
Abstract
Introduction
Given the variety of the DNA lesions, different repair mechanisms exist (Fig. 1). In greater detail, the double-strand break repair (DSBR) is deputed to restore the sugar backbone of both DNA filaments after their break (71, 79); the mismatch repair (MMR) principally occurs when the replication and the recombination can cause base–base mismatches and insertion-deletion loops (IDL) (38); the nucleotide excision repair (NER) recognizes bulky, helix-distorting lesions, involving the excision of a single-stranded, lesion-containing oligonucleotide fragment (55); the base excision repair (BER) corrects base lesions arising from oxidation, alkylation, deamination, and depurination/depyrimidination reactions (77); and, finally, the single-strand break repair (SSBR) reconstitutes the sugar backbone of the broken single DNA filament (13).

The first step for an efficient DNA-damage response is the recognition of the lesion, with this response being mediated by sensor proteins, which specifically recognize the damage and coordinate other molecules (mediators and effectors) that are responsible for the transduction of the signal from the nucleus to the cytoplasm (21, 60). As a result, the cell cycle is arrested, in order to avoid lesion inheritance. In the meantime, DNA-repair mechanisms set about healing the damage. However, if the genome integrity is irreparably compromised, the cell is forced to die. Indeed, when the DNA damage is too severe, cells can undergo permanent cell-cycle arrest (senescence) or cell death (apoptosis/necrosis). Despite the well-established link between unsustainable DNA-damage and induced cell death, the molecular mechanisms responsible for this crosstalk are largely unknown.
In this review, we will focus on the various DDRs and on their link to the apoptotic pathway, emphasizing the role of some proteins as both key players in DNA repair and direct mediators of the apoptotic response, such as Ku. In addition, particular attention has been paid to novel mechanisms that are responsible for cell fate determination on DNA damage, such as the oscillatory activation of p53, the proteasomal degradation, and the redox-mediated regulation of apoptotic and DDR proteins.
DSBR: The p53 Signaling Network and Apoptotic Functions of Repair Proteins
DNA double-strand breaks (DSBs) are among the most serious and lethal types of DNA damage; they can be induced by ionizing radiation, chemical agents, reactive oxygen species (ROS), or as by-products of DNA replication. In fact, during replication, collapsing replication forks leave the DNA backbone broken on a single strand (SSB) that can be often converted to double-strand damage (DSB) (10). The cell has evolved two types of DNA DSBs' repair: homologous recombination (HR) and nonhomologous end joining (NHEJ), on the basis of different fidelity and template requirements (71). HR needs the sister chromatid as a DNA template to repair the break and reconstitute the original sequence, and, for this reason, it occurs during the late S and G2/M phase of the cell cycle (34, 51). It involves several proteins that accumulate at the DNA ends, including the nucleases C-terminal binding protein interacting protein (CtIP)/exonuclease 1 (Exo1) and the Rad51 recombinase complex formed by breast cancer 1 and 2 (BRCA1/2), Rad51/52, Xrcc2/3 (Fig. 1) (34). On the other hand, NHEJ can be activated either in G0/G1 phase or throughout the cell cycle (51). The latter can repair DSBs by preserving the phosphodiester backbone and the molecular integrity of the chromosome without restoring the sequence information in the DNA (44). The formation of the DNA-protein kinase (PK) complex, by the interaction between the catalytic subunit of the DNA-PK (DNA-PKcs) and Ku, is the fundamental step involved in order to activate the DNA repair by NHEJ (Fig. 1) (44). However, in both DSBR pathways, DSB recognition is the first step that is mediated by the Mre11/Rad50/NBS1 (MRN) complex composed of meiotic recombination 11 (Mre11), Rad50, and Nijmegen breakage syndrome 1 (NBS1) (17). Both Mre11, which exhibits exo- and endonuclease activity, and Rad50 have DNA-binding capabilities; while NBS1 is responsible for shuttling the MRN complex to the nucleus. The MRN complex is important for recruiting ataxia telangiectasia mutated (ATM) or ATR (ATM- and Rad3-related) proteins, members of phosphatidylinositol-3 kinase (PI3K) family, which activate a signaling process that leads to cell-cycle checkpoints and pauses cell-cycle progression (35). Two crucial substrates of ATM and ATR are checkpoint 1 and 2 kinases (Chk1 and Chk2), respectively: Once phosphorylated, these are able to induce cell-cycle arrest, thus guaranteeing time for damaged cells to repair their DNA (35). However, the ATR/Chk1-related signaling pathway is principally activated under SSBs, as will be discussed later.
Another initial step for the DSBR is the ATM phosphorylation of the histone H2AX in the vicinity of DSBs, leading to the formation of γH2AX foci (12). These foci act as a signal to recruit other repair proteins in order to allow the assembly of DSBR complexes (42). H2AX phosphorylation can also be mediated by the DNA-PK, the kinase complex that is formed once the NHEJ pathway of DSBR is activated and which acts both as sensor and as effector of DSBR (65).
