
Abstract
Introduction
M
Previous reports have shown that chronic angiotensin-II (AT-II) treatment increases mitochondrial reactive oxygen species (mtROS) formation and triggers immune cell infiltration, all of which contributes to AT-II-induced endothelial dysfunction and subsequent hypertension. We here link both concepts by identifying mtROS-driven NADPH oxidase activation in phagocytic cells, aggravation of AT-II-mediated cardiovascular complications (e.g., eNOS uncoupling/S-glutathionylation and endothelial dysfunction) by manganese superoxide dismutase deficiency, and improvement by inhibition of the mitochondrial permeability transition pore (mPTP) in cyclophilin-D-deficient mice or pharmacologically by sanglifehrin A therapy. Our results indicate that mPTP inhibition might be beneficial in patients with high blood pressure.
With the present study, we sought to further determine the underlying mechanism for this crosstalk with special emphasis on the activation of NADPH oxidase in isolated leukocytes as well as cardiovascular tissue by mitochondrial superoxide, hydrogen peroxide, and, subsequently, formed peroxynitrite. A detailed explanation of the rationale for the use of the investigated cellular and animal models is provided in the Extended Introduction (see Supplementary Data; Supplementary Data are available online at
Results
Crosstalk in isolated human white blood cells
Extracellular superoxide release from activated isolated human neutrophils was assessed by lucigenin enhanced chemiluminescence (ECL) and high-performance liquid chromatography (HPLC)-based quantification of 2-hydroxyethidium in the supernatant (Fig. 1A–D). Lucigenin has an octanol/buffer distribution coefficient of 0.11±0.01, which points toward extracellular accumulation of lucigenin (13). Phorbol ester and myxothiazol caused a similar pattern of polymorphonuclear leukocyte (PMN) activation that was completely blocked by PEG-SOD (but also conventional SOD [not shown]), the Nox inhibitor 1,3-Benzoxazol-2-yl-3-benzyl-3H-[1,2,3]triazolo[4,5-d]pyrimidin-7-yl sulfide (VAS2870), and the intracellular calcium chelator 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM). Only a partial decrease in superoxide formation was observed with PKC inhibition (being more pronounced in the phorbol ester dibutyrate [PDBu]-stimulated samples) and in response to the cSrc inhibitor 4-Amino-3-(4-chlorophenyl)-1-(t-butyl)-1H-pyrazolo[3,4-d]pyrimidine (PP2) (being more pronounced in the myxothiazol-stimulated samples) pointing to contribution of distinct pathways to the NADPH oxidase activation process. Likewise, inhibitors of complex III of the mitochondrial respiratory chain, antimycin A and myxothiazol, induced a ROS production in isolated human neutrophils that was blocked by the PKC inhibitor chelerythrine, the NADPH oxidase inhibitor apocynin, and the blocker of the mPTP CsA (Supplementary Fig. S2A). This is compatible with an activation of the phagocytic NADPH oxidase by mtROS. Hydrogen peroxide functioned as a chemical mimic of this process and displayed a concentration-dependent induction of the superoxide signal (it should be noted that lucigenin ECL is highly specific for superoxide and will not generate a signal with hydrogen peroxide), which was suppressed by chelerythrine, apocynin, and diphenylene iodonium (DPI), clearly indicating that the phagocytic oxidase is the source of the detected superoxide signal (Fig. 1E and Supplementary Fig. S2B, C).

8-amino-5-chloro-7-phenylpyrido[3,4-d]pyridazine-1,4-(2H,3H)dione sodium salt (L-012) ECL generally detects extracellular, cytoplasmic, and mitochondrial superoxide, hydrogen peroxide, and peroxynitrite (although when used in isolated leukocytes, the mainly detected species is extra- and intracellular superoxide); whereas lucigenin cation rather reacts with extracellular superoxide (13). To ensure the specific measurement of extracellular hydrogen peroxide from dismutated superoxide, we also used a peroxidase/amplex red fluorescence detection system conferring specific oxidation of amplex red to resorufin by extracellular hydrogen peroxide via a peroxidase-catalyzed reaction. Myxothiazol triggered a superoxide/hydrogen peroxide signal that was normalized by PKC, NADPH oxidase, and mPTP inhibition (Fig. 1F). Addition of SOD in this assay increased the resorufin yield due to inhibition of reduction of peroxidase compound I by superoxide, which leads to oxidation of the amplex red. Activation of the leukocyte NADPH oxidase by the chemotactic peptide formyl-methionyl-leucyl-phenylalanine (fMLP) was not inhibited by the PKC inhibitor chelerythrine. HPLC-based quantification of extracellular 2-hydroethidium, the superoxide-specific oxidation product of dihydroethidine (DHE), revealed that the antimycin A-induced signal was normalized by inhibitors of PKC (chelerythrine), flavin-dependent oxidoreductase (DPI), and mPTP (CsA and sanglifehrin A [SfA]) inhibitors. Mitochondria-targeted and lipophilic antioxidants (triphenylphosphonium hydroxybenzene [HTPP], triphenylphosphonium aminobenzene [ATPP], manganese(III)-tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride [MnTMPyP], and 5,10,15,20-tetraphenyl-21H,23H-porphine manganese(III) chloride [MnTPP]) normalized the signal; whereas the hydrophilic antioxidant vitamin C caused almost complete loss of the extracellular signal (Supplementary Fig. S2E). Potential redox cycling by lucigenin at higher concentrations was assessed by measurement of superoxide-specific DHE oxidation to 2-hydroxyethidium by HPLC analysis in the presence of lucigenin (5, 50, and 250 μM). Lucigenin decreased the superoxide signal in a concentration-dependent fashion, arguing against the concerned redox cycling by lucigenin in isolated leukocytes (Supplementary Fig. S2F). The main findings obtained with isolated human leukocytes were also reproduced using electron paramagnetic resonance (EPR)-based spin trapping of superoxide with the spin trap 5-(Diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide (DEPMPO, see Supplementary Fig. S3). The usefulness of the different assays for detection of superoxide/hydrogen peroxide formation in isolated white blood cells (WBCs) is further characterized by concentration-response studies for apocynin in antimycin A or myxothiazol-stimulated granulocytes and monocytes/lymphocytes as well as by phorbol ester-stimulated positive controls (see Supplementary Fig. S4).
Determination of mtROS (superoxide/hydrogen peroxide) triggered NADPH oxidase activation by nonphotometric methods (translocation of cytosolic subunits to the membrane)
In order to provide non-ROS based evidence for the mtROS-dependent activation of Nox-derived superoxide/hydrogen peroxide production, we determined the translocation of cytosolic subunits of the phagocytic NADPH oxidase to the membrane—a read out for activation of this enzymatic system. We observed translocation of p67phox, p47phox, and Rac-1 to the membrane in response not only to phorbol ester but also to myxothiazol, all of which was blocked by PKC and mPTP inhibition as well as by a mitochondria-targeted antioxidant (Fig. 2A–C).
