
Abstract
Introduction
In the United States of America, allogeneic red blood cell (RBC) transfusion has long been considered an important treatment option for patients suffering from blood loss (56). However, infectious agent viruses and other pathogens may put the blood supply at risk (71). Currently, the American Red Cross tests donated blood for Hepatitis B and C viruses, human immunodeficiency virus (HIV), human T-cell lymphotropic virus, syphilis, West Nile virus, and the agent of Chagas disease (22, 48, 144, 145). As a result, the safety of the U.S. blood supply in terms of transfusion-transmitted diseases is good. However, screening for more infectious agents will increase the cost of blood. Of more concern is the fact that donated blood may contain yet to be identified infectious agents (48). Currently, in the United States alone, about 14 million units of whole blood are collected annually, and ∼12 million are processed and given for transfusion (1, 2). Transfusion-related adverse events, both short- and long-term, are among the costliest contributors to healthcare expenditures, without including future illness, lost wages, and impact on quality of life (136). Despite the increasing cost of transfusion, its practice remains quite liberal (67). Additionally, there are new concerns regarding the safety and effectiveness of stored blood transfusions, due to RBC-induced changes induced by ex vivo aging, so called RBC storage lesions (89, 90). To further compound the problem, blood availability can be limited in emergency situations such as war scenarios or natural disasters (143). Therefore, it has been a long-term goal of scientists to develop an efficacious and safe RBC substitute (i.e., oxygen bridge) for use in transfusion medicine.
Hemoglobin (Hb)-based oxygen carriers (HBOCs) are currently being developed as RBC substitutes for use in transfusion medicine. Despite significant commercial efforts, recent late-stage clinical results of HBOC studies hamper further development (46, 73, 165). The key issues observed during clinical trials include vasoconstriction, the persistence of long-term systemic hypertension, and oxidative-stress-induced tissue toxicity (69, 138). These studies have highlighted the importance of the clinical contexts in which RBC substitutes are to be used, as these scenarios strongly affect safety and efficacy outcomes that are observed after infusion. For example, although the injury that created the need for transfusion can lead to adverse outcomes by itself, subsequent blood transfusion can trigger a second insult, which is termed ischemia-reperfusion injury. Mechanistically, sublethal ischemia and hypoxia lead to priming of secondary messengers and to the production of reactive oxygen species (ROS) (35). Injured cells are then more susceptible to further production of ROS and to active participation in inflammatory responses. ROS exposure triggers cytotoxic reactions and induces production and release of inflammatory cytokines that result in activation of leukocytes, platelets, and macrophages. Another result is acute endothelial cell dysfunction, which prevents transfusion from achieving its intended objective (111, 155).
The Pathophysiology of Blood Loss
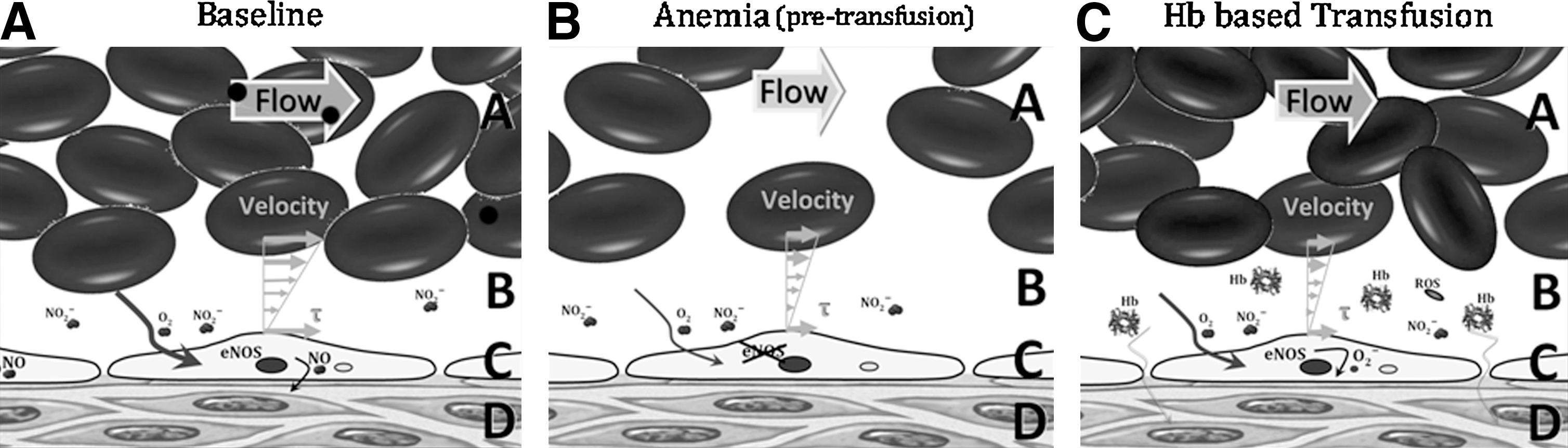
Blood substitutes have been formulated to address the deficit of blood's intrinsic oxygen-carrying capacity resulting from the loss of RBCs. However, the decrease of circulating RBCs (anemia) and blood volume are not the only factors affected during blood loss. Detailed studies of the circulation, and specifically the microcirculation, have shown that normal blood mechanical properties are optimal to maintain a functional central circulation and to insure uniformly perfused tissue capillaries (27, 84, 161). A blood loss lowers blood pressure, and thus the pressure at the capillaries decreases, and most vascular beds initiate reflexes in efforts to maintain pressure via vasoconstriction. This further reduces the pressure at the arteriolar end of the