It is worth noting that mutations of some DSBR genes have been shown to be associated with human syndromes or diseases (9). Among them, mutations of BRCA1 and 2 have been found in several breast and ovarian cancers, whereas mutations of ATM and some components of MRN complex, such as Mre11 and NBS1, have been identified as being associated with ataxia telangiectasia and NBS, as well as in cerebellar atrophy, microcephaly, radiosensitivity, and marked predisposition to cancer. These lines of evidence strongly suggest that these proteins play a crucial role in maintaining genomic integrity.
p53 at the crossroad between DNA damage and apoptosis
All the signaling pathways triggered by DSBs converge on the tumor suppressor p53 (59). p53 can be activated by all the sensor kinases already mentioned (ATM, ATR, DNA-PK, and Chk1/2) that lead to p53 stabilization and accumulation in the nucleus, where it functions as a transcription factor (Fig. 2) (59, 80). In particular, Ser15 and Ser20 of p53 are highly phosphorylated by ATM and Chk2, thus resulting in the dissociation of MDM2, a p53-negative regulator (15, 19, 62). By being modified in this way, p53 escapes proteasomal degradation and enables the activation of its target genes involved in DNA repair (e.g., damaged-DNA binding protein 2 [DDB2], XPC), in cell-cycle arrest (e.g., p21, GADD45α, and 14-3-3σ), and in apoptosis (e.g., Bcl-2-associated X (Bax), Bcl-2 homologous antagonist killer [Bak], p53 up-regulated modulator of apoptosis [Puma], Noxa, and apoptotic protease activating factor 1 [Apaf1]) (74). Moreover, recent findings demonstrate that, on DNA damage, p53 can activate some pro-autophagic genes, such as AMP-activated protein kinase subunits β1 and β2 (AMPK-β1/2), tuberous sclerosis protein 2 (TSC2), phosphatase and tensin holomog (PTEN), damage-regulated autophagy modulator (DRAM), sestrin1 and 2 (Sesn1/2), and Unc-51-like kinase 1 (ULK1) (11, 23, 29, 31).

As demonstrated by the induction of these genes, p53 activates different signaling pathways that can result in an opposite destiny for the cell. Within a short timeframe after the insult, apoptosis is avoided, thus making it possible to repair the damage. In this context, p53-mediated activation of autophagy could be functional to the energy production that is necessary for DNA repair processes, thus contributing to the pro-survival phase of the DDR. On the other hand, if the lesions accumulate, they are not compatible with the cell life, thus triggering cell death. Once p53 is activated, how can it discriminate between these two opposite scenarios? A p53-negative feedback loop could be the right answer. Active p53 can induce the transcription of its negative modulator MDM2, which has been demonstrated as being crucial in the dynamical behavior of p53 regulation on DNA-damage response (33, 43). Indeed, Batchelor et al. have shown that gamma radiation-induced DNA damage initiates a series of ATM and p53 activation pulses, consisting of p53 activation followed by its subsequent down-regulation (6). As indicated in Figure 2, p53, once phosphorylated by ATM, induces the transcription of its target genes, among them being DNA repair genes and two p53 key inhibitors, MDM2 and wild-type p53-induced phosphatase 1 (Wip1) (6). Both can directly block p53 activity, but Wip1, by dephosphorylating ATM and Chk2, can also inhibit the upstream signal of p53. These two negative feedback loops seem to underlie the regulation of ATM/p53 activation pulses. If the DNA damage is successfully repaired, ATM and p53 are no longer activated and the cell viability is preserved. If the DNA repair is unsuccessful, a round of ATM recruitment at DSBs starts again, with the subsequent activation of ATM and p53 (Fig. 2) (6). These continuous activation pulses of ATM/p53 can lead to the accumulation of several p53 targets which, on the basis of their mRNA and/or protein stability, can display themselves as a dynamic expression pattern (4). Once pro-apoptotic proteins become excessive, they prove fatal to the cell. Moreover, changes in the post-translation modification profile of p53 after its initial activation could act as a molecular barcode and direct the cell toward a specific cellular response (4, 83). In this context, it has been demonstrated that crucial acetylations and methylations at the p53 C-terminal region are essential for its activation as a transcription factor. As a result, different tolerance of p53 on transient or sustained DNA damage has been reported (49).
For the full comprehension of p53's role on DNA damage, its transcriptional-independent function should be also considered. Cytosolic p53 induces cell death by directly acting on mitochondria and favoring the release of pro-apoptotic proteins (32). In addition, it has been demonstrated that p53 has two opposite roles in autophagy regulation, depending on its sub-cellular localization (50). As discussed earlier, nuclear p53 acts as a pro-autophagic factor that induces some autophagy genes; by contrast, the cytosolic pool of p53 is implicated in the inhibition of autophagy (50). Thus, the existence of a p53-mediated interplay between apoptosis and autophagy could, hypothetically, be a response to DDR, accounting for different cellular outcomes.
In addition to the signaling network showing p53 as the major player of the crosstalk between DNA damage and apoptosis, some proteins, commonly involved in DSBR, are also implicated in mediating apoptotic signals.