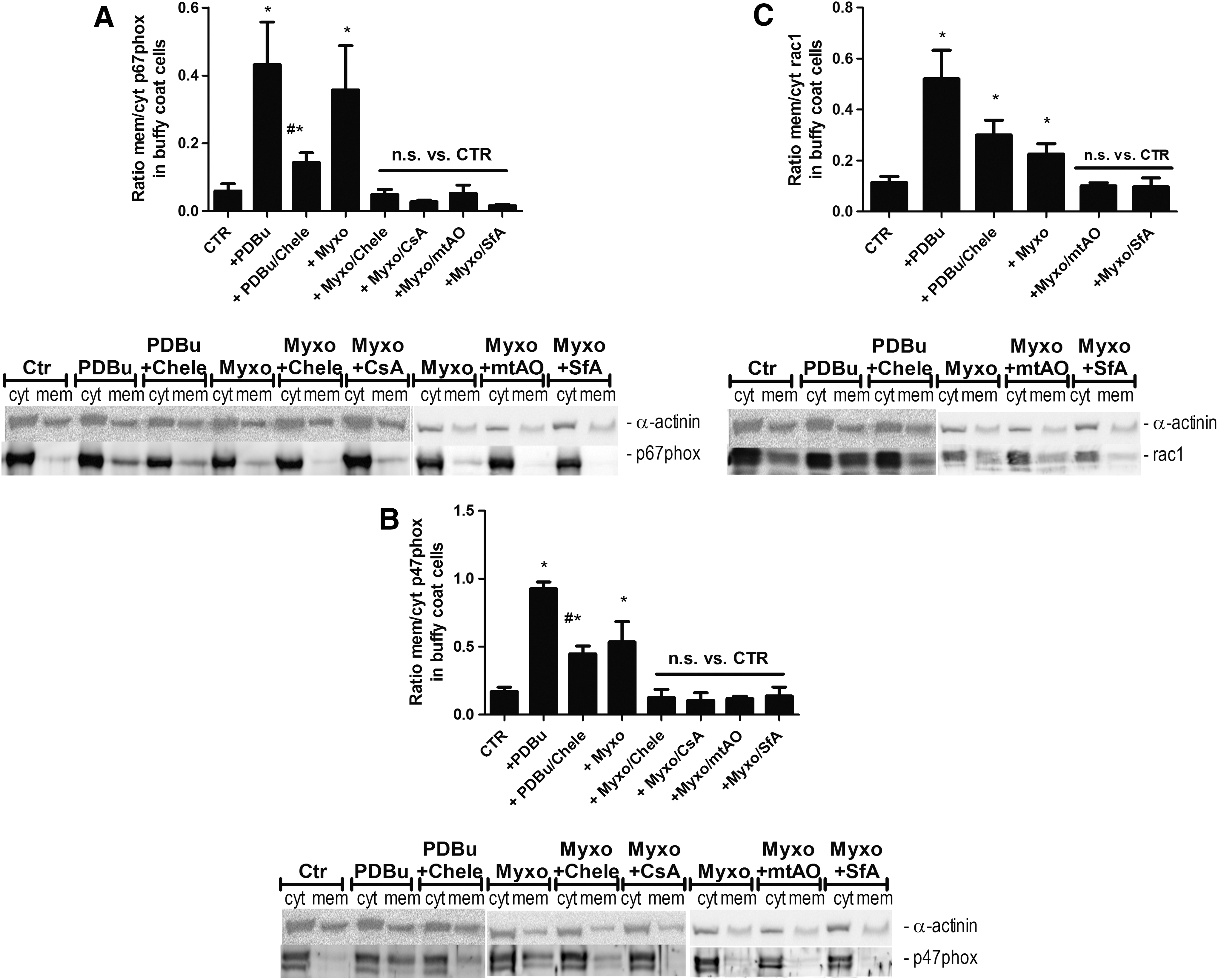
Crosstalk in isolated murine WBCs and whole blood
In addition to the pharmacological inhibitors, we used genetically modified mice to characterize the mechanism of the mtROS-Nox crosstalk. In whole blood and WBC from wild-type mice, myxothiazol induced an appreciable superoxide/hydrogen peroxide signal that was blocked by chelerythrine and apocynin (Fig. 3A). In WBC from p47phox knockout mice, the increase in the superoxide/hydrogen peroxide signal by myxothiazol was less pronounced, indicating that genetically inhibited activation of phagocytic NADPH oxidase cannot translate anymore the mtROS signal to Nox-derived oxidative burst (Fig. 3A). A similar signal pattern was observed in whole blood from wild-type versus p47phox knockout mice (not shown) after stimulation with antimycin A. Likewise, whole blood from cyclophilin D knockout mice displayed impaired mtROS-dependent activation of the phagocytic NADPH oxidase (Fig. 3B). This observation is likely due to the impaired release of mtROS (superoxide/hydrogen peroxide) from mitochondria due to decreased opening probability of the mPTP. The less pronounced response of leukocyte NADPH oxidase to myxothiazol-driven mtROS formation in WBC from p47phox knockout mice was also demonstrated by impaired oxidative burst as measured by the more specific detection of extracellular hydrogen peroxide release from superoxide dismutation by luminol/peroxidase ECL and HPLC-based resorufin quantification (Fig. 3C, D).

Studies with old MnSOD+/− mice
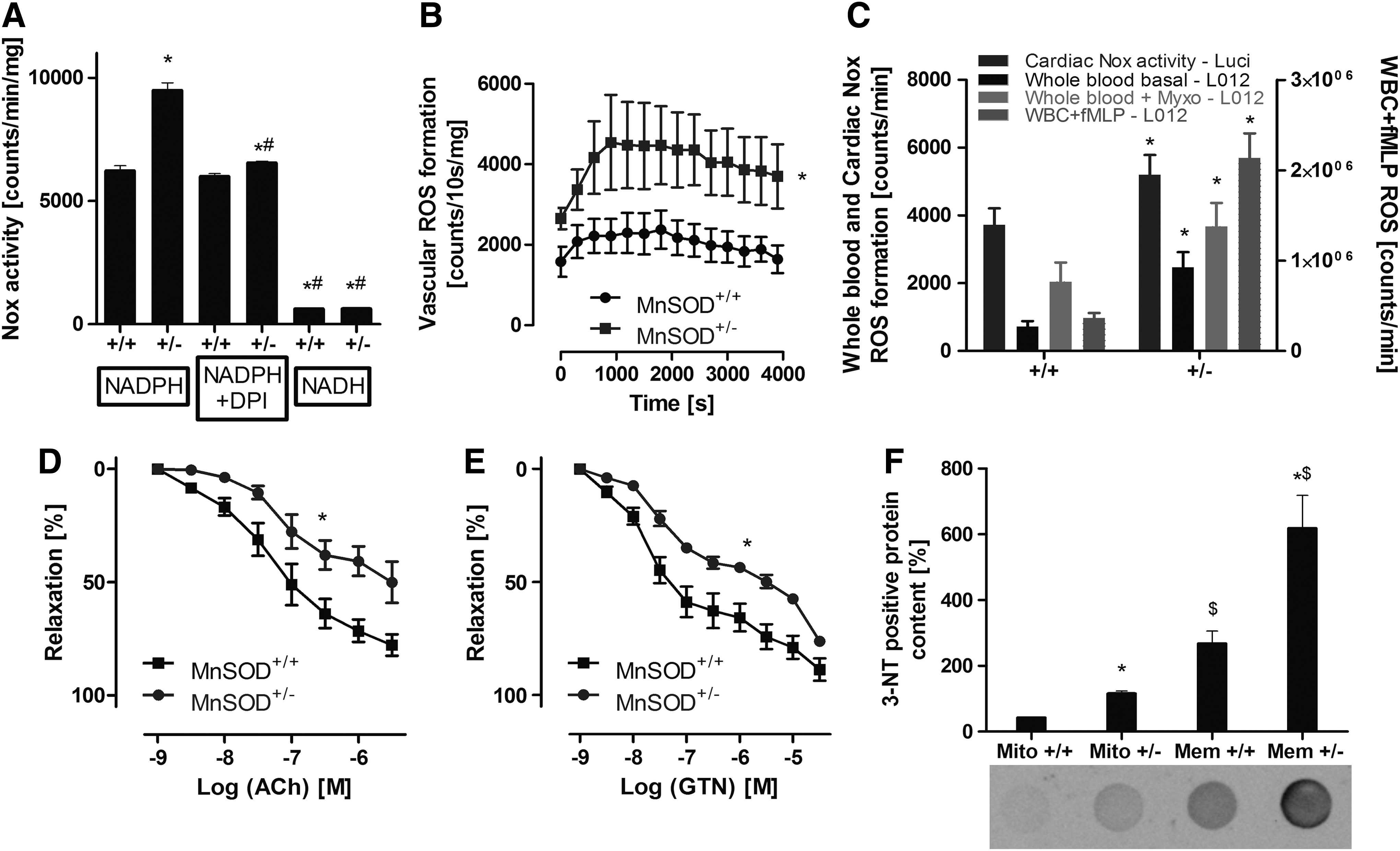
Old (age: 12 months) MnSOD+/− mice were used, as young (age: 3 months) mice with partial MnSOD deficiency display no vascular phenotype under basal conditions (see also Fig. 6C) (62). The NADPH oxidase activity in cardiac membranous fractions from old mice with partial MnSOD deficiency (MnSOD+/−) was significantly higher compared with their control littermates (MnSOD+/+) (Fig. 4A). In addition, the aortic NADPH oxidase activity, detected by Diogenes™ ECL (specific for extracellular hydrogen peroxide) in phorbol ester-stimulated aortic ring segments, was significantly increased in these old MnSOD+/− mice (Fig. 4B). Likewise, the whole blood superoxide and hydrogen peroxide (or peroxynitrite) production (basal and myxothiazol activated) as well as the fMLP triggered oxidative burst in isolated leukocytes and Nox activity in cardiac membranous fractions was increased in old MnSOD+/− mice (Fig. 4C). The increased burden of oxidative stress in old MnSOD+/− mice was also apparent by an increase in mitochondrial and membranous 3-nitrotyrosine (3NT) positive proteins (Fig. 4F). Most impressively, endothelial dysfunction (acetylcholine [ACh]-dependent relaxation) of the aorta of the old MnSOD-deficient mice was quite substantial, suggesting efficient adverse signaling of mitochondrial superoxide, hydrogen peroxide, and/or peroxynitrite to the cytosol with subsequent eNOS dysfunction or even uncoupling (Fig. 4D). Likewise, impairment of endothelium-independent relaxation (nitroglycerin [GTN] response) was observed (Fig. 4E), in accordance with our own published data on aging-induced endothelial dysfunction and the role of mitochondrial superoxide, hydrogen peroxide, and/or peroxynitrite (62). An attractive explanation for the decreased GTN potency in old MnSOD+/− mice is based on our previous findings that GTN is bioactivated by the mitochondrial aldehyde dehydrogenase (ALDH-2), a redox sensitive enzyme that is inhibited in MnSOD+/− mice (15). The cardiovascular side effects of AT-II and also tolerance development in response to prolonged GTN treatment in aged MnSOD+/− mice were almost completely prevented by in vivo administration of the mPTP blocker SfA (see extended results and Supplementary Figs. S5 and S6 in Supplementary Data).
