
Abstract
The global protein thiol pool has been reported to play a major role in the defense against oxidative stress as a redox buffer similar to glutathione. The present study uses a novel method to visualize cellular changes of the global protein thiol pool in response to induced oxidative stress. Unexpectedly, the results showed an uneven distribution of protein thiols in resting cells with no apparent change in their level or distribution in response to diamide as has been reported previously. Further analysis revealed that thiol pool oxidation is artificially high due to insufficient activity of the widely used sample quencher trichloroacetic acid (TCA). This suggests that previously published articles based on TCA as a quencher should be interpreted with caution as TCA could have caused similar artifacts. Overall, the results presented here question the major role for the global thiol pool in the defense against oxidative stress. Instead our hypothesis is that the fraction of proteins involved in response to oxidative stress is much smaller than previously anticipated in support of a fine-tuned cell signaling by redox regulation. Antioxid. Redox Signal. 18, 795–799.
Introduction
Innovation
Trichloroacetic acid (TCA) is widely used as a quencher of protein thiol oxidation during sample preparation in many different kinds of studies. Our findings show that TCA is not quenching the oxidation of diamide adequately, and therefore the use of TCA as a quencher needs to be reconsidered and previous data based on TCA as a quencher interpreted with caution. Our data also contradict a major role for the global thiol pool in the defense against oxidative stress and instead suggest that the fraction of proteins involved during oxidative stress is more specific supporting the concept of signaling by a redox mechanism.
The Distribution of Protein Thiols Shows No Apparent Chances in the Response to Oxidative Stress
It was recently reported that diamide, one of the most commonly used agents to induce strong and reproducible levels of oxidative stress in experimental setups, causes major global protein thiols oxidation (5). We therefore decided to further explore this finding by analyzing how this would affect individual cellular compartments. To visualize the level and distribution of the global protein thiols a new protocol was developed. In principal, cells were fixed and protein thiols were labeled by alkylation using fluorescence conjugated maleimide, for subsequent analysis of the distribution and level of protein thiols in cells by fluorescence microscopy. As shown in Figure 1A, untreated cells fixed with methanol and labeled with Alexa Fluor 488 C5 maleimide (NEM-Alexa) generated a strong signal. The distribution of protein thiols in the cells was unevenly spread with the strongest signal in the perinuclear area. Exposing the cells to N-ethylmaleimide (NEM) before labeling with NEM-Alexa blocked the signal, indicating high specificity for NEM-Alexa (Fig. 1B). The analysis of possible changes in distribution and level of protein thiols in cells exposed to the strong oxidant diamide showed no apparent decrease of protein thiols in response to 15-min incubation with 0.1 mM diamide at 37°C, even though the intensity of fluorescence varied (Fig. 1C). Increasing the amount of diamide to 0.5 mM resulted in a significant change in cell morphology, making comparative analysis impossible (Fig. 1D).

The finding of no apparent chances in the distribution and levels of protein thiols was unexpected since numerous studies have shown that a large fraction of the global thiol pool is oxidized in response to oxidative stress (4, 5). The most recent study indicated that even more than 50% of the protein thiol pool is oxidized in response to diamide-mediated stress (5). Since our findings were based on a semi-quantitative fluorescence microscopy assay, too low sensitivity for detection of minor redox changes could explain the discrepancy in the results. An alternative explanation would be that the previously reported global thiol pool oxidation was artificially high due to insufficient activity of the sample quencher.
