
Abstract
Introduction
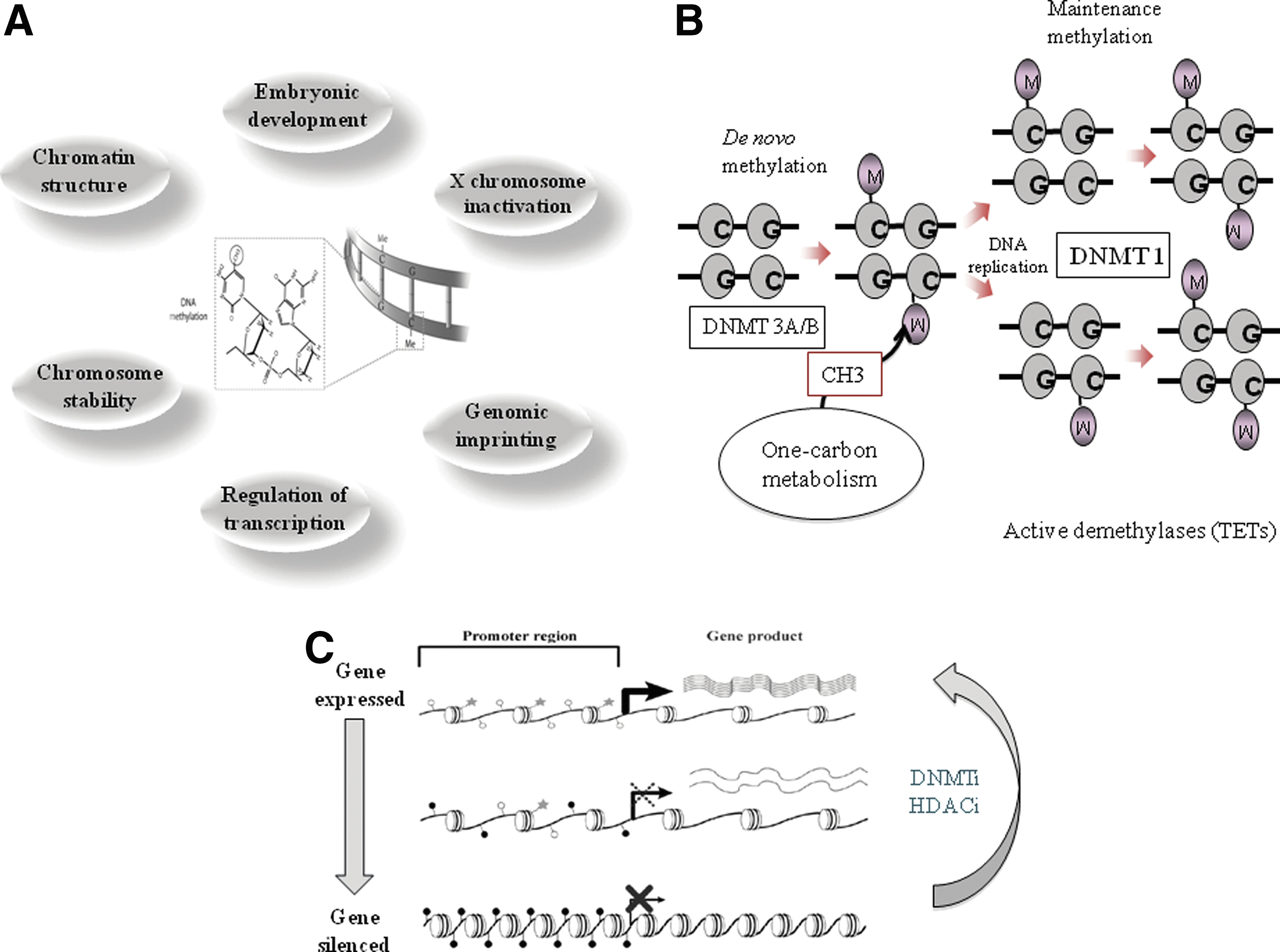
DNA methylation is the most extensively studied epigenetic mechanism, and it plays multiple roles in key cellular processes, including regulation of gene expression, embryonic development, genomic imprinting, and chromosome stability (Fig. 1A). Although methylation of other nucleotides does exist, at least for this article, DNA methylation refers to the attachment of a methyl group to the 5-carbon (C5) position of cytosine, mostly in the context of so-called CpG dinucleotide. This process is mediated by two main categories of DNA methyltransferases (DNMTs): de novo DNA methyltransferases (DNMT3A and DNMT3B) and the maintenance methyltransferase (DNMT1) (Fig. 1B). DNA methylation changes, like other epigenetic changes, are in principle reversible, which makes them attractive targets for the epigenetic therapy (Fig. 1C).
A plethora of experimental studies demonstrate that deregulation of DNA methylation is intimately linked with many human diseases, most notably cancer. In tumor cells, DNA methylation changes are found in two forms: gene promoter-associated (CpG island [CGI]-specific) hypermethylation and concurrent global loss of 5-methyl-cytosine (global hypomethylation) (Fig. 2).

) indicate unmethylated cytosines. Filled stars ( ) indicate methylated cytosines.
) indicate methylated cytosines.