Effects of MnSOD deficiency on AT-II triggered mtROS release and subsequent activation of NADPH oxidase
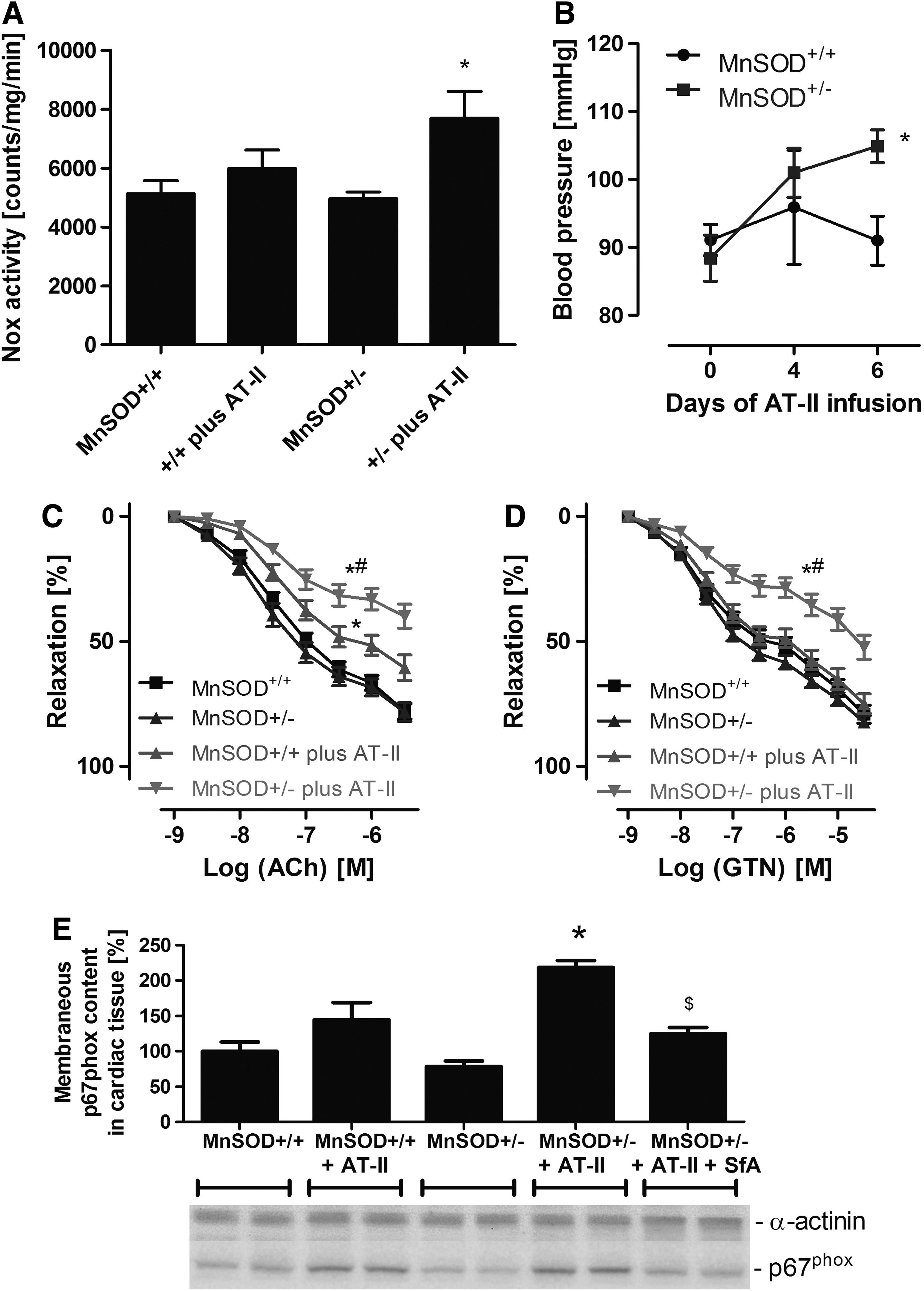
According to previous results of Dikalov and coworkers, AT-II via Nox-derived superoxide or down-stream hydrogen peroxide and peroxynitrite stimulates mitochondrial dysfunction with subsequent mtROS release contributing to endothelial dysfunction and hypertension, which was prevented by mitochondria-targeted antioxidants (23, 24). Here, we demonstrate for the first time that young mice with partial MnSOD deficiency treated with low/subpressor dose of AT-II had an increased cardiac membranous NADPH oxidase activity, more pronounced endothelial dysfunction, and higher arterial blood pressure, which was clearly less pronounced in wild-type littermates (Fig. 5A–C). The GTN-vasodilator response was more attenuated in the AT-II-treated MnSOD+/− mice (Supplementary Fig. S5D). Importantly, we observed a significantly higher degree of translocation of p67phox to the membrane in cardiac tissue (Fig. 5E) that was compatible with higher NADPH oxidase activity.
Effects of cyclophilin D deficiency on AT-II-triggered mtROS release and subsequent activation of NADPH oxidase
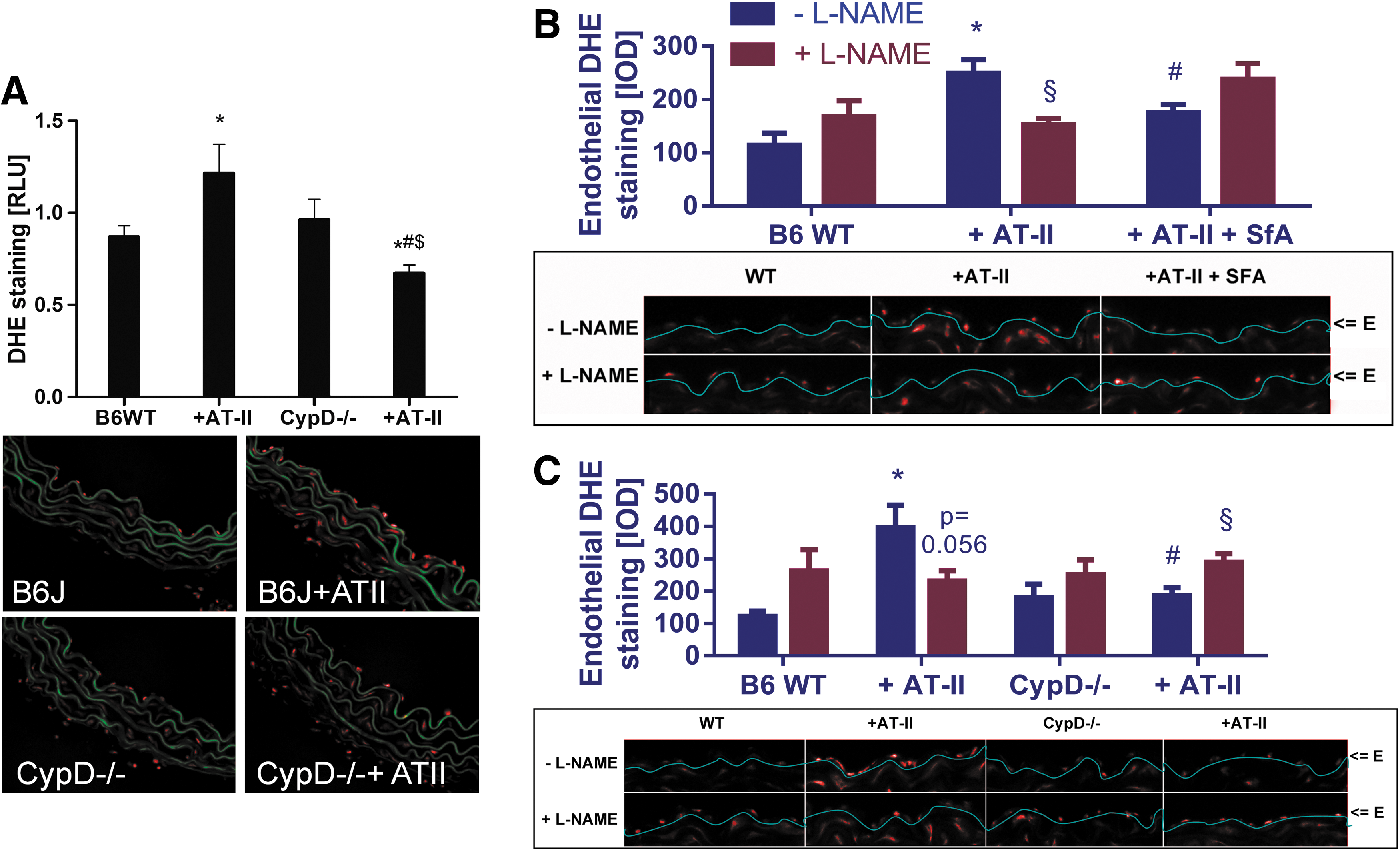
We studied the role of the mPTP for mtROS (superoxide, hydrogen peroxide but also subsequently formed peroxynitrite)-triggered NADPH oxidase activation in response to AT-II treatment at a molecular level by using cyclophilin-D-deficient mice (CypD−/−). CypD−/− mice, displayed a pronounced vascular phenotype characterized by a significant degree of vascular dysfunction (impaired ACh- and GTN-response, not shown). The further deterioration of endothelial dysfunction by additional AT-II treatment was less pronounced in CypD−/− mice (ΔpD2 [Ach]=31.5±7.0 for wild type±AT-II and ΔpD2 [Ach]=11.3±6.8 for CypD−/−±AT-II; p<0.05). As an important proof of concept, the AT-II-induced increase in blood pressure in wild-type mice was significantly retarded in AT-II-treated CypD−/− mice and at day 4 of treatment was significantly improved by mPTP inhibition (Fig. 6A). In vivo treatment with AT-II resulted in a significant increase in whole blood and WBC NADPH oxidase activity, which was almost completely absent in whole blood and WBC from cyclophilin D knockout mice and was blocked by apocynin (Fig. 6B). In cardiac tissue, the AT-II treatment increased the membranous NADPH oxidase activity, which was inhibited in AT-II-treated CypD−/− mice, by SfA in vivo infusion and by in vitro incubation with the Nox2 inhibitor VAS2870 (Fig. 6C). Vice versa, AT-II treatment increased the aortic hydrogen peroxide formation, which was virtually absent in AT-II-treated CypD−/− mice, was prevented by SfA in vivo infusion and by in vitro incubation with the Nox2 inhibitor VAS2870 (Fig. 6D). The activation of the p47phox-dependent NADPH oxidase by AT-II treatment was demonstrated by phosphorylation at serine 328, which was prevented in aorta from CypD−/− mice (Fig. 6E).

