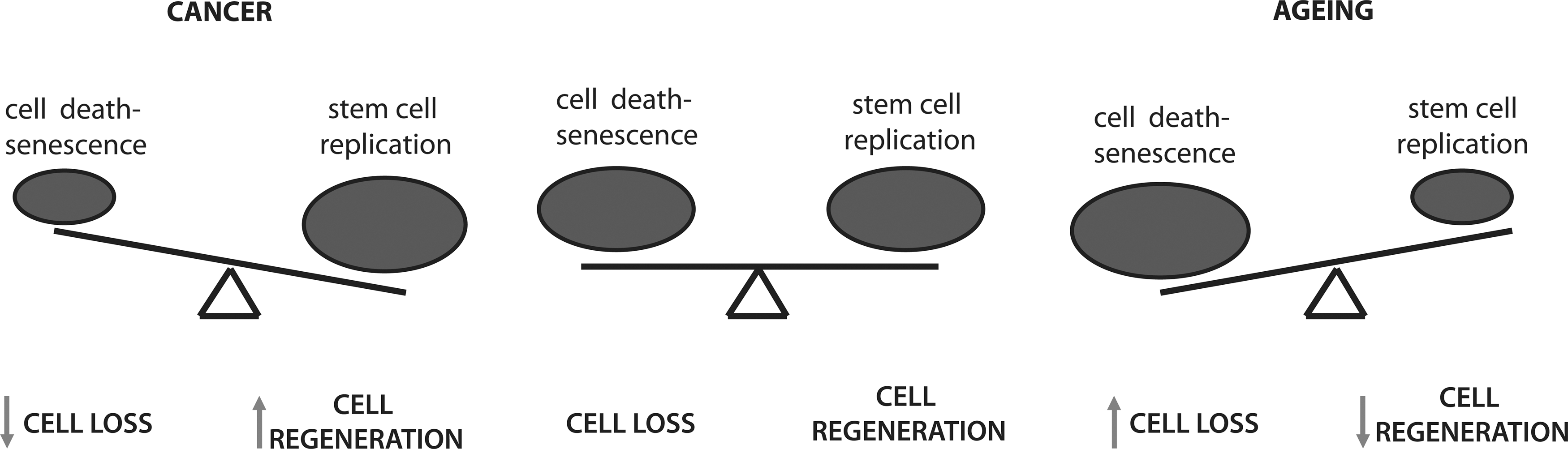
The theories of aging discussed above, although attractive to explain certain aspects of aging and age-associated diseases, certainly do not provide all answers and have been challenged by experimental data. Irrespectively, these theories share the perspective that the aging process is driven by progressively disturbed organismal homeostasis. Under normal conditions, there is an established balance between cell removal and regeneration in tissues; damaged and transformed cells are constantly removed and replaced by new functional cells. However, during aging, homeostasis is disturbed (Fig. 3). When clearance of dysfunctional cellular components is impaired, these damaged components (lipids, proteins, and organelles) will accumulate and cause cellular dysfunction (e.g., cellular senescence). If not eliminated, they will maintain tissue mass, but also contribute to aging (discussed later). Alternatively, when eliminated, cells are no longer replaced and tissues are progressively becoming more atrophic. Also, tissue regeneration upon trauma is not as efficient and different organs are more labile to malfunction; all the above constitute hallmarks of aging.
The insulin-dependent regulation of the Forkhead box O (FOXO) transcription factors serves as a paradigm for our understanding as to how disturbed homeostasis interconnects with age-related diseases and lifespan. Insulin regulates the FOXO activity through the PI3K/protein kinase B (PKB) pathway (16, 94). In human cancer, the PI3K/PKB pathway is often deregulated. Activating mutations in PI3K and PKB occur at moderate frequency in various types of cancer, while deletion or loss of function mutations within phosphatase/tensin homolog deleted on chromosome 10 (PTEN), the lipid phosphatase that counteracts PI3K function, occur at relative high frequency [for review see ref. (112)]. Furthermore, several receptor tyrosine kinases (e.g., epidermal growth factor receptor) are also frequently overexpressed in certain types of human cancer and in these settings efficiently activate PI3K signaling. Thus, on average over 50% of human tumors express somehow hyperactive PI3K/PKB signaling. On the other hand, reduced PI3K/PKB signaling is a hallmark of diabetes (163). The importance of the PI3K/PKB pathway in insulin action toward metabolic end points, such as glucose regulation, is illustrated by the observation that within a family showing autosomal dominant inheritance of severe insulin resistance and diabetes mellitus, a mutation in the PKBβ/AKT2 gene was identified and that expression of the resulting mutant kinase in cultured cells disrupted insulin signaling to metabolic end points (50). Thus, either gain- or loss-of-function of PI3K/PKB signaling contributes significantly to two important age-related diseases that is, cancer and diabetes (Fig. 4).

The relevance of the PI3K/PKB pathway toward FOXO to age-associated disorders is, ultimately, supported by the fact that this pathway has an evolutionary conserved role in lifespan determination. In 1993, Kenyon et al. reported that a mutation in daf-2, the homologue of the insulin and insulin-like growth factor (IGF) receptor in C. elegans, increases lifespan by twofold and this is dependent on daf-16, the FOXO homologue (87). Likewise, in Drosophila melanogaster, overexpression of the dFOXO protein was shown to increase lifespan (51). Importantly, FOXOs appear to regulate lifespan also in mammals, including humans; genome-wide association studies using centenarian cohorts, reveal polymorphisms in FOXO1 and FOXO3a loci that associate with increased longevity (47, 97, 189).
FOXOs are integral components in the responses to ROS and DNA damage
FOXOs regulate cell fate decisions, including cell proliferation, cell death, and cell metabolism. To provide cellular homeostasis toward stress signals that disturb homeostasis, the FOXO-induced response should combine and fine-tune these distinct processes into one.