A plethora of experimental studies demonstrate that epigenetic deregulation is intimately linked with many human disease, most notably cancer. The ubiquity and early appearance of epigenome alterations in human malignancies make them attractive targets for biomarker discovery, therapeutic intervention, and prevention (37, 48). In recent years, we have witnessed an emergence of powerful technologies in epigenomics that allow a high-throughput detection of epigenetic changes with a genome-wide scope. (Fig. 3) These remarkable advances, notably those linked to the development and application of massively parallel sequencing technologies, have tremendously accelerated epigenomic research and opened up new perspectives. In this review, we summarize methodologies for DNA methylation analysis and recent developments in genome-wide and high-throughput methods that are increasingly available and discuss their potential for use in cancer research. A comparative summary of methods discussed in the article is given in Table 1. For more comprehensive overviews of methodologies for epigenetic/epigenomic analysis, readers are directed to these excellent recent reviews (21, 35).

HELP, MeDIP and MIRA all have been used with downstream sequencing as well and except for the general superiority of sequencing over microarray, advantages and disadvantages of all these enrichment methods remain almost the same.
RRBS employs both restriction digestion and bisulfite conversion.
FFPE, formalin-fixed paraffin-embedded; Res, restriction endonucleases; WGA, whole-genome amplification.
Preparing DNA for Methylation Analysis
Owing to inability of standard molecular biology techniques such as hybridization and sequencing in differentiating cytosine from its methylated counterpart, and erasure of methylation information during PCR amplification, sample DNA has to undergo certain pretreatments. These pretreatments are methylation dependent and fall under three categories: (i) restriction endonuclease (RE) treatment, (ii) affinity enrichment, and (iii) bisulfite treatment.
RE treatment
REs are very important tools in molecular biology research. Activity of a few restriction enzymes is dependent on the methylation status of particular nucleotides in their recognition sequence. Some enzymes are inhibited (methylation sensitive), while some are only active if their recognition site is methylated (methylation dependent) at a particular base. Methylation-sensitive and insensitive isoschizomer pairs have long been handy tools in methylation research. HpaII (sensitive)/MspI (insensitive) and SmaI (sensitive)/XmaI (insensitive) pairs are among the most widely used. McrBC is one of the most commonly used methylation-dependent REs. (cut only if cytosines in recognition sequence are methylated). Its unique recognition sequence [RmC(N)55–103RmC] helps indirect enrichment of unmethylated sections of genome.
Affinity enrichment
Affinity enrichment of the methylated DNA fraction exploiting 5-methylcystosine (5mC)-specific antibodies or in other case proteins that have particular affinity for methylated CpGs (methyl-binding domain [MBD]-containing proteins) provides a powerful unbiased alternative overcoming constraint of recognition site dependence related to RE-based strategies. Methylated DNA immunoprecipitation (MeDIP) utilizes 5mC-specific antibodies to immunoprecipitate methylated DNA (58). Among MBD family proteins, MeCP2 was the first one to be used (12). Multimerized methyl DNA-binding domains of another MBD protein, MBD1, have also been widely used (31). Methylated CGI recovery assay (MIRA) is another MBD protein-based approach that utilizes a high-affinity protein complex made up of short isoform of MBD2 (MBD2B) and its binding partner MBD3L1 (47).
Bisulfite treatment
Although it was long known that sodium bisulfite treatment of DNA deaminates unmethylated cytosines into uracil at a rate much faster than the methylated ones, it was not until the description of the method by Frommer et al. in 1992 that bisulfite treatment was introduced into methylation research (24). By using optimal conditions, all unmethylated cytosines are converted to uracils, while 5mC remains unconverted. Subsequent PCR amplification will replace all unmethylated cytosines (uracil) with thymine, and this way an epigenetic methyl mark is converted into a genetic difference.
Global Estimation of 5mC Content
Global methylation content of DNA can provide basic information about the disease process and progress and can be useful in various clinical settings as biomarker as well drug screening (32). Global content of 5mC can be quantified by high-performance capillary electrophoresis (HPCE) or high-precision liquid chromatography (HPLC)-based separation of individual nucleosides (generated by enzymatic hydrolysis of DNA) coupled with electrospray ionization/mass spectrometry. Both HPCE and HPLC can separate unmethylated cytosines from the methylated ones (2).
Based on one of the most common isoschizomer pairs used in DNA methylation analysis, HpaII/MspI Karimi et al. reported a luminometric method for global DNA methylation quantification (luminometric methylation assay [LUMA]). Using EcoRI as an internal control, the DNA is digested in two separate reactions (MspI + EcoRI and HpaII + EcoRI). EcoRI leaves a 5′ overhang—AATT—while HpaII/MspI leaves a 5′ overhang—GC. Using a pyrosequencing platform, these overhangs are filled during the sequential addition of nucleotides. A failure in digestion by HpaII owing to methylation at the recognition site will result in an absence or relative reduction (in comparison to MspI-digested aliquot) of peaks during G and C nucleotide dispensations. Using mathematical calculations, the degree of methylation at all HpaII/MspI sites across the genome is quantified and is represented as a% methylation (32). Other enzyme combinations can also be used.
Repetitive elements are spread across the genome and are normally heavily methylated. The degree of methylation at repetitive elements such as long interspersed element-1 (LINE-1) is directly proportional to global methylation content (60). Bisulfite pyrosequencing of repetitive elements can provide a global picture of DNA methylation levels and has been widely used.