Finally, AT-II-mediated increase in vascular superoxide, hydrogen peroxide, and/or peroxynitrite formation was abolished by cyclophilin D deficiency (Fig. 7A) and also AT-II-induced, L-NG-nitroarginine methyl ester (L-NAME) inhibitable endothelial DHE staining in wild-type mice (indicative for eNOS uncoupling) was completely normalized in CypD−/− mice and improved by pharmacological in vivo mPTP inhibition by SfA (Fig. 7B, C). The enantiomer D-NAME was applied as a proof of the specificity of the assay for superoxide formation by uncoupled eNOS. D-NAME shares similar direct antioxidant properties with L-NAME but does not bind to eNOS. L-NAME but not D-NAME increased the signal in aorta from control animals and vice versa, L-NAME but not D-NAME decreased the signal in aorta from AT-II-infused animals (Supplementary Fig. S7).
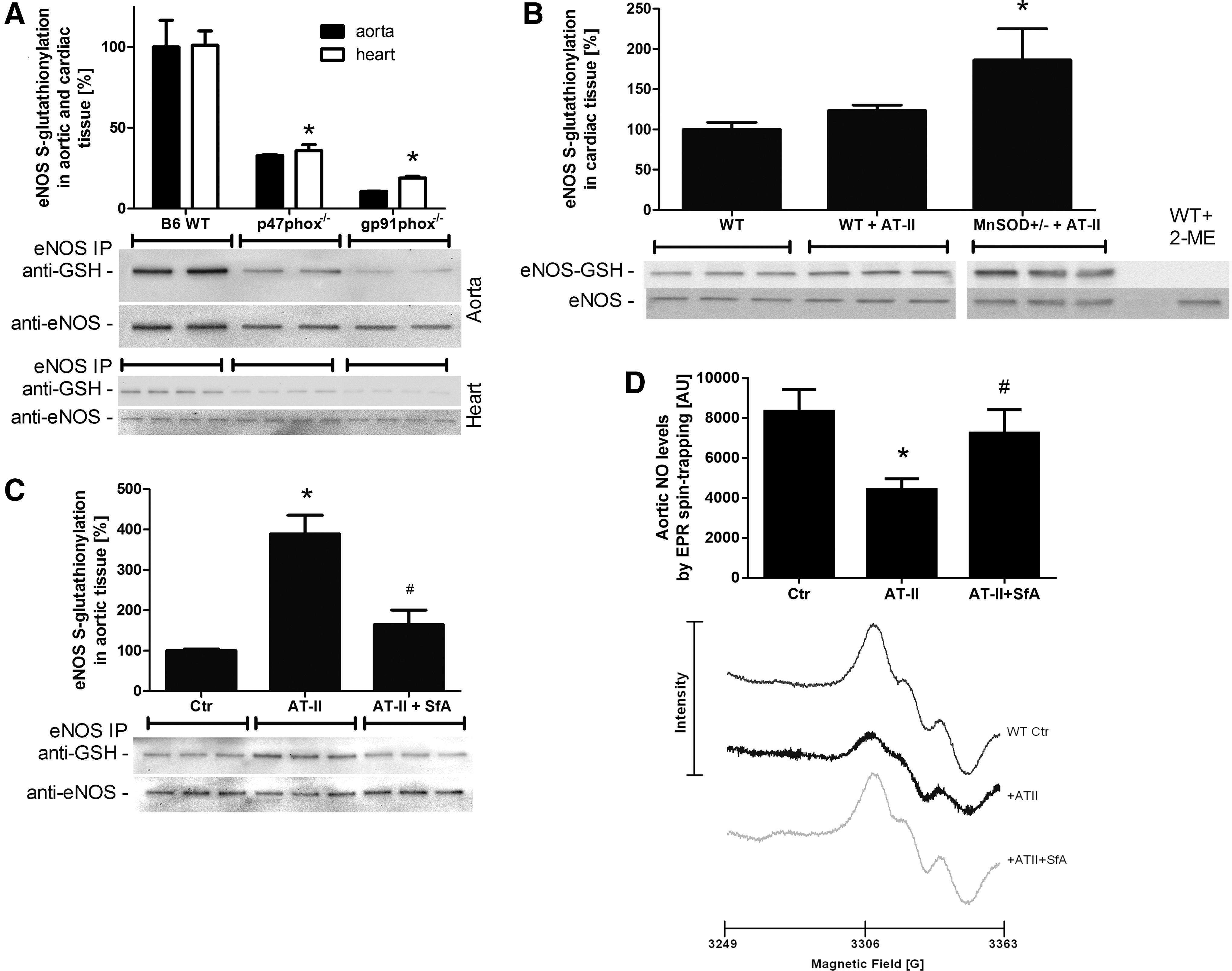
eNOS dysregulation/uncoupling by S-glutathionylation as a potential link between mtROS-triggered NADPH oxidase activation
To address the potential role of mtROS-NADPH oxidase crosstalk in causing eNOS uncoupling, eNOS S-glutathionylation in the aorta and heart from control mice in response to varying stress conditions was determined. To specifically address the role of the NADPH oxidase, p47phox and gp91phox-deficient animals were used. p47phox and gp91phox deficiency clearly decreased eNOS S-glutathionylation in wild-type animals, suggesting that baseline eNOS S-glutathionylation, interestingly in whole heart and aorta homogenates, is strongly determined by the NADPH oxidase (Fig. 8A). S-glutathionylation in heart tissue was significantly increased in MnSOD+/− mice treated with AT-II (Fig. 8B). The AT-II-induced increase in eNOS S-glutathionylation in aorta from wild-type mice was prevented by pharmacological in vivo inhibition of the mPTP by SfA (Fig. 8C). Importantly, EPR-based measurements of aortic NO formation showed a direct correlation with the S-glutathionylation pattern in Figure 8C, revealing a decrease of the EPR signal in response to AT-II infusion and a significantly greater signal in response to SfA in vivo infusion (Fig. 8D).
Discussion
With the present study, we sought to determine the underlying mechanism of the activation of NADPH oxidase in inflammatory cells and cardiovascular tissue by mtROS (superoxide, hydrogen peroxide but also subsequently formed peroxynitrite). The present data show that induction of mitochondrial superoxide and hydrogen peroxide (or subsequent peroxynitrite) formation is able to trigger the activation of phagocytic NADPH oxidase in isolated human leukocytes and in murine WBC. The increase in overall superoxide, hydrogen peroxide, and peroxynitrite signal in response to inducers of mtROS was prevented by pharmacological blockade of the mPTP (CsA and SfA) as well as by the inhibitors of PKC (Chele) and inhibitory of the NADPH oxidase (Apo and DPI). Likewise, various mitochondria-targeted antioxidants such as HTPP/ATPP as well as lipophilic, positively charged manganese porphyrins, and cytoplasmic superoxide, hydrogen peroxide, and peroxynitrite scavengers such as vitamin C inhibited the observed mtROS-driven Nox2 activation. Exogenously applied hydrogen peroxide mimicked the activation of phagocytic NADPH oxidase in accordance to previous reports on redox-sensitive activation of the PKC via thiol oxidation in the phorbol ester/diacylglycerol binding domain of PKC with two zinc-sulfur clusters (ZnCys3) that function as redox switches in all PKC isoforms [reviewed in Daiber (11) and Schulz et al. (54)].