capillaries, which affects the fluid movement across the capillary membrane that occurs as a result of filtration. Low pressure results in a reduction in the region of the capillary where fluid is lost (filter out) from the plasma into the tissues and in an increase in the region of the capillary where fluid returns (filter in) back to the plasma, producing a net gain of fluid to the plasma. This fluid gain, taken over most of the vascular beds, leads to fluid autotransfusion that partially compensates for volume loss during blood loss. Therefore, in addition to reducing the intrinsic blood oxygen-carrying capacity, blood viscosity is reduced. Historically, this has been regarded as a positive outcome, since lower viscosity reduces vascular resistance (93) (Fig. 1). Viscosity is a commonly overlooked parameter in the transfusion medicine. The loss of mechanotransduction on the endothelial cells hampers the production of nitric oxide (NO) and prevents the normal vascular signaling and regulation, thus increasing peripheral vascular resistance (30, 85, 118). Vessel wall shear stress (WSS) is the product of blood viscosity and shear rate determined by the flow velocity profile. WSS modulates peripheral vascular resistance and blood flow, because WSS is the local signal for the endothelium to produce vasorelaxation. Blood losses lower blood viscosity, thus increasing blood flow velocity; however, this effect is not able to preserve WSS due to the two-phase nature of blood (RBCs and plasma). When blood flows, an erythrocyte cell-free layer (CFL) is interposed between the RBC blood column and the vessel wall (97, 140). This hydrodynamic layer is generated by the axial migration, resulting from the flexibility of red cells that causes them to migrate toward the center of the flow stream (117, 134, 135, 163). Blood losses (and hemodilution) increase the CFL width and the concentration of all molecular species dissolved in plasma. Thus, the physiological shortcomings induced by hemodilution tend to lower the NO concentration in the arteriolar wall, preventing vasorelaxation and creating a proinflammatory environment.
Transfusion of Stored Blood and the Potential for Toxicity
Considerable progress has been made in improving blood product safety protocols during the past 20 years, significantly reducing the risk of infection and the immune response to transfusion (2, 92). Blood transfusion in some patients has been shown to provide no benefit, and in fact is associated with increased morbidity and mortality (90, 122). Several studies have indicated that the transfusion of stored blood for more than 14 days decreased oxygen delivery (95, 148), contributed to tissue hypoxia (64), and increased mortality (53). A recent systematic review and meta-analysis that evaluated the efficacy of stored blood transfusion (including 272, 596 patients) revealed that in 42 of 45 studies, the risk of transfusion outweighed the benefit by increasing the mortality rate by 70% (107). Additionally, several studies have shown that the survival rate decreases with the mean blood storage duration, and that the storage duration is a strong predictor and risk factor of multiple-organ failure (107).
From the moment that blood is withdrawn, erythrocytes begin to exhibit changes on their membrane due to stress during collection, which only increases upon manipulation, storage, and reinfusion, consequently affecting the degree of intravascular hemolysis post-transfusion (91, 96). Stored blood transfusions release small amounts of Hb in the plasma, inducing vasoconstriction, reducing blood flow and oxygen delivery (86, 162). Low levels of Hb in plasma severely disrupt NO bioavailability, via NO dioxygenation (NOD) reactions (87, 124, 156). At the smooth muscle, this process primarily decreases intracellular NO concentration by limiting the activation of the NO receptor, soluble guanylate cyclase (sGC), which results in decreased production of cyclic guanosine monophosphate (cGMP), and ultimately causes vasoconstriction (9, 123).