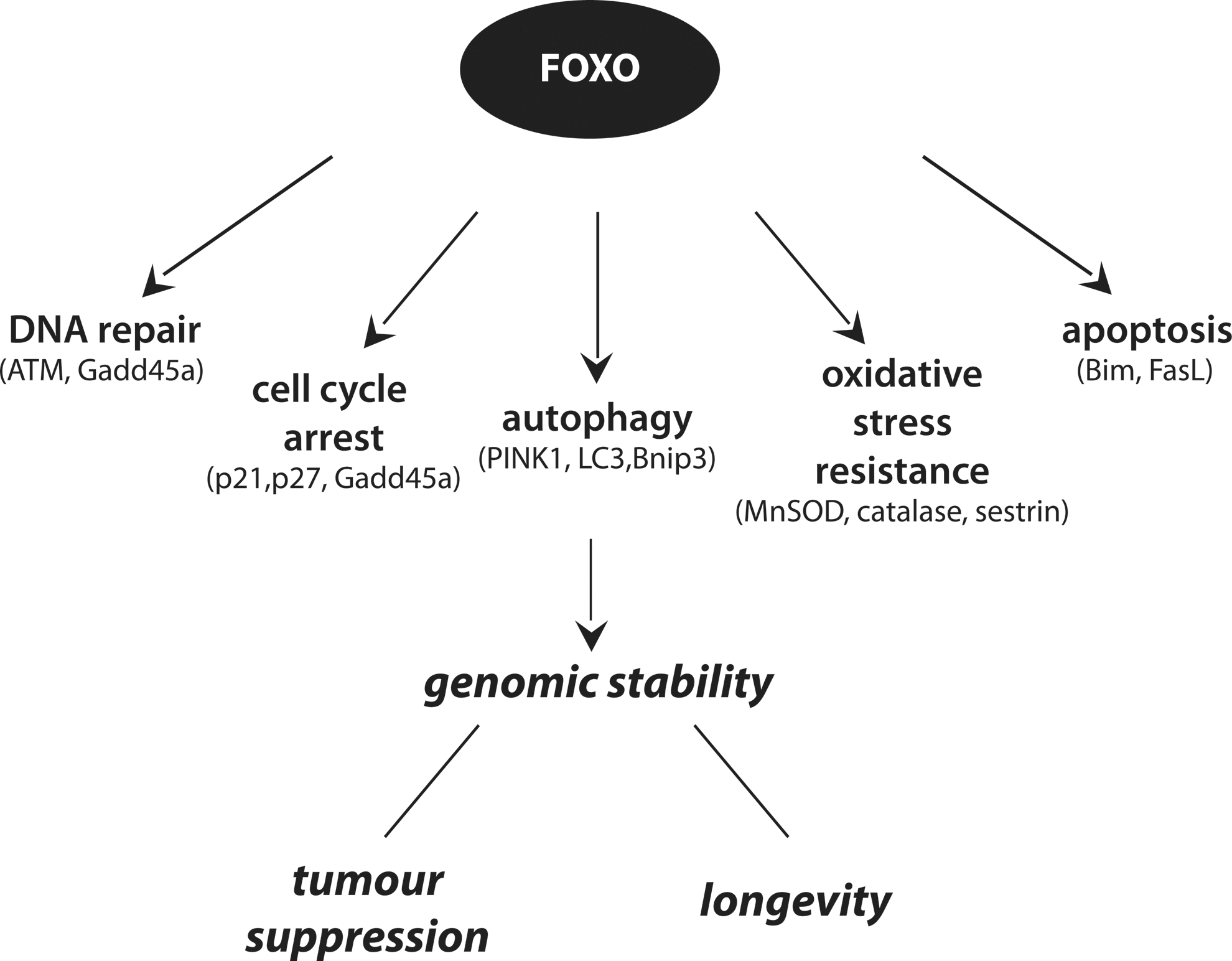
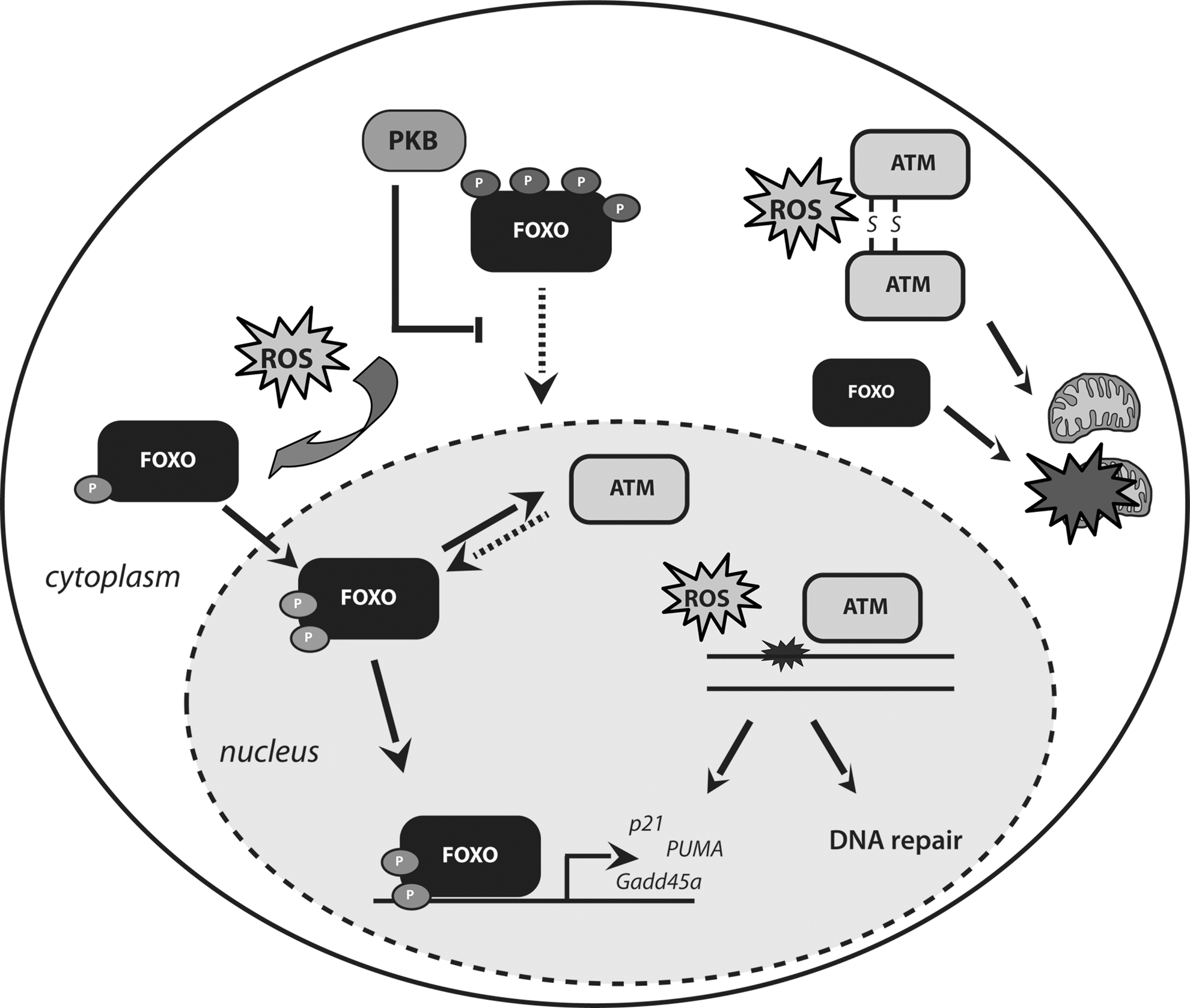
FOXO activity is regulated by a number of signaling cascades activated in response to increased intracellular ROS (21), as well as by direct ROS-induced changes [cysteine oxidation (34)]. FOXOs, in turn, act as homeostatic regulators because they do not only respond to oxidative stress, but also protect cells from oxidative stress through the transcription of ROS scavenger genes, including SOD2 (or manganese superoxide dismutase [MnSOD]), catalase, peroxiredoxin 3, and members of the sestrin family (26, 28, 93, 130). Mice engineered with inducible deletion of the three predominant FOXO members (FOXO1, FOXO3a, and FOXO4, thereafter called FOXO triple knockout [FOXO TKO] mice) display increased intracellular ROS in the stem cell (SC) compartment, associated with decreased expression of certain ROS detoxification enzymes. The associated effects, including hematopoietic stem cells (HSCs) depletion, were alleviated by concomitant treatment with antioxidant agents (134, 167).