Until now, there were conflicting data as to what extent superoxide, hydrogen peroxide, or peroxynitrite formed in mitochondria directly contribute to the opening of the mPTP, although there was good evidence that thiol oxidations in the adenine nucleotide translocase and tyrosine nitration in the voltage-dependent anion channel may increase the opening probability of mPTP [reviewed in Daiber (11) and Radi et al. (46)]. With the present studies, we could demonstrate that genetic deletion of the phagocytic NADPH oxidase p47phox (p47phox−/− mice) as well as the regulator of mPTP opening, cyclophilin D (CypD−/− mice), markedly suppressed the mtROS-dependent oxidative burst of the phagocytic NADPH oxidase. This superoxide/hydrogen peroxide-driven activation of phagocytic NADPH oxidase in leukocytes was also evident in response to chronic AT-II treatment and was suppressed in CypD−/− mice. Likewise, MnSOD deficiency aggravated the basal and myxothiazol driven oxidative burst in whole blood of old MnSOD+/− mice and increased the chemotactic stimulation of oxidative burst in isolated WBC from these mice. MnSOD deficiency not only led to an increase in mitochondrial superoxide levels (62) and formation of cytosolic as well as mitochondrial peroxynitrite (see Supplementary Fig. S4F) but also increased the overall burden of mitochondrial superoxide and hydrogen peroxide (or subsequent peroxynitrite) formation in response to the impairment of mitochondrial respiration and electron flux via oxidation of iron-sulfur centers in complexes I and II (11). It is important to note that mitochondrial superoxide and hydrogen peroxide in phagocytes via this signaling cascade not only play an important role for the adhesion and infiltration of leukocytes to the vascular wall [as demonstrated by a lower aortic WBC population in AT-II-treated mice with p47phox or gp91phox-deficient WBC (26, 60)], but are also of great importance for phagocyte apoptosis and necrosis to avoid chronic tissue inflammation. Very recently, this concept of mtROS-triggered Nox2 activation in immune cells was established in cultured human lymphoblasts, however, missing the demonstration of the in vivo relevance (21). Of interest, these authors showed a correlation between mtROS release to the cytosol and cytoplasmic increase in calcium levels, suggesting that activation of phagocytic NADPH oxidase requires both increased mitochondrial superoxide and hydrogen peroxide and also calcium concentrations (21). A more detailed discussion on the connection between AT-II-induced hypertension, inflammation, and the crosstalk between mitochondria and NADPH oxidases is presented in the extended discussion section (see Supplementary Data).
The in vivo relevance of mitochondrial superoxide and hydrogen peroxide (or subsequent peroxynitrite) in triggering NADPH oxidase activation was demonstrated by increased cardiac, aortic, and leukocyte NADPH oxidase activity in old MnSOD+/− mice, which was associated with severe vascular (endothelial) dysfunction. These abnormalities were strikingly improved by chronic blockade of the mPTP by SfA, indicating the detrimental role of mitochondrial superoxide and hydrogen peroxide escaping to the cytosol. Likewise, SfA therapy improved GTN-induced endothelial dysfunction, a process that requires the escape of GTN-triggered mitochondrial superoxide, hydrogen peroxide, and peroxynitrite to the cytosol (61). In contrast, SfA treatment failed to prevent the development of nitrate tolerance in response to chronic GTN, a process that is largely based on the oxidative inhibition of the mitochondrial aldehyde dehydrogenase (ALDH-2) (14, 56), the GTN bioactivating enzyme (10). These beneficial effects of mPTP blockade is in accordance with our previous findings on mitochondrial superoxide and hydrogen peroxide (or peroxynitrite)-triggered NADPH oxidase activation in the setting of GTN-induced tolerance and identification of this “crosstalk” as the driving force for so-called cross-tolerance (GTN-induced endothelial dysfunction) (61). These potent protective effects of mPTP blockade (it should be noted that SfA therapy in old MnSOD+/− mice was only maintained for 7 days) go hand in hand with a report by Piot et al. on a significant decrease in infarct size in patients who were treated post MI with CsA, an inhibitor of the mPTP (45). In accordance with previous reports, these data underline the therapeutic potential of targeting mitochondrial channels in particular, and mitochondrial superoxide and hydrogen peroxide formation in general (17, 18).
A final set of experiments helped us demonstrate that MnSOD deficiency substantially increased the adverse effects of AT-II in vivo treatment on the circulation; for example, the use of subpressor doses of AT-II failed to induce hypertension in control mice but significantly increased blood pressure in MnSOD+/− mice. Likewise, MnSOD deficiency aggravated AT-II-induced cardiac Nox activity and translocation of p47phox to the membrane, endothelial and vascular dysfunction, as well as eNOS S-glutathionylation and, therefore, eNOS uncoupling. The last parameter provides an attractive explanation and read-out of how mtROS trigger endothelial dysfunction via NADPH oxidase activation and eNOS dysfunction. S-glutathionylation of eNOS was characterized as an important “redox switch” in eNOS that impairs NO formation or even contributes to uncoupling of eNOS (9) [reviewed in Schulz et al. (54)]. Importantly, pharmacological inhibition of the mPTP by SfA rescued the impaired aortic NO formation under AT-II treatment, identifying S-glutathionylation of eNOS as an important predictor of its enzymatic function. Likewise, mitochondrial superoxide and hydrogen peroxide (or subsequent peroxynitrite) formation either directly or via activation of NADPH oxidase may contribute to oxidative depletion of the eNOS cofactor tetrahydrobiopterin (BH4). As a proof of concept, vascular oxidative stress and endothelial dysfunction in response to AT-II in vivo treatment were improved by genetic modulation of mPTP opening in CypD−/− mice.
A limitation of the present studies is, that although we used many different in vivo and in vitro models, we are not able to specifically answer the question, which NADPH oxidase isoform essentially contributes to the here described crosstalk between mtROS and NADPH oxidase, although the results obtained with the Nox2 inhibitor VAS2870 (Figs. 1 and 6) or with cells and tissue from p47phox−/− mice as well as data on p47phox phosphorylation by AT-II and its prevention by CypD deficiency point toward the phagocytic NADPH oxidase being the key enzyme responsible for the crosstalk. The large number of different cellular and in vivo models also forced us to restrict parts of the study to a limited number of parameters.
In light of previous reports on AT-II-induced hypertension demonstrating that genetic deficiency in p47phox, a regulatory cytosolic subunit of not only the phagocytic NADPH oxidase but also the catalytic subunit gp91phox itself, almost completely prevented the adverse effects of angiotensin infusion in mice (e.g., hypertension, endothelial dysfunction, and vascular oxidative stress) (7, 36), and the absence of these adverse effects in leukocyte-depleted mice even on adoptive cell transfer of monocytes from gp91phox-deficient mice (in contrast to reconstitution with monocytes from wild-type mice) (60), one may expect a significant contribution of the Nox2 isoform to mitochondrial superoxide and hydrogen peroxide (or peroxynitrite)-triggered endothelial dysfunction. Since p47phox can also contribute to Nox1 activation and it has been reported that AT-II-induced hypertension and adverse effects on the vasculature are also prevented by genetic Nox1 deficiency (22, 39), it may be assumed that both isoforms can contribute to the phenotype of AT-II infusion. Based on these data, it may be assumed that mtROS can trigger the activation of Nox1 in a similar process as observed for Nox2, basically via translocation of p47phox. The contribution of Nox4, which was previously also found to be localized in mitochondria (5, 25) and largely contribute to processes that are associated with mitochondrial oxidative stress (1, 2, 35), was not addressed in the present study. However, the present design of our study that was mainly focused on the oxidative activation of Nox2 (or Nox1) by PKC or cSrc-driven translocation of cytosolic regulatory subunits is most probably not shared by the Nox4 isoform, and it remains to be established whether our concept of mitochondrial superoxide and hydrogen peroxide (or peroxynitrite)-driven crosstalk between mitochondria and NADPH oxidase can be extended to Nox4 as well.
One may also criticize that we used inhibitors with a low specificity. For example, apocynin, which was used as an inhibitor of NADPH oxidase, was previously shown to also inhibit Rho kinase pathway (50) and even act as a direct antioxidant or pro-oxidant when used at higher concentrations (8, 28). DPI, which was used as another inhibitor of NADPH oxidase, inhibits almost all flavin-dependent oxidoreductases (e.g., xanthine oxidase and nitric oxide synthase), as well as cholinesterases and the internal calcium pump (65). These nonspecific actions were at least, in part, compensated by the use of more specific compounds in several experiments (e.g., inhibitor chelerythrine for PKC, VAS2870 for Nox2, PP2 for cSrc, and BAPTA-AM for intracellular calcium). We also have to acknowledge that CsA and SfA are well-known immunosuppressive drugs due to targeting of cyclophilin A, an intracellular protein that has peptidyl-prolyl cis-trans isomerase (PPIase) enzymatic activity which is important for cytoplasmic protein folding (69). Thus, the pharmacological inhibition of cyclophilin D is not specific and CsA as well as SfA in vivo therapy, in contrast to acute in vitro incubation, could potentially display immunosuppressive effects. The disadvantages of the mPTP blocking drugs were overcome by the studies in cyclophilin D knockout mice with AT-II treatment, as they enable a study of the effects of highly specific mPTP inhibition on AT-II-induced hypertension, eNOS dysfunction, and oxidative stress.