Locus-Specific DNA Methylation Analysis
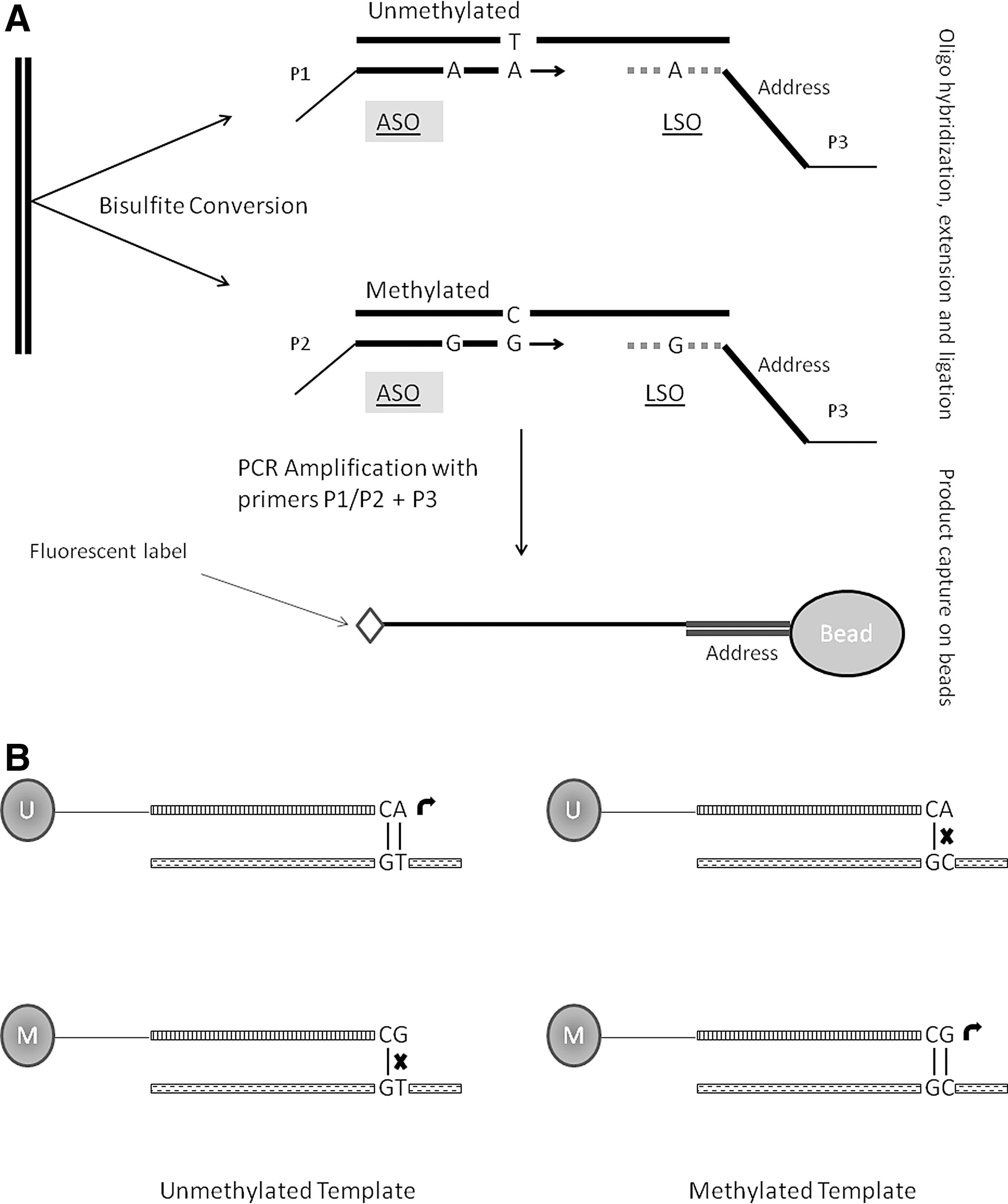
Numerous methods have been reported for locus-specific analysis. While the earlier ones relied exclusively on restriction enzymes, application of bisulfite conversion has revolutionized the field. Methylation at one or more CpG sites within a particular locus can be determined either qualitatively (presence or absence) or quantitatively. DNA samples are usually derived from a heterogeneous population of cells, in which individual cells may vary vastly in their DNA methylation patterns. Hence, most of the methods aimed at quantitative measurement of DNA methylation determine the average methylation level across many DNA molecules. The results are presented as percent methylation—percentage of DNA molecules, in a given sample, methylated at the specific cytosine position under investigation (see more details in the following paragraphs and also Fig. 4). There is a long list of locus-specific methods available; hence, only a few will be discussed here keeping in mind the extent and ease of their usage.

Methylation-specific PCR (MSP) is one of the most widely used methods in DNA methylation studies. Using primers that can discriminate between methylated (M primer pair) or unmethylated (U primer pair) target region after bisulfite treatment, DNA is PCR amplified. One primer in both M and U primer pairs necessarily contains a CpG site near its 3′ end. This CpG site is the one under investigation, and both M and U primers contain the same site. A forward primer in M pair having the C nucleotide in its sequence for the CpG position under investigation will fail to amplify the region if that particular cytosine is unmethylated (hence, converted to uracil during bisulfite reaction) and vice versa. Success or failure in amplification can qualitatively determine the methylation status of the target site (26). Although rapid and easy to use, MSP suffers from various disadvantages such as reliance on gel electrophoresis and the fact that only a very few CpG sites can be analyzed using a given primer pair. The method reported originally by Herman et al. (26) was at best qualitative. However using TaqMan® technology, Eads et al. reported a quantitative MSP-based method for DNA methylation analysis (17). In this method, named as MethyLight, bisulfite-converted DNA is amplified and detected by methylation-state-specific primers and TaqMan® probes in a real-time PCR. As the two strands of DNA no longer remain complimentary after bisulfite conversion, primers and probes are targeted for either of the resulting strands. Initial template quantity can be measured by traditional real-time PCR calculations. Incorporation of various quality controls for bisulfite conversion and recovery of DNA after bisulfite treatment have improved the quantitative reliability of this method. MethyLight has many advantages over MSP and other locus-specific DNA methylation analysis methods. It avoids gel electrophoresis, restriction enzyme digestion, radiolabeled dNTPs, and hybridization probes, yet there are a few shortcomings as well to this method, the biggest one being PCR bias. As mentioned above, individual DNA molecules originating from a mixed population of cells can vastly differ from each other in their methylation status. Such a variable population will result in DNA molecules differing widely in their cytosine content after bisulfite treatment; highly methylated molecules will be C-rich, whereas unmethylated ones will be T-rich. These two populations can sometimes amplify with varying efficiencies, a phenomenon termed as PCR bias. Warnecke et al. reported preferential amplification of T-rich unmethylated sequences (57). This PCR bias can potentially affect the accurate quantitative estimation of DNA methylation.