A growing body of evidence suggests that FOXOs also facilitate DNA repair, by regulating defined transcriptional programs and executing transcription-independent functions. Induction of ultraviolet (UV) damage activates FOXO transcriptional activity toward growth arrest and DNA damage inducible 45a (Gadd45a) (168), a protein implicated in DNA damage checkpoint maintenance (138, 188) as well as in certain types of DNA repair (82, 157, 158). However, how GADD45a partakes in DNA repair has remained fully enigmatic. A role for GADD45 in DNA demethylation has been suggested (8), but remains debated [see e.g., ref. (100)]. In fact, there is still no consensus as to how active enzymatic DNA demethylation is achieved in mammalian cells, but recent studies implicate BER in genome-wide DNA demethylation in germ cells and early embryos. Thus, GADD45a may somehow interact with, or be part of the BER machinery. Considering that UV irradiation induces, inter alia, oxidative damage that requires BER for its removal, this may account for the observed increase in DNA repair following FOXO activation reported by Tran et al. (168). However, FOXOs may also have additional functions in genotoxic stress protection. Cells deficient for PTEN or expressing hyperactive PKB show increased genomic instability due to inhibition of HR (39, 123, 137, 191). Considering that FOXOs are important downstream components of PI3K/PKB signaling, one could speculate that FOXOs could also play a role in faithful DNA repair via HR and, thereby, contribute to genomic stability. Furthermore, FOXO3 was recently reported to physically interact with ataxia-telangiectasia mutated (ATM) kinase and this interaction was suggested to be required for full activation of ATM in response to DNA damage and the subsequent DNA damage checkpoint activation (170). Importantly, this physical association is in agreement with the observed FOXO1 phosphorylation by ATM, reported in a study by Matsuoka et al. (119). In addition, FOXOs were also reported to regulate the transcription of ATM in HSCs (194).
Under conditions of genotoxic stress, FOXOs have been shown to regulate the expression of the cyclin-dependent kinases (CDK) inhibitors, p27kip1 and p21waf1/cip1, to induce cell cycle arrest (90, 104, 120). Importantly, sustained p21waf1/cip1 expression is associated with senescence induction and recent studies on oncogene-induced senescence suggest FOXO activation to mediate senescence (36) at least, in part, through regulation of p21waf1/cip1. Under conditions of excessive cellular damage, FOXOs were shown to regulate induction of apoptosis, by initiating the transcription of the Bcl-2 family proteins, Bim and PUMA (114, 199). Interestingly, FOXO-mediated apoptosis appears to have practical implications in cancer therapies. In particular, FOXOs appear to sensitize cancer cells to chemotherapy that acts through genotoxic stress, such as doxorubicin and cisplatin, thereby reducing the effective dosage of these drugs and, accordingly, the risk of side effects (42, 53, 154, 159).
Overall, FOXOs appear to contribute to longevity by regulating processes that are both antiaging (ROS scavenging, DNA repair) as well as proaging (inducing senescence and apoptosis). Below, we will discuss how these FOXO roles relate to the main cellular events associated with excessive cellular elimination—and therefore disturbed homeostasis—during aging, which are increased genomic instability, telomere attrition, and accumulation of damaged and dysfunctional macromolecules and organelles. Furthermore, we will elaborate on the association between ATM and FOXO because of their corresponding functions in aging and cancer and the increasing evidence for reciprocal regulation of FOXOs and ATM.
Tumor suppression and aging: FOXOs and ATM are bona fide tumor suppressors
Aged tissues accumulate unrepaired (or not properly repaired) DNA lesions caused by both excessive ROS and exogenous insults (e.g., UV irradiation) (142, 149, 184). The accrual of DNA mutations increases the potential for cellular transformation and cancer. Paradoxically, the aging process is characterized by a gradual functional decline attributed mostly to cell loss, whereas cancer, a major age-related disease, is characterized by the opposite; that is an uncontrolled increase in cell numbers. On the other hand, responses activated to prevent cancer and limit cell number are inducing aging phenotypes.
Recognition of DNA lesions by the cellular surveillance mechanisms initiates signaling cascades for cell cycle arrest and DNA repair or, in cases of severe and persistent damage, senescence and apoptosis (30). Senescence is the irreversible withdrawal from the cell cycle and it constitutes a prominent tumor suppressor mechanism, since it leads to the elimination of aberrantly proliferating cells. What determines entry into senescence or apoptosis is still unclear; however, the cell type appears to be important. It is hypothesized that senescence occurs more often in less proliferative tissues and tissues in which the physical presence of the cell is crucial for their integrity, for instance, the skin. Contrary to senescence, apoptosis is more often induced in highly proliferative tissues, with the exception of neuronal cells that although postmitotic commit to apoptosis in response to damage accumulation. Importantly, although senescence is employed as a mechanism for tumor suppression, senescent cells produce and secrete inflammatory cytokines that have been shown to stimulate tumorigenesis in neighboring cells (35). Both senescent and apoptotic cells are cleared by the immune system, providing efficient mechanisms to prevent disease. However, the efficiency by which the immune system clears senescent cells appears to become less during aging, thereby contributing to aging associated diseases (19). In line with this, a recent study showed that improved removal of senescent cells in a mouse model of premature aging alleviated the aging process (5).
Senescence and apoptosis are typical responses activated downstream of the DNA damage response. ATM is a central kinase in the DNA damage response and it regulates the pathway for the induction of both senescence and apoptosis, mainly through activation of the p53 tumor suppressor. In fact, deletion or inhibition of ATM kinase activity in vitro and in vivo abolishes both of these processes, resulting in uncontrolled proliferation of damaged cells and carcinogenesis (153). Moreover, A-T patients suffer from cancer predisposition, with increased prevalence of lymphomas and lymphoid leukemias (152). Also, after induction of replication stress due to oncogene activation, ATM and Rad3 related (ATR) kinase regulates similar responses, again through p53 (9, 64). Early research on FOXO function already implied a potential role for FOXOs as tumor suppressors, because FOXOs regulate growth inhibition and cell death in response to cellular stress (33). Importantly, FOXO TKO mice suffer from thymic lymphomas and hemangiomas (134) and this established FOXOs as bona fide tumor suppressors. FOXOs inhibit cancer progression by affecting several processes, including inhibition of tumor vascularization [for review see ref. (33)]. At the cellular level, their main contribution is by inducing cell cycle arrest (also senescence) and apoptosis in response to oxidative stress and other genotoxic insults (33, 55).
DNA damage-induced transcription block and progeria
Accumulated DNA lesions are not only deleterious with respect to oncogenic transformation, but are also responsible for cellular dysfunction and induction of cytostatic events. Transcription blocking lesions (25, 49) cause defects in protein production and the subsequent performance of vital cellular functions. Seminal work by Hoeijmakers and associates revealed that genetic disruption of transcription-coupled repair components results in premature aging and age-related pathologies, associated with defects in transcription progression (3, 131, 148, 179).
Interestingly, transcriptome analysis of progeroid mice with DNA repair dysfunctions revealed a suppression of the somatotropic axis (growth hormone and IGF-1) (131, 179). This is quite paradoxical, considering that previous studies linked reduced insulin–IGF signaling (IIS) signaling to increased longevity in C. elegans and mice (10, 73, 87). However, in cases of chronic DNA damage due to repair defects, attenuation of the somatotropic axis can be considered as an adaptive response that shifts the natural resources from growth to maintenance. By attempting to reduce metabolic rates, this response adds to the reduction of intracellular free radicals and the ensuing genomic instability. At the same time, increased transcription of ROS detoxification enzymes is also observed, as a mechanism to further reduce ROS-induced damage. Importantly, these mice also exhibit increased transcripts of proapoptotic proteins, probably due to the inhibition of the prosurvival IIS axis (131).