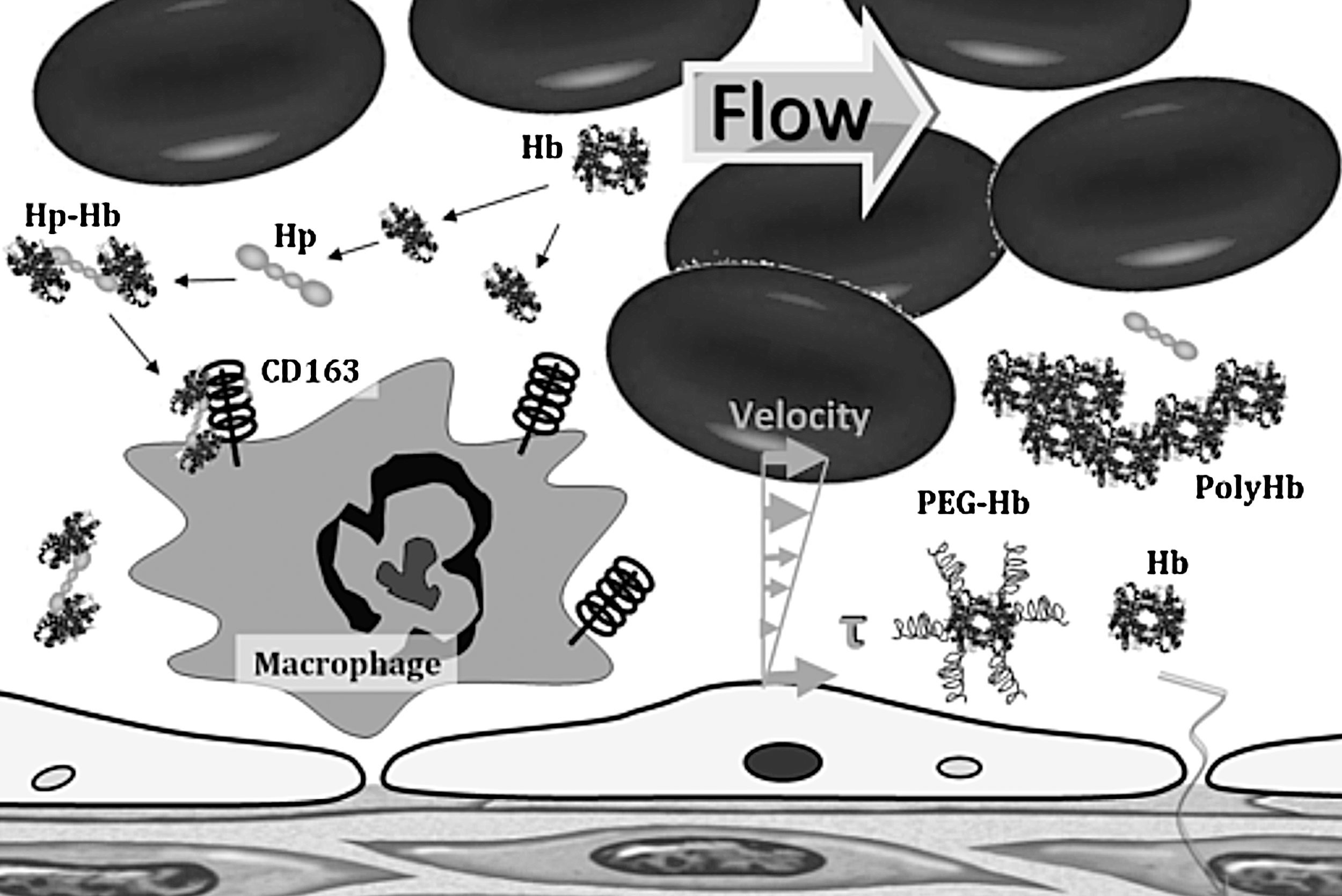
Transfusion of HBOCs and the Potential for Toxicity
Acellular Hb is present in the circulation in minute quantities due to the natural breakdown of RBCs that are readily disposed of without consequence in the healthy organism (113, 130, 147). However, elevated levels of Hb in the plasma can be observed in the blood and/or urine. Small amounts of plasma Hb are removed from the circulating plasma by haptoglobin (Hp). Other plasma proteins such as hemopexin, α1-microglobulin, and albumin have a role to detoxify free heme (11). Plasma circulating Hp transports the Hb to be removed by the reticuloendothelial system in the liver and spleen (11). The Hp-Hb complex is also removed from the plasma by deposition in various body tissues, but chiefly in the liver and renal cortex (18). Further accumulation of Hb in the plasma, beyond the combining power of Hp, is filtered through the glomeruli of the kidneys and reabsorbed by tubular cells, where it may lead to precipitate formation, termed hemosiderin, during conditions with Hb overload, causing oxidative damage and glomerular and tubular necrosis (18). Exceeding this threshold has toxic consequence during trauma, acute hemolytic anemia, breakdown of stored blood in transfusion, or the deliberate introduction of Hb as an HBOC (78, 128, 147).
Hb is also the precursor for synthesis and formulation of HBOCs to be used as blood substitutes (55, 57, 75). The first intent to develop an HBOC was stroma-free Hb (8, 51). However, transfusion of stroma-free (i.e., cell-free) Hb led to several major side effects (6, 19, 36, 45, 176). In the circulation, extracellular tetrameric Hb (α2β2) easily dissociates into two pairs of αβ dimers (19, 36), which are extremely prone to oxidation (176) and increased renal excretion (19 –21). The process of Hb oxidation to methemoglobin (metHb) promotes unfolding of the globin chains and releases cytotoxic heme into the circulation, leading to kidney tubule damage and eventual renal failure (19, 36). metHb generates harmful ROS (6), which damage cell membranes, oxidize nucleic acids, and proteins (45). The extent of tissue toxicity by plasma Hb is further magnified by the extravasation of Hb through fenestrated capillaries and endothelial cell junctions (Fig. 2).