Ku
The DNA repair complex Ku, comprising Ku70 and Ku86 subunits, is specifically involved in direct binding to the two broken DNA ends (Fig. 3), thus protecting them from excessive degradation, and in the promotion of the NHEJ by the formation of the DNA-PK complex (25, 44). However, Ku has also been demonstrated as being directly linked to the apoptotic signaling pathway that is activated on DNA damage (Fig. 3). It has been shown that Ku can regulate the expression of Apaf1, a pro-apoptotic protein which is fundamental for the execution of the mitochondrial apoptotic pathway (26). Ku-mediated Apaf1 regulation is dynamically modulated on DNA damage: As soon as DNA is damaged, Ku protects cells from apoptosis by blocking Apaf1 transcription; when the DNA damage accumulates highly inside the cells, Ku leaves the Apaf1 promoter, thereby contributing both to the up-regulation of its expression and to the occurrence of apoptosis. Furthermore, the subunit Ku70 has been found to be implicated in regulating Bax translocation into the mitochondria through its direct interaction with Bax and, therefore, in mediating the intrinsic pathway of apoptosis (Fig. 3) (2, 22, 69). Ku70/Bax interaction has been shown to be dependent on Ku70 acetylation status: After apoptotic stimuli, the acetyltransferases CREB-binding protein (CBP) and P300/CBP-associated factor (PCAF) acetylate specific lysine residues on Ku70, thereby resulting in a conformational change of Ku70 and in Bax release (22, 69). Moreover, the stabilization of FLICE-inhibitory protein (FLIP), a well-known inhibitor of death receptor-mediated apoptosis, has recently been reported to be regulated by the subunit Ku70 of the DNA repair complex Ku (Fig. 3) (41). This novel interaction, dependent on Ku70-acetylation status, enables FLIP to interact with the death-inducing signaling complex (DISC) and, thereby, to inhibit the activation of the extrinsic pathway of apoptosis. In addition, the DNA-binding activity of Ku has been found to be abolished on Ku70 acetylation in the DNA-binding domains, thus leading to its decreased capability in DSBs repairing (20). It could be hypothesized that the acetylation status of Ku70 should be properly regulated in order to direct the cell toward apoptosis. Acetylated Ku70 would diminish its ability to repair DNA damage and, simultaneously, to bind Bax and/or FLIP in order to prepare the cell for death. In this scenario, the specific post-translational modification of Ku70 could account for both its dual functions, in DNA repair and in suppressing Bax-mediated apoptosis, and decide the cell fate in a condition of severe DNA damage. Moreover, it is worth mentioning that Liu et al. have demonstrated that, on DNA damage induced by kainic acid, Ku70 is phosphorylated by DNA-PKcs, resulting in Bax release and translocation into the mitochondria to initiate apoptosis (46). There is further evidence highlighting Ku's involvement in the crosstalk between the DNA repair machinery and the DNA damage signaling regulators controlling apoptosis. In particular, it has been recently demonstrated that, on DNA damage, the Ku70 vWA domain (within the N-terminal region) affects activating transcription factor 2 (ATF2)-dependent transcriptional pathway, which modulates several genes implicated in the activation of apoptosis (28). All these findings highlight a crucial role of Ku in regulating the DSBR, acting as its effector, via both its involvement in repair DNA broken termini and its modulation of the mitochondrial apoptotic pathway.
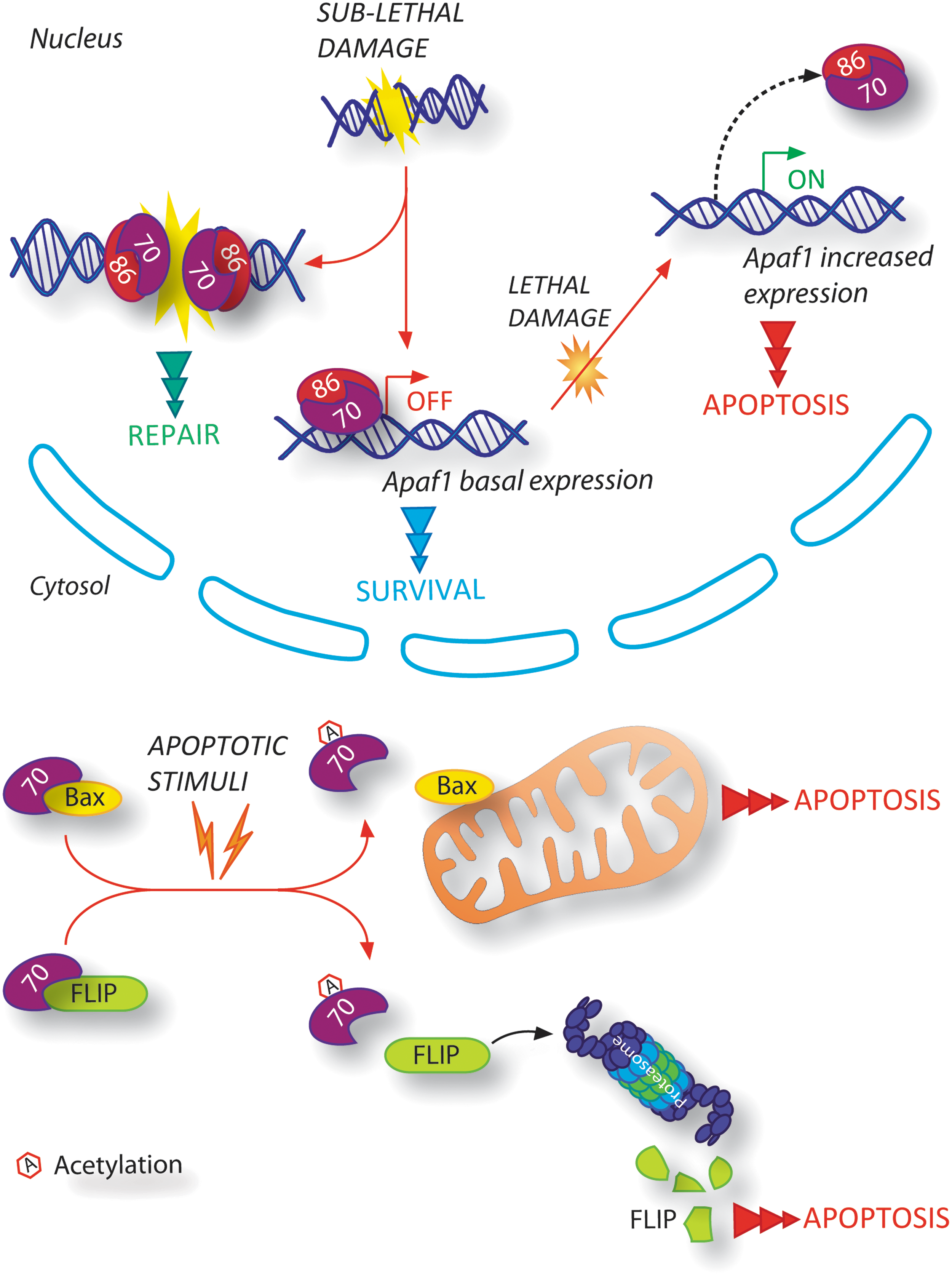
Nijmegen breakage syndrome 1