The role of FOXOs has not been studied yet in this genetic system. However, since FOXOs are expected to be active due to loss of IIS signaling, these mouse models would be particularly informative to study FOXO function under conditions of increased genomic instability.
Telomere attrition in aging
Telomeres constitute the chromosome ends and are protected by specialized structures, to prevent their attrition and fusion (24, 37, 135). Each DNA replication round results in gradual shortening of the telomeres and, in the absence of adequate telomerase activity, telomeres gradually shorten until they reach a critical length (110). At this point, telomeres lose their protective capping and are sensed by the DNA damage surveillance system as DNA breaks (37, 135). Eventually, the DNA damage response, orchestrated by ATM and ATR, results in senescence induction (replicative senescence) or, in cases that DNA repair with NHEJ is activated, chromosome fusions occur and the ensuing genomic instability leads to carcinogenesis.
Telomere erosion occurs during DNA replication, but is also modulated by other factors. Telomeres are repetitive sequences rich in G and C nucleotides, which have been shown to be particularly sensitive to oxidative damage (69), and telomere shortening occurs with higher rates under oxidative stress conditions (54, 136, 140, 183). Importantly, treatment with antioxidants or increased expression of dismutases under oxidative stress conditions appeared to rescue the telomeric length and delay the subsequent replicative senescence (151). Considering the role of FOXOs in regulating the expression of ROS detoxifying enzymes, it is tempting to speculate that another contribution of FOXOs in extending lifespan is by indirectly regulating the telomere length. Irrespective, other potential functions of FOXOs in the setting of uncapped and dysfunctional telomeres would be equally interesting to determine.
Aging and mitochondrial dysfunction: FOXOs and ATM in mitochondria protection
An important mediator of the increased ROS accrual in aged tissues is mitochondrial dysfunction (115, 136). mtDNA is especially sensitive to ROS and consequent accumulation of damage as it is directly exposed to free radicals leaking from the respiratory chain. Moreover, mtDNA is not protected by histones and the mitochondria resident repair mechanisms are less sophisticated, compared to the repair systems operating within the nucleus (99). Importantly, mtDNA regulates the expression of proteins involved in mitochondria assembly and function. As a result, aged mitochondria, with increased load of mtDNA mutations, function sub-optimally and release more free radicals (62, 155). Neurodegeneration associated with age-related disorders, such as Parkinson's and Alzheimer's disease, is connected to an increase in ROS due to accumulation of defective mitochondria within postmitotic neurons (31). Mitochondrial dysfunction is also observed in metabolic syndrome, a pathology associated with insulin resistance and eventually development of diabetes and cardiovascular disorders (124). Additionally, age-associated dysfunctions in the cardiovascular system have been connected to increased mtDNA mutations and increased ROS leakage (124, 172, 190).
Cells can mount certain protective responses to mitochondrial dysfunction and, thus, mitochondrion-induced ROS accumulation. One cellular strategy is mitophagy, an autophagic process (see below) that results in the specific removal of dysfunctional mitochondria (102, 186). Whereas mitophagy is still not fully understood, certain mediators of the process have been identified. An important mediator of mitophagy is the PTEN-induced putative kinase-1 (PINK1), which regulates the accumulation of the protein parkin in damaged mitochondria, thereby regulating mitophagy initiation (86, 129). Importantly, when dysfunctional mitochondria are not efficiently cleared and the intracellular free radical load increases significantly, cell death is induced (115).
FOXO proteins are important regulators of oxidative stress resistance and FOXO TKO mice were reported to show mitochondria dysfunction (144). FOXOs regulate the expression of free radical detoxifying enzymes, including the mitochondrial resident MnSOD, thereby regulating mtDNA integrity and, ultimately, mitochondria function. FOXOs were recently reported in two independent studies to be activated in hypoxic tissues and antagonize c-Myc function (46, 81). As a result, at least in the setting of hypoxia, FOXOs contribute to the maintenance of low mitochondria number and the reduction of intracellular ROS by inhibiting transcription of mitochondrial genes. Moreover, FOXOs were reported to regulate the transcription of PINK1, suggesting a possible involvement of FOXOs in clearance of damaged mitochondria (121). Importantly, transcription of PINK1 by FOXO was found associated with protection from apoptosis, possibly by inducing mitophagy. Furthermore, recently in Drosophila, dFOXO was shown to rescue the mitochondria dysfunction stemming from PINK deletion, suggesting PINK-independent functions as well (91). In fact, FOXOs were reported to regulate the expression of CITED2 (6), which, in turn, regulates the levels of Bmi1 (95). Interestingly, mice deficient for Bmi1 show increased stress characterized by the accumulation of dysfunctional mitochondria and increased p16INK4 expression (111), an increase also observed in FOXO TKO mice (167).
Considering the role of dysfunctional mitochondria in aging related pathologies, it is conceivable that other aging associated components and signaling pathways will also be involved in mitochondria integrity. Indeed, impaired mitophagy was recently reported in mice deficient for the ATM kinase (174). In fact, it was proposed that this deficiency causes the increased free radical accumulation in A-T patients' tissues and could account for cerebellar degeneration, a common A-T symptom (152).
Autophagy in aging: functions for FOXOs and ATM in regulating autophagy
Autophagy is an evolutionary conserved process involving the controlled degradation of cytoplasmic contents, through the lysosomic pathway. More specifically, it involves the engulfment of parts of the cytoplasm in double-membraned vesicles termed “autophagosomes” and subsequent fusion of these vesicles with lysosomes [for more detailed review see ref. (143)]. Eventually, the macromolecules are dismantled to smaller building blocks that become available for new cellular anabolic processes. Autophagy is the main mechanism for the turnover of organelles (e.g., mitophagy, see above) and it is activated in cases of nutrient starvation or in stress conditions. More specifically, oxidative stress, as mentioned, induces irreversible modifications in macromolecules (e.g., proteins), as well as whole organelles (e.g., mitochondria) affecting their function. Autophagic removal of these defective molecules/organelles maintains cellular homeostasis, while facilitating the production of new, undamaged molecules.
Autophagy is a process with important implications in aging. It has been observed that expression of central autophagy components, as well as autophagy efficiency in toto is impaired with aging (143). The importance of autophagy in the aging process in multicellular organisms was primarily demonstrated by studies in C. elegans, where deletion of genes important for autophagy results in dauer defects and reduced lifespan (66, 122, 165). Most importantly, the lifespan extension observed in nematodes due to reduced insulin/IGF-1 signaling or by dietary restriction is not only dependent on abnormal dauer formation-16 (DAF-16), the FOXO homologue in C. elegans, but also on functional autophagy (122). Moreover, in D. melanogaster, defective autophagy is associated with shortened lifespan (156) and in mice tissue, specific knockdown of certain autophagic genes results in aging associated phenotypes (143). Defective autophagy is also associated with age-associated disorders in humans; the main etiology of Alzheimer's disease is the accumulation of protein aggregates (beta amyloid plaques and tau protein tangles) and the subsequent neuronal death, whereas accumulation of protein aggregates in neuronal cells is also observed in Parkinson's and Huntington's disease (27, 67, 92).