A final limitation of the present study is that we did not discriminate between distinct intracellular, mitochondrial or cyctosolic superoxide, hydrogen peroxide, and peroxynitrite formation. Although we have carefully defined the nature of the measured reactive species by each of the present assays, we sometimes used the generic term “mtROS” (referring to not only superoxide and hydrogen peroxide but potentially also secondarily formed peroxynitrite) when discussing reactive species conferring the mitochondrial-Nox signaling and leading to the damage in the cytosol (e.g., induce eNOS uncoupling) (see Fig. 7B, C) and eNOS S-glutathionylation (presented in Fig. 8). For the interested reader, previous reports have addressed the different detection methods and redox signaling properties of different ROS and RNS in greater detail (19, 20, 29, 38, 46, 58).
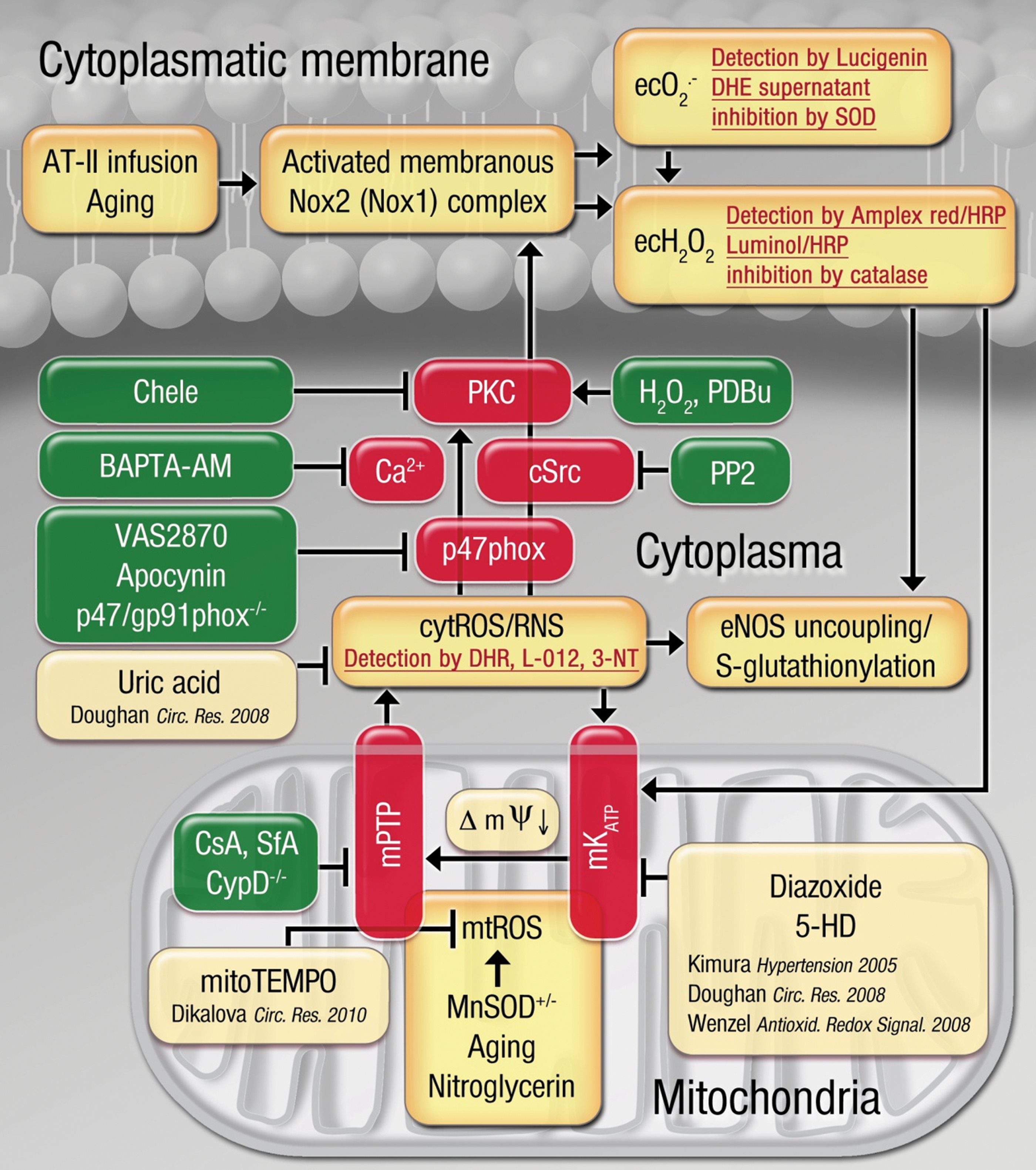
In conclusion, we demonstrate for the first time the crucial role of mitochondrial superoxide and hydrogen peroxide (or subsequently formed peroxynitrite) for the activation of NADPH oxidase in phagocytic cells as well as vascular and cardiac tissue using genetic models of MnSOD deficiency (increased mtROS levels) and impairment of the mPTP in CypD−/− mice (both in combination with AT-II infusion). The sequence of events and underlying mechanisms studied in the present work as well as the previous findings by our group and others provide the rationale to understand the mechanisms of this crosstalk and are presented in Figure 9. As shown here for the first time, MnSOD deficiency aggravates AT-II-triggered vascular dysfunction, which was prevented by pharmacological (SfA) or genetic (CypD−/−) inhibition of the mPTP opening probability. The exact identity of the mtROS species conferring this crosstalk remains elusive, but it may be reasonable to conclude that superoxide and hydrogen peroxide (or subsequently formed peroxynitrite) can trigger the activation of Nox, as previous studies demonstrated that all of these species react with zinc-sulfur centers (as present in PKC) and cause activation of other kinases (e.g., MAPK or cSrc), although peroxynitrite is the most potent candidate for sulfur-based redox sensing (58). We would also like to direct the interested reader to the recent literature on challenges and limitations of ROS and RNS measurements in biological samples (30). Based on our in vitro observations in isolated human leukocytes, we can conclude that intracellular calcium and the tyrosine kinase cSrc contribute significantly to the mitochondrial superoxide and hydrogen peroxide (or peroxynitrite)-triggered activation of phagocytic NADPH oxidase. In contrast, a contribution of the ERK1/2-MAPK pathway in this process was not evident in our experiments (lack of inhibitor 2-(2-Chloro-4-iodophenylamino)-N-cyclopropylmethoxy-3,4-difluorobenzamide [PD184352] [not shown]). The demonstration of an involvement of cSrc and intracellular calcium in this crosstalk is in accordance with previous reports by Dikalov and coworkers (21, 23). Interestingly, the cSrc inhibitor PP2 had no effect on phorbolester-mediated activation of Nox2 in isolated human PMN, pointing to distinct pathways for phorbol ester (classical PKC axis) and mitochondrial superoxide and hydrogen peroxide (mixed PKC and cSrc axis)-triggered Nox2 activation. Along with previous observations that vascular infiltration of immune cells largely contributes to the pathogenesis of AT-II-triggered hypertension (26, 34, 37, 60), the results of the present studies clearly strengthen the clinical importance of this topic by demonstrating that mitochondrial superoxide and hydrogen peroxide are able to activate the phagocytic NADPH oxidase and thereby modulate tissue activity of these immune cells, which is largely influenced by the rate of infiltration and cell death.
Methods
Materials
For isometric tension studies, GTN was used from a Nitrolingual infusion solution (1 mg/ml) from G.Pohl-Boskamp (Hohenlockstedt, Germany). L-012 was purchased from Wako Pure Chemical Industries (Osaka, Japan). mitoTEMPO was obtained from Enzo Life Sciences (Lörrach, Germany), ATPP or HTPP were purchased from Sigma (Steinheim, Germany). SfA was a gift of Novartis (Basel, Switzerland). MnTMPyP and MnTPP, VAS2870, BAPTA-AM, PP2, PD184352, polyethylene-glycolated superoxide dismutase (PEG-SOD), and Cu,Zn-SOD (bovine, 3.255 kU/mg protein, EC 232-943-0) were obtained from Sigma. All other chemicals were of analytical grade and were obtained from Sigma-Aldrich, Fluka, or Merck. The inhibitor concentrations were adjusted to the used number of cells (more cells required more inhibitor). In addition, the effect of certain inhibitors differed in whole blood versus isolated cells versus tissue and required specific adjustment.
Isolation of human neutrophils
All use of human material was in accordance with the Declaration of Helsinki and was granted by the local institutional ethics committee as well as the authorities (Landesärztekammer Rheinland-Pfalz). The procedure is described in references (12, 63). Briefly, erythrocytes in 15 ml heparin-supplemented human blood were separated by sedimentation on addition of an equal volume of dextran solution (MW 485,000, 40 mg/ml PBS). The leukocyte-containing supernatant was centrifuged on Histopaque-1077 from Sigma for 30 min at 500 g at 20°C, resulting in a neutrophil (PMN)-containing pellet and the monocyte/lymphocyte-enriched (WBCs) “buffy coat” between the aqueous and Ficoll phases. The WBC fraction was collected and purified by further centrifugation for 10 min at 500 g followed by resuspension in PBS. The PMN pellet was freed from residual erythrocytes by hypotonic lysis in distilled water and centrifugation at 500 g (two to three times). Total blood cell count and the purity of the fractions were evaluated using an automated approach using a hematology analyzer KX-21N (Sysmex Europe GmbH, Norderstedt, Germany). Typical content of WBC in each fraction was previously published (63).