Combined bisulfite conversion restriction analysis (COBRA) is based on the principle of loss or retention of a restriction enzyme site after bisulfite treatment, depending on the methylation status of targeted cytosine. Alternatively, creation of a new restriction site is also possible (7). Both of these facts can be utilized to analyze DNA methylation in a target region whose sequence is already known. Bisulfite-treated DNA is PCR amplified using primers flanking the target site and is subsequently digested with a restriction enzyme. Combined with an electrophoresis through microfluidic chips, such as Agilent™ 2100 Bioanalyzer, a procedure named as Bio-COBRA, accurate assessment of all the resulting restriction fragments can provide a quantitative measurement of the methylation status at the target region (7). Although quite useful and robust, the biggest disadvantage of this method is limited number of restriction sites that can be used.
Bisulfite sequencing of DNA using Sanger chemistry has also been used for locus-specific methylation analysis. Originally introduced by Frommer et al. (24), this method is based on PCR amplification of bisulfite-converted DNA using specific primers, followed by cloning. To determine an average picture of methylation across millions of DNA molecules, a few clones are randomly selected and sequenced. The massively parallel revolution in sequencing has also benefitted locus-specific approaches. More than 100 PCR products from different tissues were analyzed on 454 sequencing platform (Roche/454 Life Sciences, Branford, CT) in a single run (51). For each PCR product, on the average, >1600 sequences were generated, far larger than the clones usually sequenced (around 20 for each PCR product). This way more accurate measurement of average methylation levels in the given population of DNA molecules can be carried out (51). High throughput of this method can prove to be very useful in candidate region approaches, as many samples can be analyzed simultaneously in one single run that too for potentially more than one target regions.
Pyrosequencing is sequencing by synthesis approach and has been widely used for DNA methylation analysis. Pyrosequencing offers a highly reliable, quantitative, and high-throughput method for analysis of DNA methylation at multiple CpG sites with built-in internal control for completeness of bisulfite treatment. As bisulfite treatment converts unmethylated cytosines into uracils (which will be converted to thymine upon subsequent PCR amplification) leaving methylated ones unchanged, the methylation difference between cytosines is converted into a C/T genetic polymorphism and can be quantified likewise (54). Although bisulfite pyrosequencing is one of the most widely used methods for quantitative determination of methylation, it is limited by a few drawbacks. Thermal instability of enzymes used in pyrosequencing reactions, particularly luciferase, requires the reaction to be carried out at 28°C. Therefore, optimal amplicon size to be subsequently used for pyrosequencing reaction is around 300 bp or less to avoid secondary structures (13). As bisulfite conversion results in low-complexity DNA molecules (A, T, and G nucleotides, except very few methylated cytosines), designing optimal primer sets for every region of interest is a difficult task (52). For more details, see Figure 4.
In yet another high-throughput quantitative approach, bisulfite-treated DNA is first amplified with specific primers. The reverse primer is tagged at the 5′ end with T7 promoter sequence to facilitate in vitro transcription by phage RNA polymerase in the next step. Endonuclease RNase A, which cuts after every C and U in an RNA molecule, is used to generate short fragments. However, for only C-specific or U-specific cleavage, two separate in vitro transcription reactions are run. In an U-specific cleavage reaction, dCTP is used instead of CTP. This blocks cleavage after C and RNase A only cut after U. Similarly, a separate C-specific cleavage reaction is set up. This way, a complex mixture of short oligonucleotides of varying lengths is generated. Methylation-dependent C/T polymorphism in bisulfite-converted DNA is reflected as G/A in transcribed RNA molecules and results in a 16-Da mass difference for each CpG site in cleavage products, which is then analyzed by matrix-assisted laser desorption ionization–time-of-flight (MALDI-TOF) spectrometry (19).
HeavyMethyl method relies on nonextendable oligonucleotide blockers specific for bisulfite-converted unmethylated DNA. Primers flanking the region of interest are used to amplify target DNA fragment. The blockers anneal to a region overlapping with amplification primer-binding site, blocking primer binding to unmethylated DNA and inhibiting amplification. This results in selective amplification of methylated DNA only. In real-time PCR, amplification is monitored by a methylation-specific probe. This method is particularly useful for samples where the percentage of methylation might be very low (methylated sequences have been quantified in a background of 8000-fold excess of unmethylated DNA) (15).
Other important single-locus methods include single-nucleotide primer extension on bisulfite-converted DNA template with primer terminating immediately 5′ of cytosine to be assayed (methylation-sensitive single-nucleotide primer extension [MS-SnuPE]) and methods based on differential melting properties of amplicons generated from bisulfite-converted methylated and unmethylated DNA (methylation-sensitive melting curve analysis [MS-MCA] and methylation-sensitive high-resolution melting [MS-HRM]) [see Ref. (34) and references therein].