FOXO proteins are involved in autophagy, both by regulating certain transcriptional programs, as well as mediating transcription-independent functions. FOXOs were reported to regulate the transcription of important autophagy regulators, including Atg8/LC3b, Bcl2/adenovirus E1B 19 kDa protein-interacting protein 3 (Bnip3), GABA(A) receptor-associated protein like 1, Beclin, and Atg12 [reviewed in ref. (180)]. In agreement with this, in neuronal cells, c-jun N-terminal kinase (JNK) deficiency was shown to activate FOXO transcriptional activity toward Bnip3, Atg8/LC3b, and Atg12, thereby inducing autophagy and neuronal cell survival (192). Autophagy induction has been linked, however, to both cytoprotection and cytotoxicity. The latter is better illustrated in a recent report by Zhao et al., in which cytosolic FOXO1 was shown to mediate autophagy, independently of its ability to regulate transcription (203). In particular, cytoplasmic FOXO1 was shown to be acetylated in response to oxidative stress or nutrient deprivation and subsequently interacting with Atg7, an autophagy mediator, thereby regulating autophagy and cell death of neoplastic cells.
In agreement with the reported function of ATM in mitophagy, ATM is also associated with autophagy regulation. In response to oxidative stress, cytosolic resident ATM was shown to activate adenosine monophosphate-dependent kinase (AMPK), thereby activating tuberous sclerosis protein 2 (TSC2) (2). TSC2 is found in complex with the TSC1 protein, regulating the function of the target-of-rapamycin kinase (TOR), via the inactivation of the small GTPase Rheb (117). TOR is an inhibitor of autophagy (143); therefore, ATM activity, via AMPK, results in inhibition of TOR and subsequent enhancement of autophagy, in response to oxidative stress.
Overall, apart from their association in response to DNA damage, FOXOs and ATM appear to regulate similar functions in protecting cells from diverse stress insults. It is, therefore, important that future research addresses whether these proteins function in distinct or the same signaling pathways to confer protection against ROS (Fig. 5).
Aging and Cell Regeneration
Aging, as previously mentioned, can be considered the result of disturbed homeostasis, which, in part, can be due to a reduced capacity to replenish damaged and dysfunctional cells. Tissue regeneration is achieved by tissue-specific adult SCs, which are resident in most tissues and are characterized by their unique abilities to self-renew, to replenish the SC pool and to differentiate to other types of cells (multipotency). Considering the importance of the SCs for the lifetime maintenance of tissue homeostasis, it is conceivable that these cells should be equipped with very strictly controlled mechanisms for their protection against exogenous and endogenous insults.
Free radicals and DNA damage responses in SCs
Recent studies on the intestinal SCs characterized by LGR5 expression have challenged the concept that all adult SCs are maintained in a slow cycling or quiescent state for most of their lifetime (7). To reconcile most of the experimental data, a model has been put forward proposing that for most tissues basically two adult SC types exist (108); one is maintained in quiescent state and contributes to the maintenance of the SC pool, while the other type comprises the highly proliferative SCs whose function contributes to cell replenishment and tissue regeneration.
Quiescence is a state of reversible growth arrest also referred to as G0; cells can accumulate in the G0 phase of the cell cycle following an arrest in G1; however, upon mitogenic stimuli, they can resume cycling. Contrary to cycling cells, which have to devote the majority of their metabolism toward synthesis of cellular constituents (DNA, proteins, and lipids) to replicate, quiescent cells lack this demand and, thus, can decrease and/or divert their metabolic rates toward different requirements. Important in the context of aging is the accompanying ability of quiescent cells to shift metabolism toward increased stress resistance. In agreement, numerous studies have reported that quiescent cells are reduced in size, show lower rates of nucleotide synthesis, increased stress resistance, increased autophagy to recycle cellular components, and increased glycolytic flux and, hence, reduced oxidative phosphorylation. However, the latter paradigm stems largely from studies using lymphocytes and it may not be, in fact, a general hallmark of quiescence, since a recent report showed a similar glycolytic flux in quiescent as well as proliferating human primary fibroblasts (105). Irrespectively, quiescent SCs are considered to be less metabolically active and experience less mitochondrial respiration and thereby ROS accumulation (107). Moreover, by remaining in the G0 phase of the cycle, SCs are not subjected to erroneous DNA replication. At the same time, antioxidant defenses are enhanced in SCs, thereby minimizing the risk of ROS-induced damage. The requirement for ROS modulation in the establishment of quiescence is further illustrated by the fact that fibroblasts from MnSOD−/− mice exhibit increased superoxide steady state levels and failure to exit the proliferative cycle (145). Therefore, in general, quiescent SCs are expected to be less exposed to genotoxic insults and accumulate less DNA damage. Nevertheless, DNA damage was found to accumulate in SCs with age, as measured by the accrual of phosphorylated histone variant H2AX foci, a marker of DNA damage response, suggesting lack of mechanisms for faithful DNA repair in these cells (142, 193). Importantly, SCs are largely dependent on their microenvironment for their maintenance and fate determination. Excreted factors, including cytokines, from cells comprising the SC niche were shown to induce free radical accumulation and DNA damage in HSCs, leading to SC transformation or dysfunction (13).
DNA repair is significantly impaired in quiescent SCs, in which cell cycle checkpoints as well as certain types of repair are inactive (12, 116). Importantly, in case that quiescent SCs exposed to genotoxic stress re-enter the cycle, they will first proceed to the G1 phase. As previously mentioned, in G1, DSBs are repaired by the error-prone NHEJ, resulting in the accumulation of mutations or genomic insertions, deletions, and translocations that can be subsequently transmitted to the daughter cells. Indeed, it has been experimentally demonstrated that the quiescent state and the prevalence of NHEJ as the major DNA repair mechanism, are associated with DNA damage accumulation in HSCs during aging (126, 141). The high-fidelity HR is largely inhibited in SCs and only in the case that the cells were mobilized before damage infliction, they were found to preferentially employ HR for DNA repair (126, 164). Therefore, quiescence for the SCs can be considered as a double-edged sword, keeping damaging insults to the minimum on the one hand, while elevating genomic instability due to inefficient repair on the other.
Accumulated genomic instability in SCs elicits certain responses depending on the nature and extent of the damage. The majority of the cycling SCs (for instance, intestinal SCs) as well as quiescent SCs re-entering cell cycle after their exposure to genotoxic insults, are eliminated by apoptosis (12). Another response to accumulated genomic instability in SCs is the induction of cellular senescence (12). The tumor suppressor, p16INK4, is strongly associated with senescence induction in transformed cells. Expression of the INK4a/ARF locus (encoding for p16INK4 and ARF) is repressed early in life, mainly by function of the Polycomb repressor complex; however, the repression is alleviated during aging (14). Importantly, overexpression of p16INK4a has been experimentally linked to functional decline of SCs, whereas p16INK4a-deficient mice show improved SC function with age, compared to their wild-type littermates (80). Interestingly, p16INK4 was suggested to drive apoptosis with stress in SCs. Genomic damage accrual in SCs is further associated with increased SC differentiation. An elegant study by Wang et al., suggests that DNA damage in HSCs results in the preferential differentiation of HSCs to the myeloid lineage instead of the lymphoid (185), a skewing widely observed in aged HSCs as well. This skewed differentiation toward the lymphoid lineage is associated with the aging associated decline in the adaptive immune system. Importantly, whereas the gradual removal of dysfunctional SCs is a potent tumor suppressor mechanism, it eventually results in the exhaustion of SC reserves. Ultimately, the reduced capacity of SCs for tissue regeneration upon injury and the tissue atrophy—both hallmarks of aging—are stemming from SC exhaustion.