NBS1 is a key regulator of DSBR, as it is a component of the MNR complex, the sensor of DSBs, and the activator of ATM (17). It has been reported that NBS1 can also activate Bax and induce apoptosis on DNA damage (36). In particular, Iijimaa et al. have demonstrated a direct association of NBS1 with DNA damage-induced apoptosis, as NBS1 directly interacts with Ku70, allowing Ku70 acetylation and Bax translocation to the mitochondria (36). Petrini's group also discovered that NBS1 C-terminal domain is necessary for apoptosis induction on ionizing radiations (68). In fact, its deficiency interferes with ATM activity by preventing the ATM-mediated phosphorylation of structural maintenance of chromosomes protein (SMC1) and BH3 interacting domain death agonist (Bid) protein (40, 68). It is worth noting that after ionizing radiations, NBS1 C-terminus-depleted mice displayed a severe apoptotic defect without pro-apoptotic genes induction being impaired due to the normal transcriptional activity of p53 (68). Thus, it can be speculated that NBS1 acts at different steps of the DDR: either as a sensor of DSBs or as an effector of DSBs, by activating the apoptotic pathway.
MMR: Direct or Indirect Correlation to the Apoptotic Signaling?
When the cell is in the S-phase of the cell cycle, base–base mismatches can be caused by errors of DNA polymerases, yet have to be removed in order to ensure a correct transmission of genetic information to daughter cells. In addition, microsatellites, which are repeated-sequence motifs within DNA, frequently undergo several mismatches, called IDLs, due to re-annealing incorrectly after DNA synthesis. The MMR is deputed to remove these mispairs, and this involves different proteins: hMSH2 and hMSH6 (human MutS Homologous 2 and 6), which recognize the site of mismatch and, in a form of heterodimer (MutSα), monitor newly synthesized DNA for mispairs (38). hMLH1 and hPMS2, in the form of the MutL heterodimer, interact with MutS at the site of a mismatch (38). Of note, mutations in MMR genes are linked to several cancer types, such as hereditary nonpolyposis colorectal cancer (or Lynch syndrome) (21), further underlining the tight link between alteration of DDR and cancerogenesis.
When MMR is “futile”
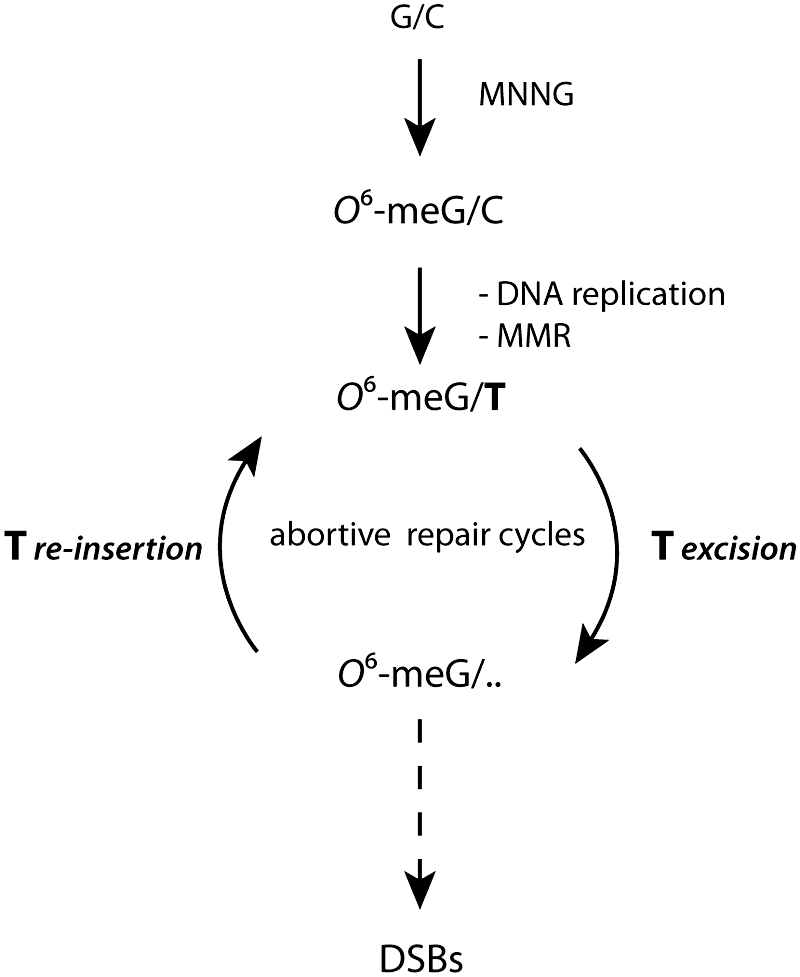
Methylating agents, such as N-methyl-N’-nitro-N-nitrosoguanidine, can induce DNA damage repaired by MMR (60). Among different DNA lesions produced by such a methylating agent, there is the methylation of guanine at the O6 -position in G-C base pairs. In this context, a well-established theory explaining the link between the MMR and the activation of cell-cycle checkpoints and apoptosis is “the futile cycle model” (Fig. 4) (73). According to this model, during DNA synthesis in the S-phase, O6 -meG mispairs with thymine, thus constituting a substrate for MMR, which excises thymine (Fig. 4). However, since O6 -meG is still present in the DNA, thymine is reinserted during DNA re-synthesis, thus leading to the futile removal and re-insertion of thymine, and to the formation of DSBs (Fig. 4) (60, 73). In this scenario, both G2 arrest and lethality responses are dependent on DSBR and independent of MMR. Recently, Quiros and collegues have demonstrated that the processing of O6 -meG into DSBs requires two rounds of replication and that only in the consequent cell cycles can apoptosis occur (58). MMR still functions solely as DNA repair, by detecting DNA lesions and creating DSBs due to futile cycles of repair or repair-replication fork collision. If the DSBs are not repaired by HR, the cells die by ATR/Chk1-dependent apoptosis (58).
MMR proteins as signaling molecules in DDR