Assessment of the activation of phagocytic NADPH oxidase activity by mitochondrial superoxide and hydrogen peroxide in isolated human leukocytes
To characterize the activation of NADPH oxidase by mitochondrial superoxide and hydrogen peroxide in detail, we used isolated human leukocytes. Mitochondrial superoxide and hydrogen peroxide in isolated neutrophils or monocytes/lymphocytes (1–5×105 WBC/ml) was induced by antimycin A (20 μg) or myxothiazol (20 μM). Classical PKC-mediated activation of NADPH oxidase was mediated by the phorbol ester derivative PDBu (0.1–10 μM) (12). PKC-independent but phospholipase D dependent activation of leukocytes was mediated by fMLP (100 μM) (49). The mtROS stimuli was also mimicked by the addition of exogenous hydrogen peroxide (10–1000 μM). NADPH oxidase-derived superoxide was measured by L-012 (100 μM) or lucigenin (100 or 250 μM) ECL using a single vial luminometer Lumat or an ECL plate reader Centro (Berthold Technologies, Bad Wildbad, Germany). Extracellular hydrogen peroxide (from superoxide disproportionation) was determined by luminol (100 μM)/horseradish peroxidase (HRP) (0.1 μM) ECL (Lumat or Centro) or amplex red (100 μM)/HRP (0.1 μM)-derived fluorescence (Twinkle plate reader, Berthold). Authentic hydrogen peroxide was used as a chemical mimic of mtROS (it should be noted that hydrogen peroxide is not detected by lucigenin), and subsequent Nox2-derived superoxide formation was measured by lucigenin ECL (see above). Furthermore, we applied a number of inhibitors to assess the role of mitochondrial superoxide and hydrogen peroxide in the activation of phagocytic NADPH oxidase such as chelerythrine (10 μM) to inhibit PKC (41), apocynin (100 μM) to inhibit NADPH oxidase, CsA (0.2 μM) to inhibit the mPTP, DPI (30 μM) to inhibit flavin-dependent oxidoreductases, VAS2870 (10 μM) to inhibit Nox2, PP2 (10 μM) to inhibit cSrc tyrosine kinases, PD184352 (2 μM) to inhibit ERK1/2-MAPK signaling, BAPTA-AM (100 μM) to deplete intracellular calcium, and PEG-SOD (100 U/ml) and different antioxidants at various concentrations (e.g., mitochondria-targeted antioxidants).
Likewise, extracellular superoxide formation was measured by HPLC-based quantification of 2-hydroxyethidium or resorufin in the supernatants of incubated leukocytes according to a previously published protocol (51, 61). Briefly, the leukocytes (1×106 WBC per sample) were incubated for 20 min with 50 μM DHE or 100 μM amplex red/0.1 μM HRP, were centrifuged for 10 min at 500 g, and 50 μl of the supernatant were subjected to HPLC analysis. 2-hydroxyethidium was quantified as described (61, 64) and was also used to investigate potential lucigenin-dependent redox cycling in PDBu-stimulated PMN at concentrations of 5, 50, and 250 μM. Resorufin was quantified using an HPLC set-up as follows: The system consisted of a control unit, two pumps, a mixer, detectors, a column oven, a degasser, an autosampler (AS-2057 plus) from Jasco (Groß-Umstadt, Germany), and a C18-Nucleosil 100-3 (125×4) column from Macherey & Nagel (Düren, Germany). A high-pressure gradient was employed with the organic solvent (90 vv% acetonitrile/10 vv% water) and 50 mM citrate buffer pH 2.2 as mobile phases with the following percentages of the organic solvent: 0 min, 41%; 7 min, 45%; 8–9 min, 100%; 10–12 min, 41%. The flow was 1 ml/min, compounds were detected by their absorption at 300 nm, and resorufin was also detected by fluorescence (Ex. 570 nm/Em. 590 nm). Typical retention time of resorufin was 2.8 min, and its formation was sensitive to the presence of catalase. Some key experiments were also verified by EPR-based DEPMPO spin trapping of superoxide anion radicals in isolated human leukocytes (experimental conditions are described in the legend to Supplementary Fig. S3).
Finally, as a measure of NADPH oxidase activation that does not rely on the measurement of ROS formation by HPLC or ECL, we determined the mitochondrial superoxide and hydrogen peroxide-triggered translocation of the cytosolic subunits (p67phox, p47phox, and Rac1) of the phagocytic NADPH oxidase in WBCs from the buffy coat from 500 ml human whole blood (courtesy of the blood preservation, University Medical Center Mainz, Germany). For separation of the cytosolic and membranous fraction, we used a commercial plasma membrane protein extraction kit (BioVision, Mountain View, CA). Briefly, Buffy coat cell suspensions were diluted 1:1 with dextran solution for red blood cell sedimentation within 30 min (see above), centrifuged at 500 g for 15 min at room temperature, and the pellet was resuspended in PBS. Next, the cells (10×106 WBC per sample) were incubated for 30 min with stimulators and inhibitors of oxidative burst as indicated. WBC were incubated with the lysis buffer and sonicated followed by a number of centrifugation and incubation steps as detailed in the manufacturer's instructions. The protein content in cytosolic and membranous fractions was determined by Bradford method, and 22 μg protein was loaded into each well followed by standard SDS-PAGE and Western blotting procedures. For specific staining, we used a mouse monoclonal p67phox antibody (dilution 1:500; BD Bioscience, San Jose, CA), a polyclonal rabbit p47phox antibody (dilution 1:500; Upstate, Billierica, MA), and a mouse monoclonal Rac1 antibody (dilution 1:1000; BD Bioscience). Detection and quantification were performed by ECL with peroxidase-conjugated anti-rabbit/mouse (1:10,000; Vector Lab., Burlingame, CA) secondary antibodies. Densitometric quantification of antibody-specific bands was performed with a ChemiLux Imager (CsX-1400M; Intas, Göttingen, Germany) and Gel-Pro Analyzer software (Media Cybernetics, Bethesda, MD). All signals were normalized to stainings with a monoclonal mouse α-actinin antibody (1:2500; Sigma-Aldrich). This Western blot procedure was also applied to some in vivo samples using the membranous fraction from cardiac samples as previously described (42, 64). For one experiment, total aortic homogenates were blotted and stained for p47phox Ser328 phosphorylation using a specific polyclonal rabbit antibody from antibodies online (Atlanta, GA) at a dilution of 1:500.
Assessment of the activation of phagocytic NADPH oxidase activity by mitochondrial superoxide and hydrogen peroxide in isolated WBCs from mice
WBCs were isolated or whole blood was used from different genetically modified mice to further establish the role of mitochondrial superoxide and hydrogen peroxide (or subsequently formed peroxynitrite) in the activation of NADPH oxidase: p47phox−/− mice with dysfunctional Nox2 activation (bred in our animal facility), cyclophilin D knockout (CypD−/−) mice with dysfunctional opening of the mPTP [obtained from and generated by Michael A. Forte and Paolo Bernardi (4)], and manganese superoxide dismutase partially deficient (MnSOD+/−) mice [obtained from and generated by Karin Scharffetter-Kochanek (55)]. Oxidative burst in isolated WBCs or whole blood was stimulated and measured as described for human leukocytes or blood (including L-012, luminol/HRP ECL, amplex red/HRP fluorescence, and 2-hydroxyethidium HPLC). Murine WBCs were isolated with a similar protocol as described for human leukocyte isolation (see above), but the total leukocyte fraction was directly precipitated on dextran-mediated erythrocyte sedimentation at 400 g for 30 min without separating the neutrophils and monocytes/lymphocytes with the Ficoll gradient centrifugation.
Animal model
All of the animals were treated in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the National Institutes of Health and were granted by the University Hospital Mainz Ethics Committee and the authorities (Landesuntersuchungsamt Rheinland-Pfalz). C57/Bl6 control and CypD−/− mice were bred in the central animal facility of the University medical Center in Mainz and had free access to water and food. The MnSOD+/− and mice were on a C57/Bl6x129/Ola mixed background and used at a different age (3 [young] and 12 [old] months) to study the effect of age-induced increase in mitochondrial superoxide and hydrogen peroxide (or subsequently formed peroxynitrite). We also used in vivo treatment with high/pressor (1 mg/kg/day) and low/subpressor (0.2 mg/kg/day) dose of AT-II for 7 days in order to test vascular function and Nox activity in C57/Bl6 control, CypD−/−, and MnSOD+/− mice. In vivo treatment with the mPTP inhibitor SfA (10 mg/kg/day, s.c. for 7 days) was used as a another proof of concept of the role for mtROS in the activation of NADPH oxidase. SfA therapy [a potent mPTP inhibitor (27)] was also performed in nitrate-tolerant C57/Bl6 mice treated for 4 days with GTN (16 μg/h) using osmotic minipumps (15). In the in vivo studies, we assessed NADPH oxidase activity, cytosolic superoxide, and hydrogen peroxide (or subsequently formed peroxynitrite) formation and whole blood as well as isolated WBC ROS/RNS (ECL, 2-hydroxyethidium, DHR 123 oxidation, protein tyrosine nitration by dot blot analysis, and DHE staining), endothelial function (isometric tension recordings), eNOS dysregulation/uncoupling (eNOS S-glutathionylation by immunoblotting and endothelial superoxide formation by DHE staining) and in two models, also blood pressure [tail cuff measurements as described (3)].