Genome-Scale Approaches for DNA Methylation Analysis
Technological advances in the past two decades have geared up research in the fields of genomics and molecular biology ushering into an era of rapid discovery, -omics-scale approaches, and high-throughput analysis. Although locus-specific and global analyses of DNA methylation have contributed important insights into biology and wide ranging role of DNA methylation, a comprehensive understanding requires methodologies that provide higher coverage, ideally up to single-base resolution spreading over whole genome.
Earlier approaches
Among the earliest forays into broad DNA methylation profiling, restriction landmark genomic scanning proved to be the most comprehensive and has been widely used, though labor intensive. Two-dimensional electrophoretic separation of methylation-sensitive restriction enzyme (MSRE)-cut genomic DNA, with cut ends labeled with radionucleotides, produces a radiograph of thousands of spots (∼2000), essentially lacking methylated sites, which failed to be cut and hence radiolabeled. Differences in methylation between two samples are inferred from the differences in patterns of these spots. This method is CGI biased, as mostly those enzymes are used as landmarks that have recognition sites more prevalent in CGIs such as NotI and AscI (11).
Two similar PCR-based methods, namely methylation-specific arbitrarily primed PCR (MS-AP-PCR), and amplification of intermethylated sites (AIMS) have also been widely used. Both methods rely on methylation-sensitive isoschizomer digestion followed by PCR amplification, which produces a distinct fingerprint when electrophoresed. However, they differ in the REs and primers used. MS-AP-PCR uses the HpaII/MspI pair for digestion followed by amplification with arbitrary primers under low-stringency conditions (36). On the other hand, AIMS use the SmaI/XmaI pair. Both enzymes share the same recognition sequence (CCCGGG). The DNA is first treated with methylation-sensitive SmaI, which cuts between third C and G leaving a blunt end, thus eliminating unmethylated sites. Treatment with methylation-insensitive XmaI cleaves between first and second C of methylated-hence-uncut sites, leaving a four base overhang—CCGG, for adaptor ligation. Adaptor-ligated fragments are PCR amplified and resolved to produce a fingerprint of anonymous DNA bands representing methylome of the cell (23).
Methyl CpG Island Amplification (MCA) is an approach similar to AIMS, and has been used in combination with downstream dot-blot or representational difference analysis (RDA) (53). Methylation-sensitive RDA (MS-RDA) employs MSREs (e.g., HpaII) to enrich unmethylated fraction, followed by PCR amplification and RDA (55)
Microarray-based DNA methylation analysis
Introduction of microarray technologies opened unprecedented horizons in methylome research. In contrast to hitherto discussed methods, thousands of regions of interest can be analyzed simultaneously. Various platforms have been used for studying DNA methylome differing in their resolution and regions targeted. Ranging from CGI or promoter region-specific platforms to oligonucleotide-tiling arrays virtually covering whole genome with high resolution, various arrays have been custom designed or are commercially available.
The earliest approaches to this end coupled restriction digestion with downstream hybridization to microarrays. Differential methylation hybridization (DMH) was among the first of such kind (29). Fragmented with a frequent cutter MseI, linker-ligated DNA is either digested with an MSRE such as BstUI or left untreated. This is useful to indentify methylation spots in a particular sample. Alternatively, using a combination of MSREs (BstUI, HpaII, and HhaI) to increase genomic representation, two samples can be compared for differential methylation (59) Using linker-specific primers, only uncut fragments, methylated or with no recognition site, will be amplified. Radioisotope (32P) labeling of DNA fractions (BstUI treated and untreated) in the former case (29) or differential fluorescent labeling in later case (59) allows quantification of methylation upon hybridization of sample to microarray. Based on same principle, but using a methylation-dependent enzyme McrBC, Nouzova et al. studied methylation changes during leukemia cell differentiation (44). In this case, only unmethylated fragments will remain intact to be amplified, and methylation status of a particular locus can be studied in the same manner as discussed above. Use of McrBC increases sensitivity to densely methylated regions. Further improvements were made to McrBC-based microarray analysis by proposing a novel array design and optimized data processing–comprehensive high-throughput arrays for relative methylation (CHARM) (30).
HpaII tiny-fragment enrichment by ligation-mediated PCR (HELP) (33) is another restriction enzyme-based strategy. Using HpaII and its isoschizomer MspI, two separate aliquots of DNA are fragmented. This way, the methylated fraction of DNA is eliminated from an HpaII-digested sample. Fragments are ligated to adaptors and amplified by ligation-mediated PCR. Both fractions are labeled with different fluorescent dyes and hybridized to an oligonucleotide array. Absence or relative weakness of signal in the HpaII fraction as compared to the one treated with MspI will show the methylation status of a particular position. HELP assay was further improved by using dual-adaptor approach to amplify smaller fragments (50–200 bp) (45). MCA (discussed above) was later combined with downstream CGI microarray hybridization and named as MCAM (20). Differential labeling of two samples and subsequent array hybridization will yield comparative picture of DNA methylation profile.
Only a very small portion of methylome can be studied using restriction enzyme-based approaches. Due to this limitation, affinity enrichment of DNA has emerged as an effective alternative. DNA can be precipitated by either of the methods discussed above and has been widely used in combination with microarrays. As a general approach, DNA is first sheared randomly (either by sonication or a frequent cutter enzyme), and a portion of DNA is set aside to be used later as reference (input control). The resulting enriched fraction and input control are differentially labeled and hybridized to a custom-made or commercially available array platform. Using a custom array, Weber et al. developed MeDIP-chip (58). MIRA has also been used in variety of studies; for example, when combined with a whole-genome tilling array, a 100-bp resolution methylation profile human B cells was generated (47).