It has been postulated that the signaling cascade of cell-cycle arrest and apoptosis could be also activated by the direct interaction of the damage-bound MMR proteins: hMutSα and hMutLα, with proteins of the ATR-Chk1 pathway. In greater detail, ATR, Chk1, and the Chk1 activator Claspin have been identified as hMutSα-binding partners (Fig. 5) (47, 81). In this scenario, MMR acts as a DNA damage sensor by directly signaling G2 arrest and cell death. This represents “the direct signaling model,” which proposes MMR as having two different functions in DDR: DNA repair and signal transduction.

PMS2, another MMR protein, has been demonstrated as interacting with proteins regulating apoptosis, highlighting other possibilities in the crosstalk between DDR of MMR and apoptosis. The subunit of MutL heterodimer PMS2 is required for cisplatin-induced activation of p73 (Fig. 5) (63). p73 is a member of the p53 family of transcription factors with proapoptotic activity (56). Shimodaira and coworkers have demonstrated that PMS2 specifically interacts with p73, causing its stabilization, and that exposure to cisplatin enhances the association between PMS2 and p73. Many years later, it was also shown that the polymorphic variant of PMS2 (R20Q) is defective in activating p73-dependent apoptotic response to cisplatin, thus contributing to the modulation of tumor responses to cisplatin in cancer patients (52). These results suggest that PMS2 contributes to genome integrity not only through DNA repair but also by directly contributing to the activation of apoptosis. In addition, Lim and colleagues recently demonstrated that a multiprotein complex, comprising MAPO1, folliculin (FLCN), and AMPK, is responsible for apoptosis induction on base-mispairing (Fig. 5) (45). When base-mispairing occurs, activated AMPK levels gradually increase in an MAPO-dependent manner, and such activation correlates with induced cell death. In this context, the molecules responsible for the delivery of the signal from the DNA lesion to the pro-apoptotic complex are still unknown. A direct interaction between the MMR-components and AMPK-MAPO1-FLCN could be supposed, although mass-spectrometry experiments do not provide any evidence that supports this hypothesis.
Furthermore, AMPK activation has previously been correlated to both apoptosis and autophagy induction (16). For this reason, a role for autophagy as a stress response on DNA damage has also to be taken into account. In the light of this evidence, Lim's work allows us to speculate that phosphorylated AMPK could be considered the key molecule triggering autophagy flux in this scenario (45).
NER: When a Repair Protein Functions Twice
The NER is able to repair a wide range of DNA lesions, such as pyrimidine dimers and 6-4 photoproducts produced on UV lights, intra-strand crosslinks, DNA-protein crosslinks, and some DNA adducts caused by oxidative damage (55). The NER machinery involves two sub-pathways: the global genome NER (GG-NER) and the transcription coupled NER (TCR). GG-NER recognizes and removes lesions throughout the entire genome, while TCR is a repair system occurring in parallel to transcription (30). Transcriptional inhibition has a pro-apoptotic effect per se, as genes essential for life are no longer transcribed. In this context, a defect in NER is responsible for impaired gene expression and results in triggering apoptosis (48). Otherwise, defective TCR correlates with nonrepaired lesions, which possibly cause DNA breaks. For this reason, to what extent gene expression blockade is relevant to apoptosis induced by impaired NER/TCR is still a matter for debate.
