
Abstract
Introduction
I
The steady-state cellular redox status is controlled by several well-balanced oxidant and antioxidant systems that involve a multitude of enzymes. In the nervous system, superoxide dismutase, catalase, glutathione S-transferase, heme oxygenase, NAD(P)H:quinone oxidoreductase-1 (40), and sestrins (Sesn) (26) act in concert to keep ROS levels balanced. Sestrins are believed to reduce intracellular ROS by regenerating overoxidized and catalytically inactive peroxiredoxins, which are ubiquitously expressed antioxidant proteins involved in sequestering H2O2 (11). In mammals, the Sestrin family comprises three members, Sesn1-3, which are tissue-specifically induced by oxidative stress and DNA damage in a p53-dependent manner. Notably, Sestrin up-regulation in response to synaptic activity has been observed in neuronal cell culture (26), but a possible contribution of Sestrin to pain processing has not been investigated so far. In the present study, we asked whether Sestrin might control the pronociceptive activity of ROS. We analyzed the expression of Sesn1-3 in the spinal cord, dorsal root ganglia (DRGs), and peripheral nerves of mice, and thereby identified Sesn2 as a putative modulator of pain processing. Characterization of mice lacking Sesn2 revealed that Sesn2 controls ROS-mediated neuropathic pain processing after peripheral nerve injury.
This is the first study that demonstrates a functional role of the Sestrin (Sesn) family member Sesn2 in the processing of neuropathic pain in vivo. Our results indicate that Sesn2 contributes to the neutralization of exaggerated reactive oxygen species (ROS) production in sensory neurons after peripheral nerve injury, thereby inhibiting pain-sensitizing effects of ROS. The data provide new insights into specific ROS-dependent mechanisms of pain sensitization and could offer new therapeutic avenues for the treatment of neuropathic pain in the clinic.