The two major theories to explain the occurrence of acellular Hb-induced vasoconstriction are that plasma Hb scavenges NO produced by the endothelium and oversupplies oxygen to vasculature via Hb-facilitated diffusion of oxygen and oxygenated Hb (170). Limiting NO prevents the relaxation of the smooth muscle (77, 149), and hyperoxygenation triggers regulatory response to limit blood flow and to reduce the surface area for oxygen transport (3, 4). HBOC-induced vasoconstriction has been clinically observed as hypertension. Scavenging of NO can occur either through binding to deoxyhemes, resulting in the highly stable ferrous nitrosyl Hb derivative, or through the NOD reactions between NO and oxyHb to produce metHb and nitrate. NO binding to deoxygenated Hb and NO reaction rate with oxyHb are virtually identical for most HBOC formulations (129); however, the vasoconstrictive effects vary between HBOC formulations (32, 129). Recombinant Hbs (rHbs) in which NO accesses to the oxyheme is sterically hindered showed correlation between NO reaction rates and the level of hypertension (42), although in vivo vasoconstriction was not studied. The rHbs reduced Hb NOD reaction rates by a maximum of one order of magnitude (42, 44, 124). However, mathematical models suggest that reaction rates need to be reduced by several orders of magnitude to obtain an NO concentration of 100–150 nM at the abluminal side of a blood vessel (82, 139), which is required to activate 50% of sGC (39, 142).
NO scavenging does not yield a unifying explanation to the vasoconstriction for all HBOCs. Polyethylene glycol (PEG)-conjugated Hbs (PEG-Hbs) show minimal levels of vasoconstriction, but they lower perivascular NO concentration, to a similar extent, as do vasoconstrictive HBOCs (149). Insight into the mechanisms behind these effects was obtained by direct measurements of microvascular pressure and resistance (27, 32). After hemodilution with PEG-Hb, capillary pressure was maintained; blood flow increased; and vascular resistance was lowered, when compared to other HBOCs (27, 32). The increase in blood flow with PEG-Hbs compared to other HBOCs has been consistently documented in other studies of hemodilution and hemorrhagic shock resuscitation (152, 167, 169, 172). PEG-Hb is a good plasma expander because of its high colloid osmotic pressure (COP) (170); when PEG-Hb is infused, it affects capillary filtration and promotes reabsorption of water, increasing plasma volume and reducing blood protein concentration, although lowering perivascular NO (149).
In the second theory, it is hypothesized that HBOCs oversupply oxygen to the surrounding vasculature via Hb-facilitated diffusion of oxygen and oxygenated Hb (168). Therefore, an autoregulatory response induces vasoconstriction to limit blood flow and the available surface area for oxygen transport through the blood vessel wall (3, 4). The oxygen-facilitated diffusion hypothesis is not entirely satisfactory, because while it is operational in the vascular compartment, it ceases to work across the vessel wall, as Hbs cannot diffuse across the vessel wall, and the oxygen flux across the vessel wall is determined by the oxygen tension gradient. Thus, facilitated diffusion only increases the amount of oxygen transported by the blood to the tissue interface. Studies using an artificial capillary system compared various acellular Hb and HBOCs and showed that unmodified Hb and small HBOCs increased oxygen release, relative to larger chemically modified HBOCs regardless of their oxygen affinity (109). Experiments demonstrating increased fluxes due to facilitated diffusion have used infinite sinks at the system boundary through which the oxygen exits (109), and such boundary conditions are not present in blood vessels.
Acellular Hb Oxidative Damage
Hb-based transfusion strategies inevitably restore oxygenation to oxygen-deficient tissues. This often results in ischemia–reperfusion injury, which exacerbates underlying conditions and worsens clinical outcomes. Mitigating the problems associated with ischemia–reperfusion injury has proven itself to be difficult, and there have been a number of studies of complementary mechanisms aimed at increasing protection (116, 127, 177). Studies suggest that pathogenesis of ischemia-reperfusion-reoxygenation injury is strongly linked to oxidative stress and the formation of ROS. In vascular endothelial cells, active consumption of NO by ROS contributes to vascular inflammation and endothelial dysfunction (ED) (60). Restoration of vascular levels of NO has been shown to sustain mitochondrial and cellular function, thus reducing oxidative damage (112, 127). The antioxidant defenses of plasma revolve around urate and ascorbate. In the case of HBOCs, the maintenance of the ferrous form of Hb is necessary for effective oxygen delivery, and oxidation to metHb (nonoxygen carrying) leaves Hb void of efficacy and prone to degradation.
Status of Commercial Developments
Vasoconstriction, heme-derived ROS, and free iron can account for the myocardial infarction observed in clinical trials of small-sized commercial polymerized Hbs (PolyHbs) such as Hemopure® (OPK Biotech., Boston, MA) and PolyHeme® (Northfield Laboratories, Inc., Evanston, IL) (120). Hemopure is developed and manufactured as a chemically stabilized, cross-linked bovine Hb suspended in an electrolyte solution for human use, with a P50 (i.e., O2 affinity) of 38 mm Hg, and an average molecular weight of 250 kDa with <2% α2β2 (49, 75, 99, 108). Hemopure is also known as Hb Glutamer-250 or HBOC 201. PolyHeme is the only HBOC to reach a phase III trial, polymerized in a multistep polymerization and purification process. The final product is a pyridoxylated polymerized human Hb in an electrolyte solution with a P50 ranging from 28 to 30 mm Hg, an average MW of 150 kDa with <1% α2β2 (55, 76, 115). Glutaraldehyde has been widely employed to nonspecifically crosslink/polymerize Hb (4, 7, 58, 59). Despite commercial development of PolyHbs, hypertension and other important safety concerns remain critical impediments to their clinical use (65, 141).