The dual role of DDB2
A more direct effect of NER components on apoptosis induction is mediated by DDB2. DDB2 is a component of the NER machinery that is responsible for the recognition of UV-damaged DNA. More recently, Bagchi's group characterized DDB2 as an adapter protein of the Cul4 (Cullin4) E3 ligase complex, implicated in ubiquitin-mediated proteasomal degradation (66, 67). Remarkably, the association of DDB2 to Cul4 plays a role in the proteasome targeting of the Cullin-substrate p21 and p53 (Fig. 6). On DNA-damage, p53 is phosphorylated at the Ser15 by the activated ATM/ATR; as a result, its transcriptional activity is enhanced and the expression of its target-gene p21 is increased. In this context, DDB2 counteracts p53 activation by allowing selective delivery of phosphor-Ser15 p53 to the proteasome, thus abolishing p21 up-regulation. In addition to the indirect regulation of p21 transcription, DDB2-Cul4 complex targets p21 protein for ubiquitylation and proteolysis in cells affected by DNA damage. In this context, NER can take place and efficiently repair DNA damage, thus sparing a cell's life. This scenario demonstrates DDB2 function on low-dose UV irradiation (Fig. 6), when the genome integrity is not irreversibly compromised. On the other hand, the negative regulation that DDB2 exerts on p53 is disrupted when high-dose UV irradiation affects the cell, probably due to a post-translational modification occurring in the N-terminal region of p53 and preventing DDB2-mediated degradation (Fig. 6). In this case, p53 is fully active and p21 expression is further increased. p21 up-regulation on DNA damage inhibits the apoptotic pathway, acting as a pro-survival signal for the cell and allowing the damage to be repaired. Despite this, if DNA damage is over-extended, apoptosis is hopefully induced: At this point, p21 down-regulation is required, without affecting global p53 activity. The mechanism coupling these two effects is the DDB2-Cul4 mediated ubiquitylation and consequent degradation of p21 which still occurs on high-dose UV irradiation (Fig. 6) (66, 67). Therefore, the DDB2-dependent mechanism specifically points to the crosstalk between NER-mediated DDR and apoptosis. Of note, a recent characterization of the autophagy interactome has revealed an involvement for Cul4, the DDB2-associated E3 ligase, in the autophagy pathway (7). An investigation of Cul4-mediated switch between apoptosis and autophagy on DNA damage could therefore be of utmost interest.

Base Excision and SSBR: Chk1 Opposite Effects on Apoptosis
BER is a multistep DNA-repair mechanism whose intermediates are potentially dangerous for the cell. It is characterized by the removal of one nucleotide or 2–13 nucleotides and uses the other strand as a template to repair the specific lesion (77). During BER, both AP (apurinic/apyrimidinic site) sites and SSB occur and can cause more severe DNA damage, if they are stalled, because of a defective repair (60). These DNA lesions can be replication-dependent or -independent. In nonreplicating cells, unsolved SSBs occurring on partially overlapping DNA strands result in the formation of DSBs, which, if accumulated, are not compatible with cell life. Moreover, during DNA synthesis, AP sites and SSBs constitute an obstacle for the replication fork, which is blocked and can collapse, leading to DSBs. SSB is always integrated in the BER pathway or can occur in the absence of BER; for instance, when the hydroxyl radical attacks and directly breaks the sugar-phosphate DNA backbone, without creating a base damage.
The ATR/Chk1 signaling pathway
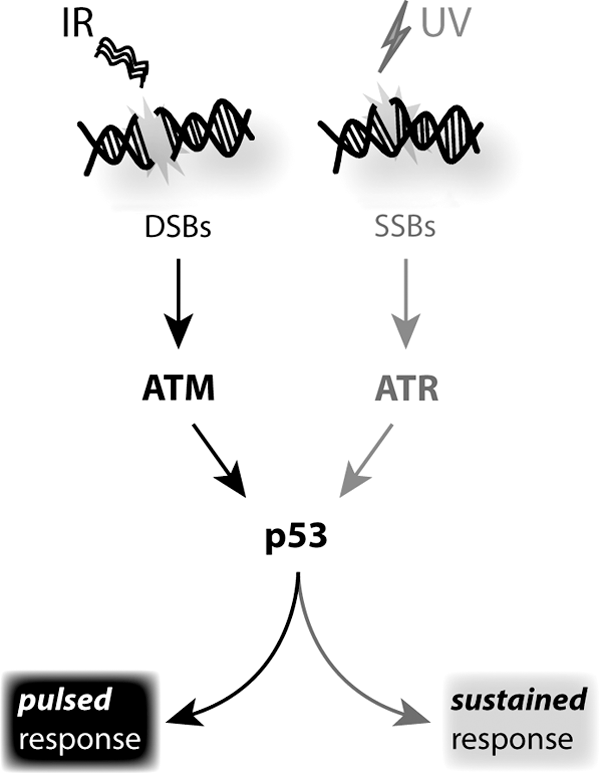
The well-established ATR/Chk1 signaling pathway links the repair system SSBR to apoptosis (54). In particular, the sensor of DNA replication stress is the ssDNA-binding protein replication protein A (RPA), which tightly binds the ssDNA and functions as a platform activating the ATR signaling pathway (24). The ATR/ATR interacting protein (ATRIP) complex is formed within RPA-coated ssDNA regions and is deputed to recruit several factors in order to induce the DNA replication responses, and, in the case of collapsed replication forks, to activate cell-cycle arrest or apoptosis (54). It is worth noting that, during DNA replication stress, the ATR/Chk1 signaling pathway plays a role in preventing apoptosis, because it is directed at the stabilization of DNA replication after stress (24). In particular, two ATR/Chk1 pathways can be activated on different DNA replication stressors: In the context of ionizing radiations, Chk1 blocks a caspase-2-mediated