Results
Sesn2 expression is up-regulated in sensory neurons after peripheral nerve injury
To assess a potential contribution of Sestrins to pain processing, we first investigated the expression levels of the three Sestrins (Sesn1–3) in sensory neurons after spared nerve injury (SNI), a model of neuropathic pain induced by injury of the sciatic nerve (3). Using quantitative real-time reverse transcription polymerase chain reaction (RT-PCR), we detected transcripts for all Sestrin family members in lumbar DRGs of naive mice. Interestingly, Sesn2 mRNA was significantly up-regulated from 1 to 7 days after SNI, whereas the levels of Sesn1 and Sesn3 mRNA remained unaltered (Fig. 1A). Based on the injury-induced up-regulation of Sesn2 mRNA in DRGs, we supposed that Sesn2 might contribute to pain processing, and, therefore, we focused on this Sestrin in the following experiments.
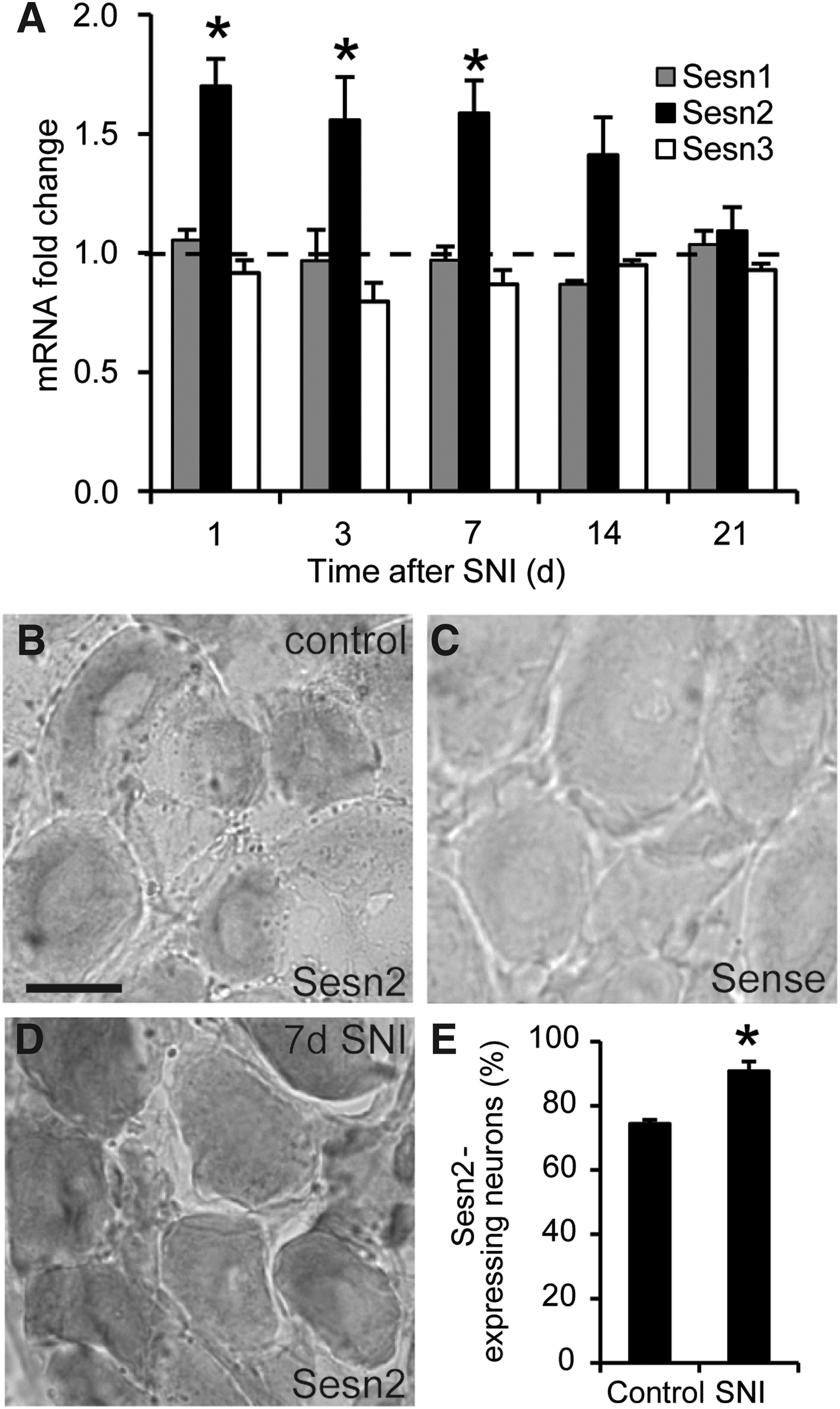
We characterized the distribution of Sesn2 mRNA in DRGs by in situ hybridization and detected Sesn2 mRNA in 74% of DRG neurons (Fig. 1B, E). No hybridization signal was detected in DRGs by the respective sense probe, which was used as a negative control (Fig. 1C). In accordance with the SNI-induced up-regulation of Sesn2 mRNA detected by quantitative real-time RT-PCR, we observed an enhanced Sesn2 hybridization signal in DRG neurons 7 days after SNI (Fig. 1D). Moreover, Sesn2 mRNA was expressed in 91% of DRG neurons 7 days after SNI (Fig. 1E). These data demonstrate that Sesn2 is expressed in DRG neurons and that its neuronal expression is increased in response to peripheral nerve injury.