Quantitative Analysis of the Global Protein Thiol Status in Cells Demonstrates the Inability of TCA to Quench Diamide Induced Oxidation
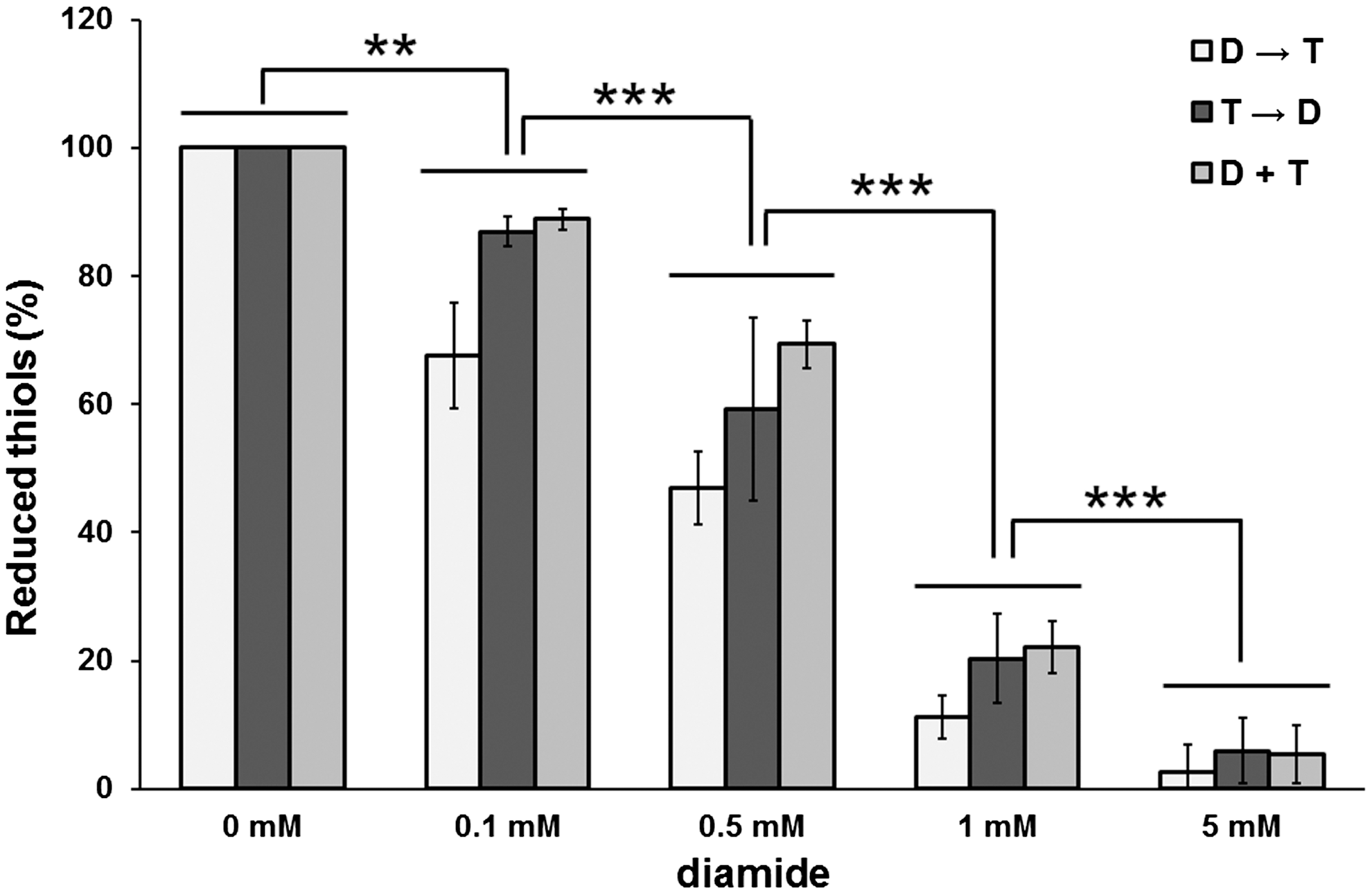
To exclude the possibility that the oxidation in response to diamide was due to insufficient quenching, the activity of the most commonly used sample quencher, TCA, to quench protein thiol oxidation in samples exposed to diamide was tested. A quantitative analysis with the possibility to detect minor level changes was selected, in which cells were exposed to a relevant range of diamide (0.1–5 mM) before or after treatment with TCA as well as in cells exposed to diamide and TCA simultaneously (Fig. 2). Diamide caused a significant (p<0.01) dose-dependent decrease of protein thiols already starting at concentrations as low as 0.1 mM diamide, an effect that was intensified in response to 1 and 5 mM diamide. Surprisingly, treatments with TCA before or simultaneously with diamide only slightly affected the extent of diamide oxidation compared to adding TCA after diamide exposure at any diamide concentration tested (p>0.05). As this inability of TCA as a quencher was already observed at low diamide concentration (0.1 mM), it indicated that TCA may not be active up to a threshold value of diamide. Our data were again unexpected, since quenching by acidification is one of the most widely used strategies and the protocol of using TCA to quench diamide-treated cells is well-established (1 –3, 5, 7, 9). In principal, TCA is supposed to mediate its inhibitory activity by protonation of thiols. However, our data show that TCA is an inadequate quencher of diamide-induced oxidative stress. This raises the question whether previously published data on large redox shifts could be artifacts, and we suggest that these should therefore be interpreted with caution.