apoptotic response independently of p53 (64); while the ATR/Chk1-mediated suppression of the caspase-3-dependent pathway is activated by replication inhibitors (53). This increases the complexity of the DNA damage apoptotic response, which can involve several signaling pathways depending on the specific forms of DNA damage. Indeed, ATR and Chk1 can also phosphorylate p53, the central downstream checkpoint signaling protein that is responsible for apoptotic responses, as mentioned earlier. In the case of UV-light-mediated SSB generation, a divergent mode of activation from that discovered on gamma-radiation-induced DSBs has been demonstrated (Fig. 7) (5). Indeed, Batchelor et al. have postulated that p53 is activated by a single continuous pulse which is dependent on UV doses, rather than by ATM/p53 no-dose dependent repeated oscillations (5,6). They explain this opposite p53 behavior by the fact that ATM/p53 inhibitor Wip1 is switched off and, thus, cannot contribute toward regulating p53 activation in a pulsatile fashion. Moreover, ATR is not able to induce a rapid MDM2 degradation as ATM does. Taken together, these results involve the same central molecule p53, as it plays a key role in the transcriptional activation of cell cycle and apoptotic proteins. Simultaneously, they also represent an interesting example of how different inputs (UV or gamma radiations) converge to a single molecule (p53) that can display different behaviors depending on the surrounding proteome and, thus, producing different cellular outcomes (Fig. 7). In this context, recent findings from Lahav's group have shown that a switch from pulsed to sustained p53 determines the accumulation of different target genes and, thus, different cellular responses (57). For the first time, it has been demonstrated that p53 dynamics can by themselves control the cell's fate, through a different dynamic induction of cell cycle, apoptosis, senescence, and p53 regulators (57). Moreover, in their study, Purvis et al. manipulated p53 pulses with a sequence of precisely timed drug additions, suggesting that such perturbation of p53 dynamics could constitute a new pharmacological strategy dictating cell fate in a range of diseases (57). However, future studies are necessary to understand how these signaling dynamics are regulated and how they affect cellular outcomes in physiological conditions.
Redox Implications in DNA Damage
As frequently mentioned earlier, oxidative stress is among the primary inducers of DNA damage. Indeed, ROS, along with nitric oxide (NO)-derived radicals, the so-called reactive nitrogen species (RNS), can react with DNA and produce different DNA lesions (Fig. 8). Moreover, in addition to this direct action, ROS and RNS can oxidize proteins belonging to the DDR system, thereby causing DNA damage indirectly (Fig. 8). ROS and RNS cause structural alterations in DNA, ranging from point mutations to wide chromosomal alterations. These effects are included among those underlying the cell death response. However, it should be remembered that the same effects have been shown to be involved in the induction of neoplastic transformation, thus allowing expression of the mutated phenotype (18). Many environmental insults (e.g., ionizing radiations) have been shown to produce ROS and affect DNA integrity; however, nitroxidative DNA damage also appears to occur naturally, as low steady-state levels of base damage products have been detected in nuclear DNA (1). Indeed, mitochondria and several cellular enzymes can produce ROS and RNS: (i) as by-products of redox reactions that are required for the physiological functions of the cell, (e.g., ATP synthesis or xenobiotics detoxification); (ii) as useful products, whose toxicity is leveraged for whole organism survival (e.g., inflammatory response). Both these events underlie the increased DNA damage observed during ageing and in chronic inflammatory diseases.
Direct oxidative damage
The principal DNA damaging ROS is the hydroxyl radical (OH•), although other members, such as peroxyl and alkoxyl radicals (RO2 • and RO•, respectively), peroxynitrite (ONOO−), and ozone (O3), are effective as well (78). Conversely, hydrogen peroxide (H2O2) and superoxide radical (O2 •−) have not been documented as significantly affecting DNA stability. ROS generate a plethora of by-products: oxidized bases, the most common of which is 8-hydroxyguanine (8-OHG); abasic sites; DNA–DNA intrastrand adducts; DNA strand breaks; and DNA–protein cross-links (14). All these lesions can trigger the DDR involving the DNA damage repair systems previously discussed.