Longevity factors in SC maintenance
The importance of SCs in the long-term maintenance of adult tissues and, subsequently, to the aging process is well illustrated by the observation that genetic factors associated with aging have crucial functions in SC maintenance. The FOXO transcription factors, as well as DNA repair components have all been shown to regulate SC long-term survival and repopulation capacity.
As argued above, maintenance of a low ROS load is crucial for SC function, since intracellular ROS accrual was shown to result in increased genomic instability and, subsequently, SCs differentiation (128). Recent studies in the FOXO TKO mice revealed a vital role of FOXOs in HSC maintenance via the regulation of intracellular ROS and cell cycle progression (166, 167). HSCs from the FOXO TKO animals suffered from increased ROS due to impaired expression of several ROS detoxifying enzymes. Importantly, administration of the ROS scavenger N-acetyl-cysteine (NAC) in these animals alleviated several of the SC defects stemming from FOXO deficiency. The importance of FOXOs in SCs ROS modulation is further illustrated by observations in PKBa/b deficient mice that display reduced levels of free radicals and increased quiescence (83). Interestingly, similar phenotypes to FOXO depletion were observed in mice with ATM deletion. More specifically, ATM−/− mice exhibit depletion of their HSC pool and bone marrow failure due to elevated ROS, whereas the HSC defects are relieved with concomitant administration of NAC (79). Interestingly, in both FOXOs, TKO and ATM−/− mice elevated ROS are associated with increased p38MAPK activity and p16INK4 levels. Recent studies support a direct involvement of ATM in regulation of intracellular ROS (40). ATM was described to act as a ROS sensor by the formation of intermolecular disulfide bridges (61), similar to FOXO proteins (34). However, the exact mechanism of ROS regulation by ATM is not clear still, with some reports suggesting this to be via regulation of the pentose phosphate pathway (32, 40). In addition to ATM, the Fanconi anemia complementation group D2 (FANCD2), another DNA repair protein, was also recently associated with FOXOs and free radical regulation in HSCs. FANCD2, a component of the Fanconi anemia group of proteins, was found to interact with FOXO3 to regulate the expression of ROS scavenger genes, including MnSOD and catalase (106). Importantly, Fanconi anemia patients show bone marrow failure as a result of increased intracellular ROS in HSCs and HSC pool depletion (41).
In addition to their function in ROS regulation, FOXOs appear to regulate other processes related to SC maintenance as well. FOXOs are integral factors for the quiescent state of SCs, regulating the expression of p21cip1/waf1 and p27kip1. Indeed, in FOXO TKO mice, HSCs were depleted due to increased cell cycle mobilization and subsequent differentiation, at the expense of self-renewal (166). Interestingly, HSC differentiation in these mice was skewed toward the myeloid lineage, at the expense of the lymphoid lineage, as is observed in aging. Remarkably, similar phenotypes were observed in PTEN conditional knockout mice (59, 60), in which, PKB is expected to be overly active and thereby inactivate FOXOs. PTEN deficiency in the HSCs resulted in increased HSC mobilization and eventually SC depletion, while an increased differentiation toward the myeloid lineage was also observed (198, 202). Moreover, similar effects were observed in the neuronal stem cells (NSCs) of the FOXO TKO mice. These mice exhibited increased proliferation of NSCs early in life, followed by depletion of the NSC pool later in life, thereby contributing to reduced long-term neurogenesis (133). In another SC compartment, the spermatogonial stem cells (SSC), FOXOs appear to regulate SSC self-renewal capacity and differentiation (52), whereas FOXO3 also suppresses ovarian follicle activation, since FOXO3a−/− mice exhibit oocyte exhaustion and infertility (23). ATM, in accordance to its established role in cell cycle regulation, also regulates the cycle status of SCs. In particular, ATM was recently reported to regulate the quiescence status of HSCs, via regulation of BID and intracellular ROS (118) and in ATM−/− mice, the SSC population is progressively depleted by increased quiescence exit and cell cycle re-entry (162).
Regulation of SC homeostasis is crucial for their long-term survival and re-population capacity. Eventually, maintenance of SC pools is of utmost importance for tissue regeneration in aging. Longevity factors, including FOXOs and DNA repair components, such as ATM, play important roles in SC maintenance. The major impact of these factors in SCs is in the regulation of the intracellular free radical load, thereby contributing to genomic stability in these cells. Moreover, by retaining SCs in quiescence, they contribute to their protection by severe damage insults. Importantly, however, whereas the processes they mediate are clear, the regulation of these factors in SCs is not fully elucidated. It would be particularly important to expand our understanding of protein interacting modules as well as post-translational modifications (PTMs) regulating FOXOs function in the SCs, as this holds promise for relevant interventions, and thus will be valuable for regenerative medicine.
FOXO and ATM in Diabetes, the Other Age-Associated Disorder
Diabetes is a condition stemming either from decreased insulin secretion from the pancreatic β-cells, or decreased insulin perception in peripheral tissues (insulin resistance). As mentioned, reduced PI3K/PKB signaling plays an important role not only in cancer, but also in diabetes, another typical age-related disease. In agreement with FOXOs being important downstream components of PI3K/PKB, a role for FOXOs has been established in diabetes, both in peripheral insulin resistance as well as in β-cell function [for more elaborate reviews see e.g., ref. (1, 58)]. Intriguingly, a role for ATM in diabetes has also been documented.
Patients with A-T display metabolic abnormalities, such as poor growth, insulin resistance, and increased risk of developing diabetes mellitus, which at least, in part, are due to lack of ATM activity (152). However, how ATM controls metabolic function with respect to glucose handling and insulin signaling remains unclear. Interestingly, oxidative stress appears to be an important determinant for the pathology of both diabetes and metabolic syndrome, a condition with increased risk of developing insulin resistance. As already mentioned, loss of ATM results in increased ROS and, similar to FOXO, free radicals have recently been shown to directly control ATM activity (61). Moreover, combined ATM and apolipoprotein E deficiency results in increased JNK activity and ATM has been proposed to reduce JNK activity (146), thereby relieving JNK feedback inhibition on insulin signaling (197). Furthermore, ATM appears to be activated by insulin and to subsequently contribute to the full activation of PKB, thereby regulating the translocation of the cell surface glucose transporter 4 in response to insulin (63). Thus, whereas FOXOs are inhibited by insulin signaling, ATM appears activated. This may underlie the different mode of involvement of FOXO versus ATM in peripheral insulin resistance, in which loss of FOXO improves insulin function (1), whereas loss of ATM provokes insulin resistance (152). The role of ATM is further illustrated by the observation that genetic variants of ATM correlate with treatment success of metformin, the most commonly used drug to treat type 2 diabetes (204).