Bisulfite treatment of DNA results in reduced sequence complexity and increased redundancy, thus reducing hybridization specificity (35). Therefore, coupling bisulfite treatment with array hybridization has not been much successful. However, Illumina GoldenGate® BeadArray™ and Infinium® are well suited for this purpose and have been widely used. Principle of both platforms is shown in Figure 5. GoldenGate® Human Cancer Panel I is a typical example of this assay, and up to 1536 CpG sites spanning 807 genes can be studied using this platform; however, custom-made arrays can also be used alternatively (4). An example of results obtained from GoldenGate® platform is shown in Figure 6 [adapted from Hernandez-Vargas et al. (27)]. The first version of Illumina Infinium Methylation assay called Infinium HumanMethylation27 BeadChip can investigate 27,578 CpG sites spread over the proximal promoter regions of 14,475 consensus coding sequences, including 110 miRNA promoters in the human genome (5). Improved version of this chip, called Infinium HumanMethylation450 BeadChip, can be used to investigate >485,000 CpG sites per sample. This version of chip has included more than 99% of RefSeq genes with multiple CpG sites per gene spread across the promoter, 5′ untranslated region (UTR), first exon, gene body, and 3′ UTR. In addition to 96% CGIs, the chip also includes various CpG sites corresponding to CGI shores and flanking regions (3). Both the chips can be used to analyze up to 12 samples per chip simultaneously.

Sequencing-based approaches
Sequencing a whole methylome has not been possible until very recently. Although the use of DNA-sequencing platforms to identify and quantify the methylation of individual cytosines is as old as application of bisulfite treatment for DNA methylation analysis (24), the methodology reported by Frommer et al. was at best useful for a very limited number of targeted regions even with the use of brute-force Sanger sequencing (18). Massively parallel revolution in sequencing has shifted the paradigm of genome-wide DNA methylation analysis. Whole methylome map of a particular cell type can now be generated in a matter of 3 to 5 days (38) at costs much lower than previously possible. As compared to array hybridization, sequencing-based analysis provides more detailed information with less DNA input and does not rely on laborious efforts needed to design an appropriate array. In addition, complexity of methylation patterns in DNA samples originating from heterogeneous population of cells and existence of cytosine methylation outside traditionally analyzed CpG-rich regions (30) demand an unbiased approach that can quantitatively determine the level of methylation ideally at single-nucleotide resolution throughout the genome.