Redox-mediated inhibition of DDR proteins
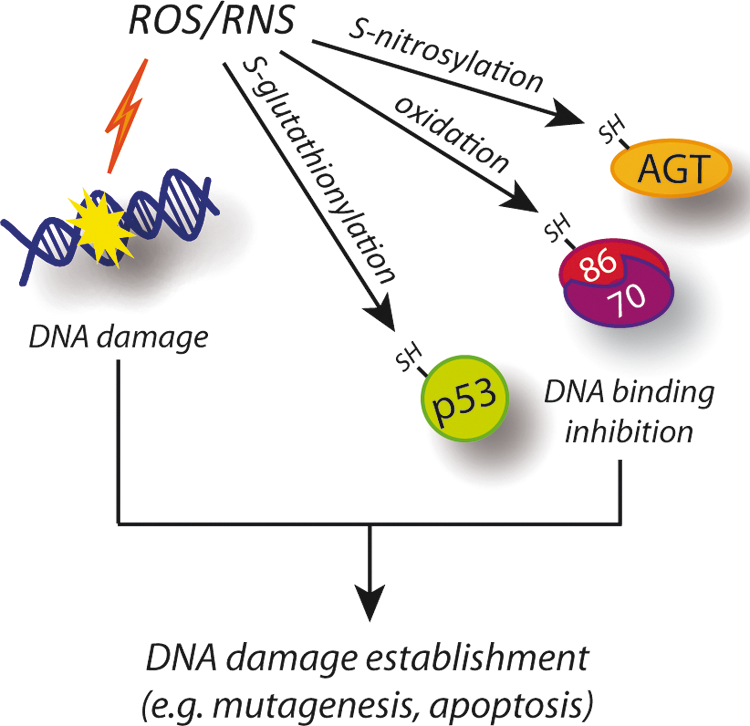
Nitroxidative stress has been carefully demonstrated as reversibly modifying proteins at the levels of reactive cysteines. Owing to their low pKa, these residues are also present as thiolate anions at physiological pH and can easily undergo a reaction with ROS and RNS, as well as with glutathione, thereby generating S-hydroxylated, S-nitrosylated, and S-glutathionylated derivatives, respectively (27). Generally, cysteine redox modifications result in a loss of function of protein activity; this effect has also been demonstrated for some of the proteins involved in DDR, thereby indirectly contributing to the establishment of DNA damage (Fig. 8). Here, we report the most representative examples of proteins required for repairing DNA lesions and whose activity has also been reported as being subjected to redox regulation.
p53
p53 has been reported to have about 10 redox-sensitive cysteine residues undergoing alkylation with N-ethylmaleimide (61). Among them, Cys182 has been suggested as undergoing S-hydroxylation, S-glutathionylation, and disulfide bond formation with Cys227, which are oxidative modifications that contribute to decreased p53 tetramerization, DNA affinity, and gene transcription (70). Moreover, Velu et al. demonstrated that Cys124 and, above all, Cys141, both located in the proximity of the DNA-binding domain of p53, are selectively S-glutathionylated, thus causing inhibition of the domain's DNA-binding activity and dimerization (Fig. 8) (72). S-glutathionylation of Cys141 of p53 could have deep consequences in oncogenesis, as reported in recent correlative studies which show that several cancer tissue and tumor histotypes known to produce ROS, to a great extent, exhibit high levels of S-glutathionylated p53 (82).
Ku
Ku has also been shown to undergo cysteine oxidation-dependent conformational change and reduced DNA binding activity (Fig. 8) (3). However, only a minor role for Cys483 of the Ku86 subunit has been hypothesized, leaving doubts as to the precise residue governing such a regulation (8).
O 6-alkylguanine-DNA alkyltransferase
Another protein involved in DDR, and whose levels have been very recently demonstrated to be regulated by nitroxidative conditions, is the O 6-alkylguanine-DNA alkyltransferase (AGT) (Fig. 8). AGT repairs highly mutagenic and cytotoxic O 6-alkylguanines (O 6-AG) by transferring the alkyl group from DNA to the enzyme active site cysteine, thus restoring guanine. O 6-AG is mispaired by DNA polymerases to thymine during DNA replication; the O 6-AG:T mispairs can subsequently result in G:C to A:T mutations and DNA DSBs, with both these having been reported as a potent trigger of cell death (39). Although it was known that mice deficient in AGT were more susceptible to tumorigenesis and to acute mortality from alkylating N-nitroso compounds (37), only recently, it has been found that AGT undergo S-nitrosylation on NO (over)production. Liu's laboratory elegantly demonstrated that, once S-nitrosylated, AGT is rapidly degraded via the proteasome system, the DNA being left unrepaired with the result that mutations are established (Fig. 8) (75, 76).
Conclusions
In this review, we have focused on the possibility of molecules involved in DNA damage repair systems being a part of the orchestra characterizing the apoptotic signaling, and how these proteins could function in possible feedback loops to finely regulate either DNA repair or subsequent cell death. Indeed, we have considered DNA repair proteins that are directly associated to “typical” apoptotic factors and, therefore, reasonably implicated in modulating their activity/function on DNA damage. Two well-characterized examples are DDB2 and Ku: Both play a dual role, by acting as DNA repair proteins and inducing the apoptotic signaling according to the severity of DNA damage. However, recent evidence suggests that specific post-translational modifications can completely change the behavior of some proteins involved in DDR, directing them toward a specific outcome: p53 is an emblematic example in this regard. One study after another have attempted to delineate the pathway(s) linking this type of cellular stress (DNA damage), made of several types of lesions and repair systems, to the signaling network dynamically orchestrating the apoptotic cell response. In recent years, the autophagic response has become a part of this scenario: A transient sub-lethal autophagic function could guarantee survival while the repair system acts on DNA. This may help the cell resist organelle-derived stress and is probably regulated by a fine cross-talk with the repair machinery. It is not by chance that p53, the Cullins, and AMPK are involved, as we discussed, in both pathways.
In the context of cancer studies, the understanding and defining of all these processes is of utmost importance: Modulating a cell's capability to better cope with DNA damage has undoubted potential for both cancer prevention and treatment.
Footnotes
Acknowledgments
The authors are grateful to Dr. G. Filomeni for scientific discussion and suggestions. F. Cecconi is supported by Grants from the Telethon Foundation (S99038TELU/TELC), AIRC (2011), FISM (2009), the Italian Ministry of University and Research (PRIN 2009 and FIRB 2011 funding programs), and the Italian Ministry of Health (Ricerca Finalizzata e Ricerca Corrente).