Polymerized bovine Hb for veterinary use (oxyglobin; OPK Biotech, formerly Biopure) was evaluated in hemorrhagic shock resuscitation and hemodilution as a function of Hb concentration administered. Although oxyglobin was vasoconstrictive, it improved survival when used in small dosage by comparison to nonoxygen-carrying colloidal fluids (29, 31). However, when used at higher dosages, tissue oxygenation was significantly impaired, and toxic effects on tissue viability were observed within 8 h (28). In a similar model, MP4OX, a PEG-Hb developed by Sangart, Inc. (San Diego, CA), was found to be a plasma expander rather than an oxygen carrier, as its high COP precludes PEG-Hb from being used at concentrations required for increasing oxygen transport (152, 169). If the benefits of PEG-Hb are plasma expansion, the Hb can be replaced with a less-toxic protein like albumin, especially when PEG-conjugated albumin appears to show no difference in terms of perfusion compared to PEG-Hb (33). The gain in oxygen-carrying capacity with MP4OX is not physiologically relevant in clinical situations.
Basic Strategies to Mitigate Acellular Hb Toxicity
Acellular Hb can arise from hemolysis, transfusion of stored blood, RBC parasitic diseases, and the infusion of HBOCs (52, 72, 126, 171). Acellular Hb toxicity is exacerbated by pre-existing vascular dysfunction found in large segments of the population who suffer from diabetes, hypertension, and obesity (68). Several approaches have been implemented to control HBOC toxicity using either chemical or genetic modifications of the Hb, and then developing strategies for mitigating pathological effects with targeted biochemical and rheological therapies.
Molecular size
Larger-sized HBOCs, unlike smaller tetrameric HBOCs, can (i) provide a greater degree of physical separation from the blood vessel wall, thus reducing NO scavenging by increasing the diffusion barrier; (ii) have a higher viscosity, and thus increase vessel WSS exerted by the blood, leading to an increased NO synthesis; (iii) reduce extravasation through endothelial cell–cell junctions; and (iv) decrease the Hb diffusion coefficient, and thus reduce the oxyHb flux to the blood vessel wall, thereby decreasing both the rate of NO scavenging and oxygen oversupply (26, 124, 149, 157) (Fig. 3). Attempts to develop large polymerized bovine Hb by increasing the cross-link density (50:1, i.e., molar ratio of glutaraldehyde to Hb) have yielded no vasoconstriction compared to smaller polymeric Hbs (26), and other toxicities have not yet been explored. Natural acellular Hbs of terrestrial (47) and marine (150) worms are acellular and have evolved in this manner. Hb from the marine worm Arenicola marina is being developed as an HBOC (150), and with similar properties, a zero-linked polymeric Hb (63) has been developed by OXYVITA, Inc., New Windsor, NY.

Other strategies to increase the molecular size of HBOCs include conjugation of PEG to the surface of Hb (157, 159) and encapsulation of Hb inside PEG-conjugated vesicles (132). The hydrodynamic radius of Hb is about 3 nm, while that of PEG-Hb is 9 nm. Thus, the flux of oxygen due to facilitated diffusion in the plasma in the presence of Hb is threefold than that of PEG-Hb. However, PEG-Hb shows a nonlinear dependence of COP with respect to concentration, reflecting colligative properties, the Donnan effect (121), and effects arising from its molecular-excluded volume (146), increasing capillary reabsorption and diluting plasma PEG-Hb concentration. A different approach to increase the size is to encapsulate Hb vesicles (HbV). This technology was initially developed by E. Tsuchida and consists of encapsulating Hb in 250-nm-diameter phospholipid vesicles coated with PEG (154). HbV must be delivered in solution in a plasma expander, thus allowing the design of its rheological properties. The rheology of HbV suspended in albumin was nearly Newtonian, whereas starch, dextran, and gelatin caused the suspensions to be non-Newtonian with shear-thinning behavior, as solution viscosity decreases as the applied shearing stress increased (131).
Rational Hb mutagenesis
The approach taken by J.S. Olson's group at the Rice University is to alleviate hypertensive effects through rational mutagenesis of the distal pocket of rHb to mitigate NO scavenging (42). Considering the ultrastability of the αβ interface (110), the first site-directed mutagenesis strategy was focused on genetically engineering an α–α-fusion Hb tetramer that was physically unable to dissociate into αβ dimers (98). The first rHb to be developed via this strategy consisted of rHb1.1 (Baxter Hemoglobin Therapeutics, Boulder, CO). Unfortunately, although rHb1.1 exhibited no renal toxicity, it elicited adverse side effects such as decreased heart rate, fever, and chills (160). This approach has been shown to reduce vasoconstriction and hypertension (44, 124, 125); however, obtaining sufficient quantities of recombinant material remains prohibitively expensive.