With respect to pancreatic β-cells, ATM and FOXO appear to be both important factors for their survival and functionality. In β-cells, FOXO1, the major FOXO member in the pancreas, is continuously kept cytoplasmic, due to the constant production of insulin. However, in conditions of increased oxidative stress, such as induced by hyperglycemia, FOXO translocates to the nucleus. Nuclear FOXO1 was shown to protect β-cells and to induce the transcription of NeuroD and MafA, transcription factors that regulate insulin production (89). Importantly, however, prolonged FOXO1 activation also induces cell growth attenuation in β-cells of mice with disturbed insulin signaling, leading to β-cell dysfunction. This effect of FOXO is likely through induction of p27kip1 and reduction of cyclinD, as it has been shown that p27kip1−/− mice are protected from diabetes (173). ATM was also found to be active in β-cells to ensure insulin secretion (125). These functions of ATM described are important determinants for the prevalence of an aging associated disorder, such as diabetes and appear independent of its established role in DNA repair. Nonetheless, we can envision additional functions for both FOXO and ATM in ROS modulation and genomic stability in β-cells.
FOXOs Balancing Cancer and Disease—Signals from Above
Tumor suppressors have been long categorized in two main groups; namely, the caretakers and the gatekeepers (22). The caretakers are proteins involved in maintenance of genomic stability, thereby minimizing the potential threats for neoplastic transformation. Typical components of DNA repair pathways and proteins otherwise ensuring survival and genomic stability are categorized in this group. Gatekeepers on the other hand are proteins, such as p16INK4, inhibiting oncogenesis by inducing the removal of the transformed cells via senescence and apoptosis. FOXOs are bona fide tumor suppressors; however, their categorization to one of the above groups appears to be more complicated. Indeed, FOXOs regulate transcriptional programs for ROS detoxification and DNA repair; yet, at the same time regulate senescence and apoptosis, typical proaging responses. These functions appear to be contradictory when considering the well-established role of FOXOs as positive regulators of lifespan extension. Thus, one may speculate on a differential regulation of cellular fate by FOXOs, depending on the input signals, that determines FOXOs to be pro- or antiaging. Under low stress conditions (young cells), FOXOs promote stress protection and survival, thus acting as antiaging factors; in cases when the ensuing instability is of great threat for tissue functionality (old cells) then they promote cell clearance and aging.
However, are the signaling inputs indeed tipping the balance in the regulation of these contradictory programs by FOXOs? A differential balance in input signals due to differential regulation of upstream components could affect FOXO PTMs and subsequent association with binding partners, eventually shifting the output effect from stress resistance to cell death. Importantly, intracellular ROS accumulate with age and upstream components regulating FOXO function, including PTEN phosphatase, share redox-sensitive cysteines that oxidize, thereby affecting enzyme functionality with aging. In fact, ROS regulation of FOXOs comprises an intricate and complex network of proteins with differing sensitivities and modes of ROS regulation [reviewed in e.g., ref. (160)]. To increase complexity, FOXOs are central nodes in ROS signaling being both regulated by, as well as regulators of the intracellular ROS load. Eventually, the balance in regulation may be found in the details of redox regulation.
FOXO regulation by stress
FOXOs activity is regulated through a variety of PTMs, such as phosphorylation, methylation, and acetylation. These PTMs regulate FOXO subcellular localization, protein stability, and protein–protein interactions. As for other transcription factors, the variety of PTMs identified for FOXOs led to the suggestion that combinatorial codes of PTMs regulate the function as well as the binding of cofactors under various conditions, toward specific functional outputs (21). Although an attractive hypothesis, there is, at present, little evidence for the existence of such codes and in this respect it is evident that detailed studies are required to fully understand the role of all PTMs and their interconnections, for FOXO function. Here we will describe PTMs identified for FOXOs in response to oxidative and genotoxic stress, as well as to growth factors and cellular nutrients.
FOXO regulation in response to ROS
Phosphorylation
In mammalian cells, oxidative stress activates JNK via the small GTPase Ral and, subsequently, JNK phosphorylates FOXO4 on multiple residues, resulting in its nuclear accumulation (45). Studies in C. elegans and Drosophila further demonstrated by genetic means that JNK activates DAF-16 and dFOXO, respectively, to mediate lifespan changes and stress resistance (132, 187). JNK was also demonstrated to phosphorylate FOXO3a in response to the cytotoxic agent Paclitaxel. However, thus far, FOXO1 appears not to be a substrate for JNK, at least in vitro (4).
Mammalian sterile 20-like kinase 1 (MST1) is a serine/threonine kinase activated by cellular stress and MST1 was shown to play an important role in apoptosis induction by a variety of stresses. Following apoptotic stimulation, MST1 activation regulates a number of downstream targets, including JNK/p38, histone H2B, and FOXO. MST1 has been shown to phosphorylate FOXO1 in vitro on Ser212 (103). In cells, MST1 phosphorylation of FOXO has been shown to induce loss of 14-3-3 binding to FOXO and consequent nuclear translocation. Also, whereas MST1 is primarily involved in apoptotic signaling [e.g., neuronal cells ref. (201)], the opposite has been reported as well, where the Mst1-FoxO signaling pathway plays a crucial role in survival, but not apoptosis, of naïve T cells (29).
Nemo-like kinase (88, 161) was also demonstrated to phosphorylate FOXO4 in response to oxidative stress and inhibit FOXO-dependent transcription, although with an undefined mechanism still.
Acetylation/de-acetylation
In response to oxidative stress FOXOs were found to be acetylated by the histone acetyl-transferases p300, CREB-binding protein (CBP), and p300/CBP-associated factor [reviewed in ref. (175)]. The result of FOXO acetylation on its transcriptional activity is still debated, as deciphering the FOXO-specific effects from the effects of histone acetylation remains tedious [reviewed in ref. (176)]. FOXO acetylation is removed by the action of Sir2/Sirt family de-acetylases (18, 178).
Ubiquitination/de-ubiquitination
FOXOs were also found to be regulated by mono- and poly-ubiquitination. In response to oxidative stress, the E3 ligase, MDM2, was shown to mono-ubiquitinate FOXO4, resulting in its nuclear translocation and transcriptional activation, whereas the de-ubiquitinating enzyme, USP7/HAUSP, removes these marks (15, 177).
FOXO regulation in response to genotoxic stress
Phosphorylation
CDKs are key regulators of cell cycle progression and, through phosphorylation, they control many essential cell cycle components. Both CDK1 and CDK2 have been shown to phosphorylate FOXO1 on Ser249 in vitro as well as in vivo (76, 113). In proliferating cells, this phosphorylation event was shown to inhibit the FOXO1 activity, via its cytoplasmic retention. Importantly, genotoxic stress activates signaling cascades eventually inhibiting the CDKs. Indeed, it was shown that under DNA damage conditions, the negative FOXO1 regulation by CDKs is alleviated, resulting in FOXO1 nuclear re-localization. Interestingly, in postmitotic neurons, FOXO1 phosphorylation by CDKs appeared to activate FOXO-dependent transcription (200). Whether this reflects a cell type-specific difference or experimental differences is still unclear.
FOXO3 was recently reported to be phosphorylated by the MAP kinase MK5 (96). This phosphorylation results in FOXO nuclear translocation and transcriptional activation, toward miR-34, to antagonize overproliferation induced by Myc oncogene. More importantly, previous work from the same group suggested that MK5 regulates the expression of miR-34 in response to genotoxic stress, suggesting DNA damage signaling to regulate FOXO toward different end points.
ATM, together with ATR- and DNA-dependent protein kinase (DNA-PK) belong to the PIKKs. ATM, ATR, and DNA-PK are activated by genotoxic stress and phosphorylate a sequence motif within proteins defined as Sp/TpQ. A proteome-wide analysis of proteins phosphorylated by the PIKKs in response to DNA damage revealed many potential ATM substrates, including FOXO1 (119). Interestingly, all FOXO members possess this Sp/TpQ motif, suggesting this to be a potential common regulatory mechanism. Whether this phosphorylation has a functional relevance in the DNA damage response, however, remains to be determined.