Vascular function
Vasodilation to endothelial-dependent (ACh) and -independent (GTN) vasodilators was assessed by isometric tension recordings in prostaglandin F2α (PGF2α) preconstricted isolated aortic ring segments as previously described (40, 44).
Vascular, blood, and cardiac formation of reactive oxygen and nitrogen species
Vascular superoxide, hydrogen peroxide, or secondary peroxynitrite formation was determined by DHE (1 μM)-dependent fluorescence microtopography in aortic cryo-sections (according to our experience, this assay mainly reflects vascular superoxide levels) (33, 53). Endothelial superoxide formation by DHE staining in the presence and absence of the NOS inhibitor L-NAME (or D-NAME) at a concentration of 500 μM was used to assess the coupling state of eNOS (43, 64). In healthy tissue, the NOS inhibitor blocks NO formation and thereby indirectly increases the DHE staining due to reduced break-down of superoxide by the reaction with NO. In diseased tissue, the NOS inhibitor directly blocks superoxide formation from uncoupled NOS, and the DHE staining is decreased. Importantly, the densitometric quantification of the DHE staining has to be restricted to the endothelial cell layer in order to specifically assess eNOS-derived superoxide. Vascular superoxide production was determined using the highly selective chemiluminescence indicator reagent Diogenes™, a luminol-peroxidase based assay (National Diagnostics, Atlanta, GA; 50% of total reaction volume) in intact aortic rings with PDBu (0.1 μM) stimulation using a Mithras Microplate Luminometer (Berthold) as described (61). Aortic hydrogen peroxide formation was measured by an amplex red/peroxidase-based HPLC assay as described earlier. Briefly, aortic ring segments of 4 mm length were incubated with 150 μl of amplex red (100 μM) and HRP (0.2 μM) in PBS with Ca2+/Mg2+ under stimulation with myxothiazol (20 μM) for 60 min at 37°C. The Nox2 inhibitor VAS2870 (25 μM) was used to inhibit the NADPH oxidase activity by preincubating the aortic ring segments for 20 min at 37°C. In addition, superoxide and hydrogen peroxide (potentially also peroxynitrite) formation was detected in isolated WBC and whole blood by L-012 (100 μM) ECL as described earlier. Superoxide formation was determined in cardiac membranous fractions by lucigenin (5 μM) ECL and HPLC-based 2-hydroxyethidium quantification as reported (61, 64). In some experiments, the Nox2 inhibitor VAS2870 (25 μM) was used to inhibit the NADPH oxidase activity by preincubating the cardiac tissue pieces for 30 min on ice before homogenization. Cardiac oxidative stress was also assessed by dot blot analysis of cardiac tissues, which was modified from a previous report (51, 52). Protein tyrosine nitration was detected using a specific antibody for 3NT.
Cardiac superoxide, hydrogen peroxide, or secondary peroxynitrite formation was also determined by eNOS S-glutathionylation, a redox modification of eNOS as recently described (33, 51). Briefly, M-280 Sheep anti-Rabbit IgG-coated beads from Invitrogen (Darmstadt, Germany) were used along with a monoclonal mouse eNOS (Biosciences) antibody. The beads were loaded with the eNOS antibody and crosslinked according to the manufacturer's instructions. Next, the aortic homogenates were incubated with the eNOS antibody beads, precipitated with a magnet, washed, transferred to gel, and subjected to SDS-PAGE followed by a standard Western blot procedure using a monoclonal mouse antibody against glutathionylated proteins from Virogen (Watertown, MA) at a dilution of 1:1000 under nonreducing conditions. All signals were normalized on the eNOS staining of the same sample.
In addition, cardiac oxidative stress was also assessed by dot blot analysis using a specific mouse monoclonal 3NT antibody at a dilution of 1:1000 (Upstate Biotechnology, Lake Placid, NY) as described (51).
Intracellular superoxide or secondary peroxynitrite levels (see Supplementary Fig. S1) were assessed in cardiac homogenates by quantification of DHR 123 oxidation using an HPLC-based assay. The stability of the formed oxidation product rhodamine during storage at −80°C was also determined (see Supplementary Fig. S1). Briefly, heart tissue was incubated with 50 μM dihydrorhodamine 123 (DHR123) for 30 min at 37°C in PBS buffer. Wet weight of heart pieces was determined; they were snap frozen, stored at −80°C, homogenized in 50% acetonitrile/50% PBS by a glass/glass homogenizer within 1 week of storage, centrifuged, and 50 μl of the supernatant were subjected to HPLC analysis. For composition of the Jasco HPLC system, see what has been described earlier. A high-pressure gradient was employed with solvent B (acetonitrile/water 90:10 v/v%) and solvent A (25 mM citrate buffer pH 2.2) as mobile phases with the following percentages of the organic solvent B: 0 min, 30%; 8 min, 65%; 8.5–9 min, 100%; and 9.5 min, 30%. The flow was 1 ml/min, and DHR was detected by its absorption at 280 nm whereas its oxidation product was detected by fluorescence (Ex. 488 nm/Em. 530 nm, gain 1×). The signal was normalized on wet weight of the heart tissue.
Aortic nitric oxide formation was measured using EPR-based spin trapping with iron-diethyldithiocarbamate [Fe(DETC)2] colloid, which was freshly prepared under argon. One murine aorta was cut into ring segments of 3 mm length (six to seven pieces) and placed in 1 ml Krebs–Hepes buffer on a 24-well plate on ice. The samples were stimulated with 10 μM calcium ionophore (A23187) for 2 min on ice; then, 1 ml of the Fe(DETC)2 colloid solution (400 μM in PBS with Ca2+/Mg2+) was added, and the plate was placed in the incubator at 37°C. After 60 min of incubation, the aortic rings were placed at a fixed position in a 1 ml syringe with removed top in PBS buffer and frozen in liquid nitrogen (in the way that the entire aortic sample was placed within a 100 μl volume of the syringe). For measurement, the frozen cylinder with the aortic sample was pressed out of the syringe and placed in a special Dewar vessel (Magnettech, Berlin, Germany) filled with liquid nitrogen. The localization of the aortic sample was adjusted to the middle of the resonator. EPR conditions: B0=3276 G, sweep=115 G, sweep time=60 s, modulation=7000 mG, MW power=10 mW, gain=9×102 using a Miniscope MS400 from Magnettech (Berlin, Germany). The A23187-stimulated NO signal was absent when the aorta were denuded, L-NAME (200 μM) was added, or when aorta from eNOS−/− mice were used (not shown). The general conditions for this assay were previously described by Kleschyov et al. (32).
Statistical analysis
Results are expressed as mean±SEM. Two-way ANOVA (with Bonferroni's correction for comparison of multiple means) was used for comparisons of vasodilator potency and efficacy as well as Diogenes and luminol/HRP ECL time course. One-way ANOVA (with Bonferroni's or Dunn's correction for comparison of multiple means) was used for comparisons of ROS and RNS detection, translocation assays, dot blot and Western blot analysis, and blood pressure. One-way repeated measures analysis of variance with all pairwise multiple comparison procedures (Holm-Sidak method) was used for p47phox phosphorylation. p-values<0.05 were considered significant.
Footnotes
Acknowledgments
The authors thank Jörg Schreiner, Angelica Karpi, Nicole Glas, Jessica Rudolph, Marcus Hortmann, Christian Otto, and Benjamin Opitz for their expert technical assistance and Margot Neuser for graphical assistance. They are also indebted to Fabio Di Lisa, Paolo Bernardi, and Michael Forte (University of Padova, Italy and Oregon Health & Science University, Portland, USA) for their kindness to provide the CypD knockout mice. The present work was supported by generous financial support rendered by the Johannes Gutenberg University and Medical Center Mainz (MAIFOR and Forschungsfonds grants to A.D.), the German Research Foundation (DFG WE 4361/3-1 to P.W. and DFG KFO142 SCHA 411/15-2 to K.S.-K.), the Stiftung Mainzer Herz to all authors, and by a grant from the Federal Ministry of Education and Research (BMBF 01EO1003) to P.W., T.M., and A.D. within the Center for Thrombosis and Hemostasis (CTH) Mainz. Yuliya Mikhed holds a stipend from the International PhD Program on the “Dynamics of Gene Regulation, Epigenetics and DNA Damage Response” from the Institute of Molecular Biology gGmbH, (Mainz, Germany) funded by the Boehringer Ingelheim Foundation. This article contains results that are a part of the doctoral thesis of Alexander Scholz.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.