Enhancing Hb physiological clearance
Pharmacological activation of the glucocorticoid pathway induces expression of the Hb-scavenging Hp in dogs and guinea pigs (15). Hp sequesters plasma Hb and neutralizes much of its vasculotoxic oxidative stress, protecting against systemic hypertension and other vasculopathic outcomes (15). The depletion of NO is compounded by the oxidative stress induced by decomposition of Hb into heme and elemental iron. The canine model of intravascular hemolysis demonstrates the induction of systemic and pulmonary hypertension, as well as impaired creatine clearance, presumably due to decreased renal blood flow (15). The acute experimental canine vasculopathy is partially reversed by administration of NO or by glucocorticoid-induced production of endogenous Hp (15).
Hp is produced mostly by the liver, but also by the skin, lung, and kidney (41). Hp, in its simplest form, consists of two α- and two β-chains, connected by disulfide bridges. Hp exists in two allelic forms in human populations, Hp1 and Hp2, the latter one having arisen due to the partial duplication of the Hp1 gene (102). Three genotypes of Hp found in humans: Hp1-1, Hp2-1, and Hp2-2. Hp of different genotypes binds Hb with different affinities, with Hp2-2 being the weakest binder (94). Hp escorts Hb to the CD163 protein, in which the Hp-Hb complex is avidly bound and cleared from the plasma, depleting plasma Hp in the process (12) (Fig. 4). Hp binding to Hb prevents the generation of oxidant species, and mitigates the hypertension and other adverse vascular outcomes (80). The Hp-Hb complex has high oxygen affinity, which could increase nitrite reductase activity, potentially producing the endogenous antioxidant NO from nitrite (14). Infusion of Hp may have potential benefits during acute sickle-cell vaso-occlusion (12, 80). Hp effects might also explain some of the life-saving effects of glucocorticoids and aggressive plasma exchange transfusion therapy in thrombotic thrombocytopenic purpura (79).
NO-scavenging inhibition
Angeli's salt (sodium α-oxyhyponitrite, Na2N2O3) has been used to compensate for NO scavenging by plasma Hb, by selectively converting plasma Hb to ferric, and ideally iron-nitrosylated heme species that do not actively scavenge NO (66). Nitroxyl oxidizes oxy-Hb to metHb, and it can also convert metHb to a more stable, less-toxic species, iron-nitrosyl Hb (66). This concept can treat intravascular hemolysis, but would not apply to HBOCs, since it will prevent oxygen transport.
NO supplementation
Various compounds have been used to compensate for the NO consumed by HBOCs and the reduction the vasoconstriction. The effects of administering nitroglycerin (NTG) during HBOC fluid resuscitation from hemorrhagic shock were to attenuate the vasoconstrictive complications of HBOC (81). NTG is a weak NO donor (an organic nitrate that requires a three-electron reduction to release NO) that causes vasodilatation (164). NTG did not eliminate the vasoconstriction and caused elevations in metHb levels, thus reducing oxygen delivery to tissues. Similarly, sodium nitrite has been used in a similar fluid resuscitation from hemorrhagic shock with HBOCs, and vasoconstriction was attenuated at high doses of nitrite (10). In addition to increased metHb levels, nitrite supplementations reduced platelet function and increased the risk of pulmonary complications (edema and congestion) (10, 114). Pure NO inhalation has been used to treat NO depletion (174, 175). Inhaled NO prevented pulmonary and systemic hypertension induced by HBOCs (175). NO inhalation restores the NO that the NOD reactions consume within the lungs, and increases metHb levels, without restoring the NO bioavailability to peripheral tissues. Of late, NO-releasing nanoparticles (NOnps) have been used to restore NO bioavailability after infusion of HBOC (24). The slow release of NO from the NOnps, as they circulate, allows small amounts of NO to diffuse abluminally, thus reducing the effects of NO scavenging and limiting metHb formation (24, 25, 118). Additionally, a minor increase in intraluminal NO affects the diffusion gradients form endothelial-derived NO, allowing endothelial-derived NO to diffuse further into tissues, restoring signaling at smooth muscle (Fig. 5).