The pioneering efforts at generating a whole-genome single base-pair resolution methylome map of eukaryotic organisms were made by two independent groups in the year 2008. By using two different shotgun bisulfite high-throughput sequencing protocols named BS-seq (10) and MethylC-seq (39), both groups generated comprehensive cytosine methylation maps of Arabidopsis thaliana genome. In the BS-seq approach, genomic DNA from A. thaliana aerial tissues was used. An interesting finding of this study was identification of a pattern of periodicity between sites of methylation, 167 nucleotides, which roughly correlates with internucleosome linker length in plants, highlighting the functional correlation of methylation machinery with nucleosomal positioning (10). Using a slightly different approach named MethylC-seq, Lister et al. sequenced DNA from A. thaliana flower buds, mapping 79% of total cytosines (16×average coverage) to the reference genome (39). MethylC-seq was also later applied to generate the first ever single-base resolution map of human methylome in embryonic stem cells and fetal fibroblasts (40). Remarkably, this study identified that nearly one quarter of cytosine methylation in embryonic stem cells is in non-CG context, emphasizing the need of unbiased, genome-wide approach (Fig. 7).
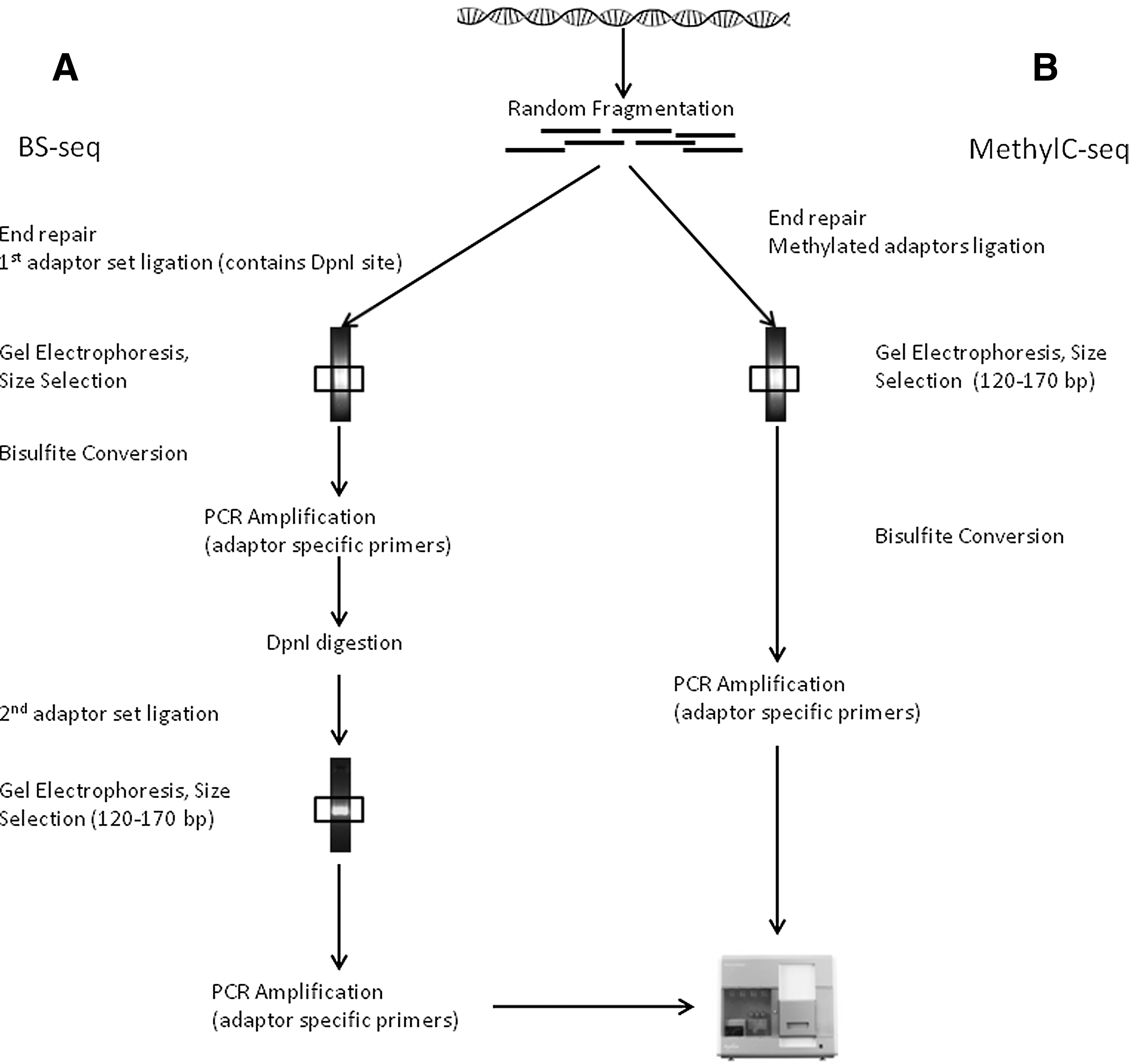
RE treatment and affinity enrichment methods have been adapted to downstream massive sequencing, with additional advantage of reduction in target DNA complexity and amount of sequencing. Improving original HELP assay by using two sets of adaptors to amplify <200-bp fragments during the LM-PCR step, and coupling HELP output with NGS, Oda et al. could analyze 98.5% CGIs in human genome. Using MspI digestion as control, methylated spots are identified by their absence in the HpaII-cut fraction's sequence reads (45). Using an essentially similar approach named Methyl-seq (9), important differences in methylation patterns between hESCs, their in vitro differentiated derivatives, and human tissues have been identified. Another widely used method named as reduced representation bisulfite sequencing (RRBS) couples RE representation followed by BS sequencing on a massively parallel platform (42). MspI digestion before BS conversion allows reducing redundancy by selecting a CpG-rich genomic subset (42). Another RE-based approach dubbed as methyl-sensitive cut counting (MSCC) (1) was used in parallel analysis of human B lymphocyte methylome along with padlock probe-captured DNA (discussed later).
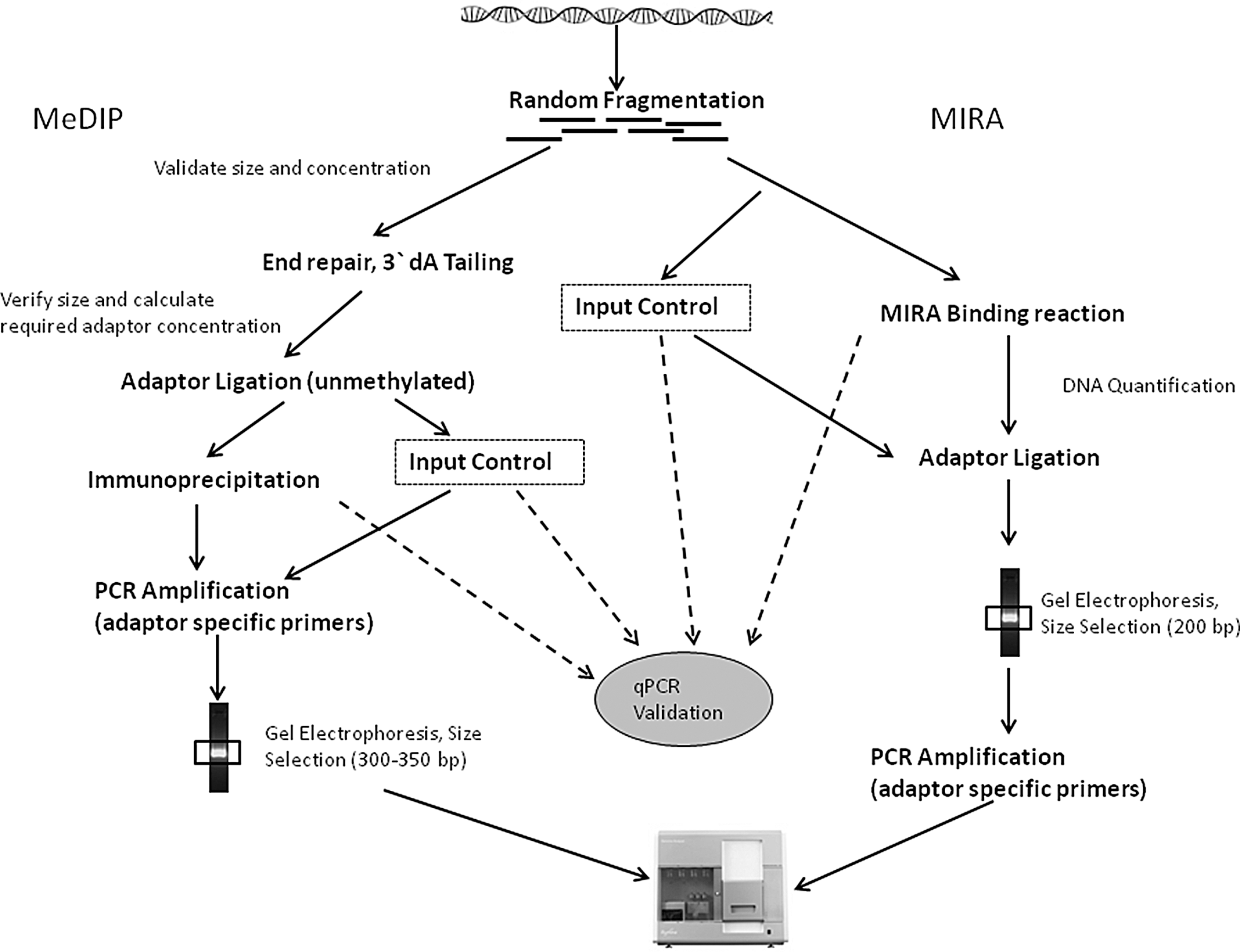
Affinity-enrichment-based methods have been adapted to downstream analysis by massive parallel sequencing. MIRA-seq (46) MeDIP-seq (16), MethylCap-seq (using MeCP2) (8), MBD-isolated genome sequencing (49) follows more or less similar protocols (except affinity enrichment itself). As an example, two of the reported workflows are summarized in Figure 8 (46, 50). Although similar in approach, enrichment methods target different compartments of the genome. While MeDIP captures methylated regions with low CpG density, MBD favors high-CpG-density regions. Moreover, inclusion of multiple elution steps with increasing salt concentrations in MBD capture protocols can enrich moderately methylated regions, making it more useful as compared to MeDIP (43).