TCA Is Unable to Quench β-Actin Oxidation by Diamide, But Can Quench β-Actin Reduction by Dithiothreitol
Having observed that TCA may have low activity to quench protein thiol oxidation during sample preparation, it was decided to confirm this finding by an alternative assay, in which the activity of TCA to quench diamide-induced redox changes of β-actin was tested. The redox status of β-actin was measured by labeling thiols through biotinylation using a derivative of the alkylating agent NEM. Nonreduced and untreated β-actin showed low activity, which can be considered the minimal value for the absorption of β-actin during the experiment (0%), while β-actin reduced by dithiothreitol (DTT) showed a strong signal (positive control; 100%) (Fig. 3). Nonreduced β-actin exposed to diamide, on the other hand, showed similar absorption as untreated β-actin (unpublished data). Reduced β-actin exposed to diamide diluted in PBSET, pH 7.4, showed a significant decrease (p<0.001) in the level of reduction. However, no significant inhibition of β-actin oxidation was observed when β-actin was exposed to diamide diluted in the redox quencher TCA (p=0.9 for 2 mM diamide and p=0.8 for 10 mM diamide).

To investigate the possibility that TCA itself could cause removal of β-actin from the plate, the levels of reduction of β-actin in response to DTT versus PBSET, both with and without subsequent exposure to diamide, were determined (Fig. 4). The levels of reduction of β-actin without DTT exposure were similar in the presence and absence of TCA, while the levels of reduction markedly increased with the addition of DTT (p<0.001) to the β-actin with an even higher level of reduction with the addition of TCA (p<0.001). Furthermore, as TCA has failed to quench the oxidation of β-actin in the plate assay, it was investigated whether TCA can quench the reduction of β-actin. A plate assay was performed similar to the one for β-actin oxidation with DTT as reducing agent. The results show that DTT reduces β-actin, but that a simultaneous exposure to TCA quenches the reaction (p<0.001) (Fig. 5).


Our observations suggest that protonation by TCA is inadequate to block protein oxidation induced by diamide; however, we have shown that TCA is active as a quencher for reduction of β-actin by DTT. The differences between the reduction by DTT and the oxidation by diamide may lie within the reducing and oxidizing agents themselves. DTT achieves the reduction of proteins through a thiol exchange reaction, while diamide dehydrogenates thiols via a two-step nucleophilic mechanism.
Conclusions and Open Questions
In summary, our findings show that TCA does not quench redox reactions as sufficiently as previously anticipated with the implication that previously published data on large redox shifts may be artifacts and therefore need to be interpreted with caution. Another important implication of this study is that the global protein thiol pool may not serve as a major redox buffer like glutathione (GSH) as previously suggested. Redox control of cellular processes has been shown to be fundamental and the number of proteins under redox control is steadily increasing. However, the simultaneous occurrence of specific cell signaling by redox mechanisms and the occurrence of a large oxidized global protein thiol pool during oxidative stress are not fully understood. The data presented here suggest that the fraction of proteins involved during oxidative stress is more specific, which supports the concept of signaling by a redox mechanism and contradicts a major role for the global thiol pool in the defense against oxidative stress. Overall, our data suggest that analysis of oxidative stress conditions is more complicated than previously anticipated and that numerous previously published observations need to be re-evaluated.
Notes
Materials and Methods
The distribution of protein thiols by fluorescence microscopy
H157 cells (ATCC-LGC) were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco, Invitrogen) supplemented with 10% fetal bovine serum (FBS; Gibco, Invitrogen) and 1% penicillin–streptomycin (Pen Strep; Gibco, Invitrogen) at 37°C and 5% CO2, and then plated on eight-well chamber slides. After 24 h, the cells were washed once with complete medium and then either kept untreated or exposed to 0.1 mM or 0.5 mM diamide (Sigma-Aldrich) in complete medium. After 15-min incubation at 37°C, the cells were washed once with phosphate-buffered saline (PBS), pH 7.4, and fixed in 100% methanol for 5 min at room temperature. The cells were then washed four times with PBS+0.05% Tween 20 (PBST) and incubated with Alexa Fluor 488 C5 maleimide (NEM-Alexa; Invitrogen) for 15 min at 37°C to label protein thiols. To test the specificity of NEM-Alexa, some cells were exposed to 50 mM NEM (Sigma-Aldrich) for 15 min at 37°C after fixation, but before exposure to with NEM-Alexa. After four washes in PBST, the cells were analyzed by fluorescence microscopy.