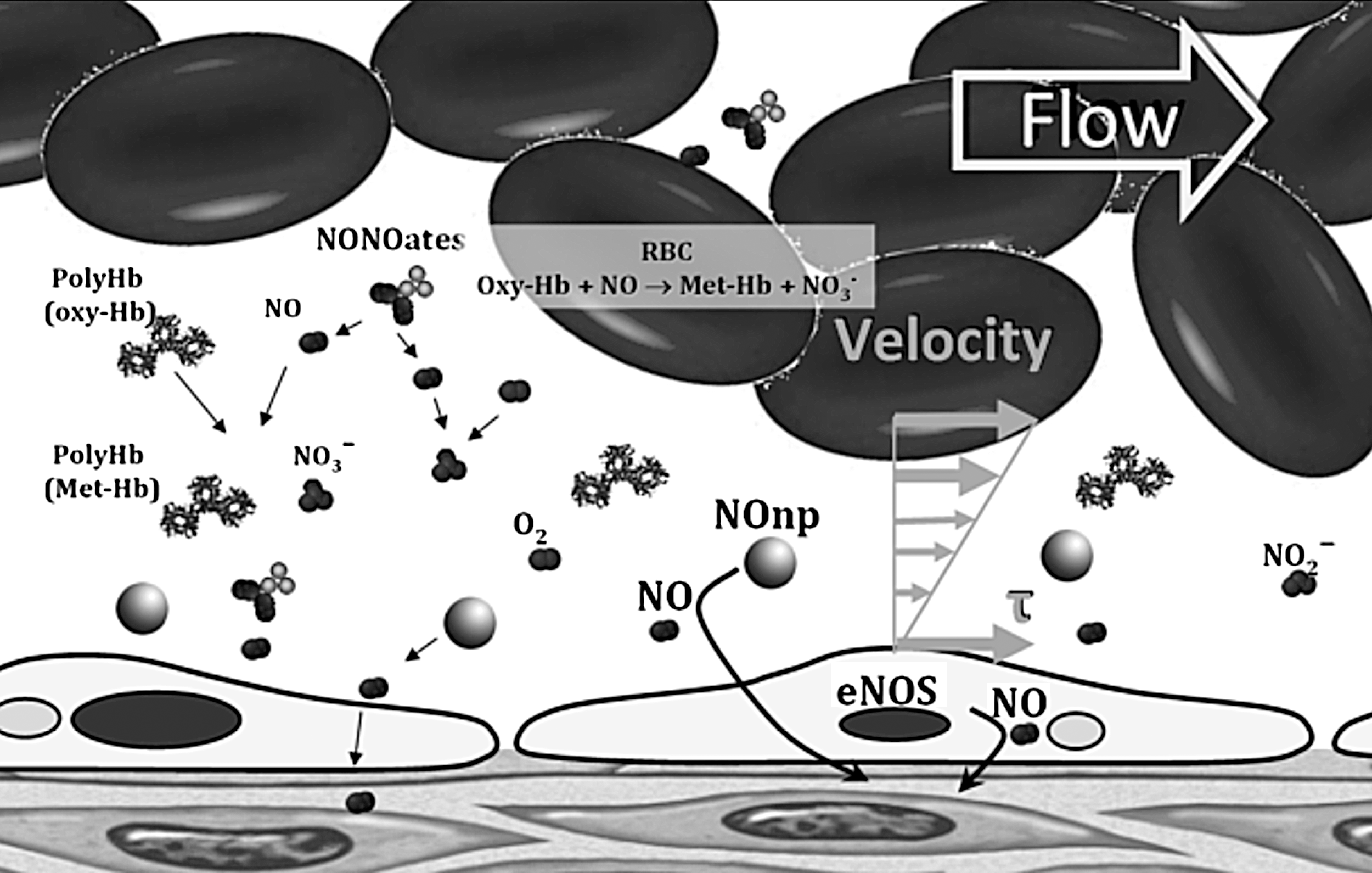
Other alternatives for intravascular NO therapy include compounds containing either NO or an NO precursor in a stable form, typically lack the capacity for controlled and sustained delivery (50, 70). The largest family of NO donors currently in use is NONOates, also known as diazeniumdiolates. The NONOate decomposition rate depends on pH and temperature (105, 106). NONOates have been used to mitigate the vascular inflammation-induced plasma Hb during malaria infection (34). They have limited application to HBOCs, due to short half-life and increased formation of metHb. Other alternatives to prevent vasoconstriction induced by HBOC include coadjunct therapy with Sildenafil, a phosphodiesterase (PDE) type 5 inhibitor (PDE5), by limiting cGMP breakdown (54). PDE5 inhibitor therapy partially prevented some hemodynamic changes (54). Therefore, a combination of PDE5 inhibitor with NOnps may increase the efficacy of a given dose of NOnps and decrease the amount of NOnps needed to counteract HBOC vasoconstriction.
Enhanced Hb nitrite reductase
It has been recently reported that PEG-Hb can convert nitrite to NO at a faster rate than does the native protein, which in part can compensate for the scavenging of NO (100). PEGylation of Hb stabilizes Hb into a conformation and hydration state that facilitates nitrite reductase to NO (103). The heme redox potential becomes more negative, making the iron a more efficient electron donor, thus increasing the rate of nitrite reductase by PEG-Hb (101). The reduction of nitrite to NO results in metHb formation, which clearly causes a negative outcome in terms of oxygen-carrying capacity (87).
Experimental Approaches to Predict Toxicity
The potential efficacy of several physiologically relevant reducing agents (glutathione, NADH, and ascorbic acid) in the redox stabilization of several HBOCs indicated that the rate of reduction by endogenous reducing agents is specific for each HBOC, and that most HBOC reduction by NADH requires the presence of another electron carrier (62). One of the main goals of preclinical toxicological evaluations for HBOCs was to select animal models that closely reflect human physiology. Knockout strains of mice have been generated that lack l-gulonolactone oxidase (LGO), which is the final enzyme in the synthesis of ascorbate. However, as with all transgenic and knockout animals, nonequivalent compensatory mechanisms may limit the predictive power of studies with these animals. Guinea pigs are a suitable species to evaluate human-relevant preclinical toxicokinetics as related to oxidative processes after Hb and HBOC exposure (17, 23). Guinea pigs, like humans and other primates, lack LGO, and hence are incapable of endogenous synthesis of ascorbate (37). They distribute antioxidant capabilities within the circulation and tissue parenchyma, presumably to counteract the reduced plasma ascorbate (119). Cooper et al. demonstrated the role for urate and ascorbate in the reduction of oxidized species of Hb and utilized the unique similarity of the guinea pig to the human to evaluate the role of ascorbate and urate after HBOC administration (40). Infusion of acellular Hb as oxygen therapeutics in animals and humans almost always induces an immediate vasoactive response, leading to elevation of both systematic and pulmonary blood pressure due to the removal of NO (40). In the presence of peroxide, Hb functions as a true enzymatic ascorbate peroxidase.