As affinity enrichment methods target methylated DNA, the unmethylated state of DNA is inferred from its absence in reads. A confidence in this inference is highly dependent on the sequencing depth. To overcome this potential issue, Maunakea et al. (41) took on a unique approach to elucidate methylome of the human frontal cortex gray matter. They used two complementary methods, MeDIP (used to enrich methylated fraction of genome) and MSRE (MRE) digestion (used to enrich unmethylated fraction), followed by high-throughput sequencing to achieve a greater genome coverage. For MRE-seq, DNA is first digested in three separate reactions with REs HpaII, AciI, and Hin6I, combined in equal quantities and size selected (100–300 bp). Sequencing DNA from both MeDIP and MRE fractions highlighted the role of intragenic and intergenic methylation in gene regulation, pointing out to the complexity of cytosine methylation code (41).
Other important sequence selection strategies used before sequencing include simultaneous capture and amplification of bisulfite-converted targeted regions using padlock probes (14) and capture of specific sequences by array hybridization (28).
Raw sequencing reads from each type of high-throughput platform need to undergo dedicated and complex bioinformatics analysis pipelines, which differ from each other according to the platform used and the particular type of experiment and protocol. Such bioinformatics tools are beyond the scope of this article. As a general approach, enzyme- and affinity-based sequencing methods determine the relative abundance of different genomic regions in enriched fraction by counting the number of reads that uniquely map to the reference genome as compared to input control. On the other hand, bisulfite sequencing extracts information directly from the sequence (35).
All the sequencing approaches discussed above have their own advantages and shortcomings. Shotgun bisulfite sequencing, though still a gold standard because of its genome coverage, is cost and effort intensive. This makes this approach unfeasible for studies involving large number of samples, for example, cancer epigenomic studies. Sequence selection strategies though useful are invariably prone to particular biases. While RE-based strategies are limited by the number and distribution of enzyme recognition sites, affinity enrichment methods cannot yield information on individual CpG dinucleotides (35). Therefore, while the whole-genome approach is useful to generate reference methylome maps, sequence selection strategies can yield useful information about most relevant regions. Relative merits and demerits of each method have been excellently reviewed elsewhere (35).
Future Challenges
Unlike genetic code, which is the same in every single cell of an organism, epigenetic code shows wide-ranging variability across different cell types and also in the same cells at different developmental stages and under the influence of various environmental stimuli. This plasticity of epigenetic code poses a significant challenge in terms of effort, resources, and technology. In our opinion, it would be necessary in the long run to develop robust methods capable of analyzing methylome of single cell in a heterogeneous population of cells. From the standpoint of translational applications of DNA methylome research, such as in clinical settings as diagnostic biomarkers, methods capable of analyzing methylation patterns accurately in minute quantities of DNA need to be developed. DNA from archived formalin-fixed paraffin-embedded samples collected as a part of epidemiological studies is a special challenge, owing to its degraded state. Although a few adaptations to previously established protocols can help [for example (56)], this area is in need of further development.
Innovation
Recent discoveries of 5-hydroxymethylcytosine (5hmC) occurrence being more widespread in mammalian cells than previously thought and the role it might play in demethylation have opened up a whole new dimension to epigenetic layer of coding. As most of the methods currently in use for 5mC detection cannot differentiate between 5mC and 5hmC, future developments in this area need to be focused upon. Affinity purification and downstream sequencing with 5hmC-specific antibodies, similar to that for 5mC, cannot determine precise location of 5hmC. Recently, two excellent methods have been reported for sequencing 5hmC up to single-nucleotide resolution (6, 61).
Footnotes
Acknowledgments
We apologize to authors whose relevant publications were not cited due to space limitation. The work of the IARC Epigenetics Group is supported by the grants from the National Cancer Institute (NIH), United States; l'Association pour la Recherche sur le Cancer (ARC), France; la Ligue Nationale Contre le Cancer, France; the Swiss Bridge Award; and the Bill and Melinda Gates Foundation (to Z.H.).