Quantification of protein thiols in cells exposed to diamide by DTNB assay
HeLa cells (ATCC-LGC) were grown in DMEM supplemented with 10% FBS and 1% Pen Strep at 37°C and in 5% CO2. Cells at logarithmic growth were trypsinized and washed with as well as re-suspended in PBS, pH 7.4. Cells were separated into three sample groups. The first group was exposed to diamide (0.1–5 mM) diluted PBS, pH 7.4, for 5 min at 37°C, subsequently treated with 100% ice-cold TCA (Sigma-Aldrich) and incubated for 30 min on ice. The second group was directly treated with 100% ice-cold TCA and subsequently exposed to diamide (0.1–5 mM) and incubated 5 min at 37°C followed by 30 min on ice. The third group was incubated with a premixture of 100% TCA and diamide (0.1–5 mM) and incubated 5 min at 37°C followed by 30 min on ice. The precipitated proteins were thereafter pelleted by centrifugation at 10,000 g for 10 min at 4°C and analyzed for their thiol content using a standard 5,5-dithiobis(2-nitrobenzoic acid) (DTNB) assay with some modifications. Briefly, the TCA-precipitated protein pellets were re-suspended in 1 ml DTNB assay solution (9 ml of 8 M guanidine hydrochloride/670 mM Tris, pH 8, and 1 ml of 10 mM DTNB [Sigma-Aldrich] in 100% ethanol) and analyzed for their absorbance at 412 nm using a Thermo Spectronic Genesys 10 UV spectrophotometer. The experiment was repeated three times.
β-actin oxidation assay
Human platelet β-actin was obtained from Cytoskeleton, Inc. Ninety-six-well Maxi-Sorp ELISA plates (Nunc, Inc, VVR) were coated with 1 μg/ml β-actin in carbonate buffer pH 9.6 for 30 min at room temperature. The assay was performed by exposing the wells to 5 mM DTT in PBS, pH 7.4, with 1 mM EDTA and 0.05% Tween-20 (PBSET) or to PBSET alone (control) for 15 min at 37°C. The wells were then washed four times with PBST. Diamide was pretreated with GSH (Sigma-Aldrich) for 15 min at room temperature. Subsequently, the wells were exposed to the indicated concentrations of diamide/GSH diluted in PBST or 100% TCA, respectively, and incubated 15 min at room temperature. After four washes in PBST, 2 μM biotin-maleimide (Sigma-Aldrich) diluted in PBST was added and incubated at room temperature. After 15 min, the wells were washed four times and exposed to alkaline phosphatase conjugated streptavidine (Mabtech) diluted 1:1000 in PBST with 1% (w/v) bovine serum albumin (Sigma-Aldrich) and incubated for 30 min at room temperature. The well was then washed four times in PBST and 1 mg/ml p-nitrophenyl-phosphate (Sigma-Aldrich) dissolved in 10% diethanolamine pH 9.8 with 0.5 mM MgCl2 was added. Absorbance at 405 nm was determined using a microtiter plate reader (Wallac, Perkin-Ellmer). The experiment was repeated twice in duplicate/triplicate.
To establish whether the TCA washes off β-actin from the plates, a plate was coated with β-actin as described above. Wells were exposed to either 5 mM DTT in PBSET or PBSET alone, incubated for 15 min at 37°C and washed three times with PBSET. Ice-cold TCA or PBSET was added to the appropriate wells and incubated on ice for 5 min. All wells were treated with 2 μM biotin-maleimide. After 15-min incubation at room temperature, the plates were washed and the biotinylated proteins detected as described above. The experiment was repeated three times in duplicate.
β-actin reduction assay
The β-actin reduction assay was performed with β-actin coated on the plate as described above. One or 5 mM DTT (Sigma-Aldrich) diluted in PBST or 100% TCA, respectively, was added to the wells and incubated for 15 min at room temperature. After four washes in PBST, 2 μM biotin-maleimide in PBST was added and incubated for 15 min at room temperature. After 15-min incubation at room temperature, the plates were washed and the biotinylated proteins detected as described above. The experiment was repeated twice in triplicate.
Statistics
All experimental data are reported as mean value, and error bars indicate standard error of the mean. Student's t-test was used to analyze differences between the mean values, and a p<0.05 was considered statistically significant.
Footnotes
Acknowledgments
This work was supported by grants from the Medical Faculty of Karolinska Institutet and the Swedish Research Council (project 66X-12162).