Buehler and coworkers have evaluated polymerized bovine Hb in guinea pigs to examine homeostatic mechanisms in a model relevant to human physiology (13). In their model, evaluation of blood pressure in response to PolyHb formulation suggested that molecular size was a critical attribute. Uptake by peripheral tissue macrophages was increased in lower-molecular-sized PolyHbs, and iron deposition was highest in the spleen and liver and was partially excreted via the kidneys, whereas the larger-molecular-sized PolyHbs were excreted exclusively by the spleen and liver (13). Future studies should include direct measurements of oxidative stress and lipid peroxidation (4-hydroxynonenal lysine adducts) as reliable indices of in vivo oxidative stress. Undiagnosed ED caused by genetic factors, smoking, hypertension, diabetes, and hypercholesterolemia may have played an important role in unforeseen clinical adverse events (transient ischemic attacks, strokes, and myocardial infarction) observed with HBOCs (137). These events cannot be predicted by toxicology studies conducted using rodent models (16). Therefore, evaluation of HBOCs in species or in transgenic animals with antioxidant states and induced ED similar to those of humans in health and in disease would increase our understanding of oxidative stress safety and efficacy of HBOC infusion.
Microvascular Assessment of Manifestation of Toxicity
Intravital microscopy is a powerful tool for the in vivo analysis of microcirculatory responses at the tissue level, especially to alterations of blood composition. The microcirculation usually is defined as that part of the vascular tree comprising blood vessels smaller than 100 μm, including arterioles, capillaries, and venules. The microcirculation plays a central role in tissue oxygenation, because it is across the walls of the microvessels that oxygen arrives to all tissues. Assessment of tissue oxygenation represents a key factor in evaluating the need for blood transfusion, as most of the clinical tools only provide information subrogate markers of global oxygenation (oxygen saturation, blood lactate, acid–base balance, and gastric pH), and many of them imply invasive monitoring. The current animal models to study the microcirculation all have their drawbacks, but as long as we lack clinical techniques to measure local perfusion and tissue oxygenation, they remain the only way to increase knowledge about the effect of transfusion tissue health and function.
In vivo measurement and characterization of the microcirculation with well-established animal models to visualize tissues in intact conditions, in parallel with systemic circulatory analysis, allow for the study of hemodynamics and vascular WSS, NO levels, oxygen transport, and the effects of strategies to control toxicity of acellular Hb. The in vivo effects of acellular Hb that are first manifested in microvessels can early detect the changes between normal vascular tissue and tissue under acute inflammatory conditions, and ED within short time scales. Vasoconstriction is observed early in the microcirculation, before hypertension and vascular resistance are systemically detected (5, 104). The worst implications of vasoconstriction are at the microcirculation, since constriction lowers capillary pressure and decreases functional capillary density (FCD), producing uneven tissue oxygenation and accumulation of metabolic by-products, causing microvascular dysfunction (83).
A major barrier to progress in the HBOC field has been the lack of a systematic analysis of the toxicity associated with acellular Hb. There is currently no universal gauge that allows comparisons of levels of toxicity between various Hb-containing solutions and whole blood (38, 74, 173, 175). The deliberate introduction of acellular Hb into the bloodstream arose with the development of HBOCs. It has been assumed that the gold standard of transfusion is blood; however, it is increasingly evident that transfused blood may itself be a source of toxicity if it has aged (i.e., >25–30 days) (130). Hb is a colloid, and it could be argued that comparable nontoxic standards should be nonheme-based colloids; however, even if such a comparison were practical, the lack of a measuring tool for toxicity prevents rational evaluation. FCD is a sensitive measurement of toxicity in the microcirculation and a marker for microvascular dysfunction (28, 61, 88, 133, 152, 166, 167); however, FCD can only be measured in experimental studies.
An optimal oxygen carrier has not been devised yet, and blood-banked blood is probably not optimal in this context. Moreover, the design of a HBOC that has the properties of native blood in the circulation may be unattainable, particularly because it is not possible to reproduce the biomechanical characteristics of native blood, and allows the microcirculation to remain functional. Nevertheless, this can be achieved by recognizing that a critical property of a blood substitute is that of maintaining the functionality of the microcirculation, which is significantly dependent on adequate levels of oxygen, NO, and hemodynamic forces on the vascular endothelium.
Footnotes
Acknowledgments
This work was partially supported by the National Institutes of Health (NIH) under the Program project P01-HL071064, and grants R01-HL52684, R01-HL62354, and R01-HL62318.