FOXO regulation in response to growth factors and nutrients
Phosphorylation
PKB has been shown to phosphorylate three evolutionary conserved residues within FOXO members, which for FOXO1 are Thr24, Ser256, and Ser319. Phosphorylation of proteins by PKB results in 14-3-3 binding, but full binding of 14-3-3 to FOXO requires only the first two PKB phosphorylation sites. The third PKB site is involved as a gatekeeper in further phosphorylation of FOXO by casein kinase 1 (CK1). CK1 phosphorylates Ser322 and Ser325 in FOXO1, following the third PKB site. Together with DYRK1a mediated phosphorylation at Ser329, this generates a patch of negative charges involved in an interaction with the nuclear export/import machinery (139). Moreover, phosphorylation of FOXOs by PKB inhibits their DNA binding potential, as the second phosphorylation site (Ser256 in FOXO1) introduces a negative charge within the DNA binding domain, thereby inducing repulsive forces. Serum- and glucocorticod-induced kinases (SGK) are distantly related to PKB, and its activation is also dependent on the action of PI3K through PDK1. The consensus sequence for SGK phosphorylation is highly similar to that of PKB. Thus, SGK can also phosphorylate the PKB sites of FOXOs. However, comparison of FOXO3a phosphorylation by PKB and SGK suggested the third PKB site (Ser315 in FOXO3a) to be preferentially phosphorylated by SGK, whereas the others are preferentially phosphorylated by PKB (17). SGK was also identified as a kinase for the PKB sites of FOXOs. Interestingly, studies in C. elegans suggest that depending on the experimental condition either PKB or SGK is the main mediator of DAF-16-dependent lifespan, and biochemical analysis suggests that PKB and SGK are components of the same protein complex (70). This would indicate that PKB and SGK are largely redundant with respect to FOXO regulation. Interestingly, leucine-rich repeat kinase 2 (LRRK2) and cyclic guanosine monophosphate-dependent kinase II (cGKII) have been shown to also phosphorylate the third PKB/SGK site in dFOXO. However, unlike PKB/SGK, both LRRK2 and cGKII activate the dFOXO transcriptional activity (84, 85). A mechanism that can possibly unify this differential regulation is at present hard to envision.
Following the notion that in human primary tumors FOXO cytosolic localization did not completely correlate with a high PKB activity, Hu et al. identified IkB-kinase (IKK) as a novel negative regulator of FOXO3a (75). IKK phosphorylates FOXO3a at Ser644 a site not conserved within the other FOXO members. IKK-mediated FOXO3a phosphorylation has been shown to result in poly-ubiquitination and proteosomal degradation of FOXO3a.
The MAP kinases p38 (71) and ERK (196) are implicated as well in FOXO regulation. Knowledge on the functional significance of these phosphorylation events is limited; however, initial studies suggest that ERK-mediated phosphorylation of FOXO3 induces its poly-ubiquitination and proteasomal degradation.
FOXOs are also positively regulated by phosphorylation, independently of oxidative stress. AMPK is a heterotrimeric serine/threonine kinase that performs a central role in cellular energy homeostasis. Under low-energy conditions, AMPK activation controls cell growth and energy expenditure by phosphorylating a number of substrates, including FOXOs. In vitro phosphorylation of FOXO3a by AMPK identified six potential AMPK phosphor-acceptor sites. Subsequent analysis of a FOXO3a mutant lacking all identified AMPK sites suggested that AMPK phosphorylation channels the FOXO activity toward activation of alternative energy sources and stress resistance (57). In agreement, it was found that a similar pathway operating in C. elegans confers lifespan extension when using an appropriate regimen for caloric restriction (56). Thus, regulation of FOXO by AMPK appears an evolutionary conserved mechanism for homeostasis of energy metabolism. In addition, this indicates FOXOs to be a nodal point for cross talk between the AMPK pathway and the insulin-PI3K-PKB pathway.
GlcNAcylation
O-linked β-N-acetylglucosamine (GlcNAc) addition, similar to phosphorylation, occurs on serine and threonine residues of proteins and this notion led to the hypothesis that O-GlcNAc modifications could directly oppose phosphorylation. FOXOs are also GlcNAcylated and this correlates with FOXO activation (74, 98). O-GlcNAc modified residues were identified by mass spectrometry, but did not correspond to any of the known FOXO phosphorylation sites. Therefore, the mechanism of activation remains to be identified.
Methylation
FOXOs are targeted for arginine methylation by the protein argininemethyltransferase-1 (PRMT1) (195). Methylated residues identified in FOXO1 are Arg248 and Arg250, which are part of the consensus PKB phosphorylation site for Ser256 phosphorylation. Methylation of these residues has been shown to inhibit PKB-mediated FOXO1 phosphorylation. In this way, PRMT1-mediated FOXO methylation appears to induce protein stabilization, nuclear localization, and transcriptional activation.
Ubiquitination
The E3 ligase, MDM2, was also shown to induce the poly-ubiquitination of FOXOs. Mdm2-dependent poly-ubiquitination was reported in response to ERK phosphorylation (196). In response to PKB phosphorylation, the Skp2 E3 ligase regulates FOXOs' poly-ubiquitination and degradation (77), whereas IKK phosphorylation of FOXO3 results in the formation of a βTrCP degron and the subsequent FOXO3 poly-ubiquitination (171). Clearly, detailed analysis has to reveal how E3 regulation is interconnected and/or whether different E3 ligases partake in mediating a specific signal toward FOXOs.
Concluding Remarks
Aging includes a multitude of processes, both at the cellular and organismal level, that interconnect and influence each other in a complex manner. The accumulation of damaged molecules and dysfunctional organelles due to endogenous and exogenous stresses appears to drive the aging process by disrupting the cellular homeostasis; however, aging in toto is challenging to fully comprehend. Nonetheless, certain factors that are known to influence aging and related disorders can provide useful answers, both on the molecular determinants of the process itself, as well as on potential interventions to ensure a healthy lifespan.
FOXOs are proteins with evolutionary conserved roles in lifespan determination and age-related disorders, including cancer and diabetes. The functions that FOXOs exert appear to be rather contradictory, in respect to their end effect on the aging process. On one hand, by transcriptional regulation of senescence and apoptosis, FOXOs can be seen as proaging factors; on the other hand, by regulating stress resistance, they function as antiaging factors. This apparent discrepancy, however, can be better understood, if seen under the prism of genomic stability. Indeed, all the processes regulated by FOXOs eventually serve in maintaining cellular genomic stability and, in turn, genomic stability ensures both increased longevity and tumor-free survival (Fig. 6). Important in this respect is also the notion that reducing oxidative stress by FOXOs not only adds in preventing genetic damage and consequent disease, but also reduces nongenetic contributions to disease, such as the contribution of redox misbalance to protein-folding diseases and diabetes. In fact, the emerging close association between FOXOs and ATM may suggest that FOXOs should have more direct implications in maintaining genomic stability. Eventually, progress on understanding FOXO regulation in the context of individual cell fates sets a basis for our understanding of FOXO regulation in the context of aging, which is the ultimate challenge.