Next, we investigated the Sesn2 protein expression after peripheral nerve injury in the lumbar spinal cord, the lumbar DRGs, and the sciatic nerve by western blot analyses. Specificity of the Sesn2 antibody was confirmed by detecting a band at the estimated molecular weight of 54 kDa in spinal cord protein extracts of wild-type (WT) mice, but not in extracts derived from Sesn2−/− mice (Supplementary Fig. S1; Supplementary Data are available online at

Sesn2−/− mice demonstrate normal basal sensitivity
To analyze the functional role of Sesn2 in pain processing in vivo, we analyzed the noiciceptive behavior of Sesn2−/− mice (42) and their WT littermates in animal models of pain. We tested both male and female mice (7, 24), but no significant main effects of sex were detected in any assay. To exclude the fact that possible phenotypic differences of the Sesn2−/− mice are caused by anatomic abnormalities, we first investigated the anatomy of peripheral nervous system using standard methodology. The distribution of terminals of mechanoreceptive and thermoreceptive primary afferents in the superficial dorsal horn (Fig. 3A) and the distribution of DRG neuron populations positive for the standard markers substance P, isolectin B4, or NF200 (Fig. 3B) were similar between WT and Sesn2−/− mice, indicating that Sesn2 deficiency does not affect sensory neuron structure. Moreover, Sesn1 and Sesn3 expression in the spinal cord and DRGs was similar to WT in the Sesn2-deficient mice (Fig. 3C and D), making a functional compensation by these proteins unlikely. Furthermore, Sesn2−/− mice displayed a normal motor coordination and balance as analyzed in the rotarod test (median fall-off latencies: Sesn2−/− mice, 89.0 s [interquartile range 78.5–104.0 s]; WT mice, 96.2 s [interquartile range 86.3–120.0 s]; p=0.801; n=10–13 per group).

To investigate the basal sensitivity in Sesn2−/− mice, we determined their immediate behavioral responses to thermal and mechanical stimulation. Sesn2-deficient mice exhibited no significant difference in the acute nociceptive behavior when compared with WT mice in both the hot- and cold-plate test (50°C–54°C and 0°C, respectively; Fig. 3E and F), and also behaved similar on mechanical stimulation with a Dynamic Plantar Aesthesiometer (Fig. 3G). Overall, these data indicate that Sesn2 deficiency does not affect the immediate responses to acute noxious thermal or mechanical stimulation.
SNI-induced neuropathic pain behavior and ROS production is increased in Sesn2−/− mice
Next, we examined the neuropathic pain behavior of Sesn2−/− mice using the SNI model. After SNI surgery, WT mice developed the typical mechanical hypersensitivity of the affected hindpaw characterized by reduced paw withdrawal latency times on mechanical stimulation (Fig. 4A). This mechanical hypersensitivity also occurred in Sesn2−/− mice. However, in Sesn2−/− mice, the degree of mechanical hypersensitivity gradually increased starting at day 7 after SNI and continuing until the end of the 42 days' observation period (Fig. 4A), implying that Sesn2 is involved in neuropathic pain control after peripheral traumatic axonal injury.

To investigate whether the increased mechanical hypersensitivity of Sesn2−/− mice is associated with an accumulation of ROS, we measured the relative H2O2 levels 21 days after SNI using the Amplex Red assay. H2O2 is the most stable endogenous ROS and is often used as an indicator of endogenous ROS production (32). As shown in Figure 4B, H 2O2 levels were significantly higher in the injured proximal sciatic nerve stump of Sesn2−/− mice as compared with WT mice, which is consistent with a reduced antioxidant activity in the knockouts. In contrast, H2O2 levels in DRGs and the spinal cord were similar in both genotypes, suggesting that the antioxidant Sesn2 effect is restricted to the injured peripheral nerve.
To directly assess the impact of ROS on nociception in Sesn2−/− mice, we administered the ROS scavenger N-tert-Butyl-α-phenylnitrone (PBN) to Sesn2−/− and WT mice 21 days after SNI, and measured mechanical hypersensitivity. Notably, after systemic administration of low-dose PBN (20 mg/kg intraperitoneal [i.p.]), the mechanical hypersensitivity significantly decreased in Sesn2−/− mice within the first 4 h after treatment; while the hypersensitivity in WT mice remained nearly unaffected (Fig. 4C). Administration of a higher dose of PBN (50 mg/kg i.p.) significantly reduced mechanical hypersensitivity in both genotypes (Fig. 4D), confirming earlier reports that higher PBN doses can inhibit neuropathic pain behavior (14, 33). Thus, we conclude that low-dose PBN treatment reverses the neuropathic pain behavior in Sesn2−/− mice resulting from elevated ROS, whereas higher doses of PBN are required to neutralize the Sesn2-independent ROS levels after nerve injury. Altogether, these data suggest that the increased SNI-induced neuropathic pain behavior in Sesn2−/− mice is caused by elevated ROS levels.
SNI-induced ATF3 expression is enhanced in Sesn2−/− mice
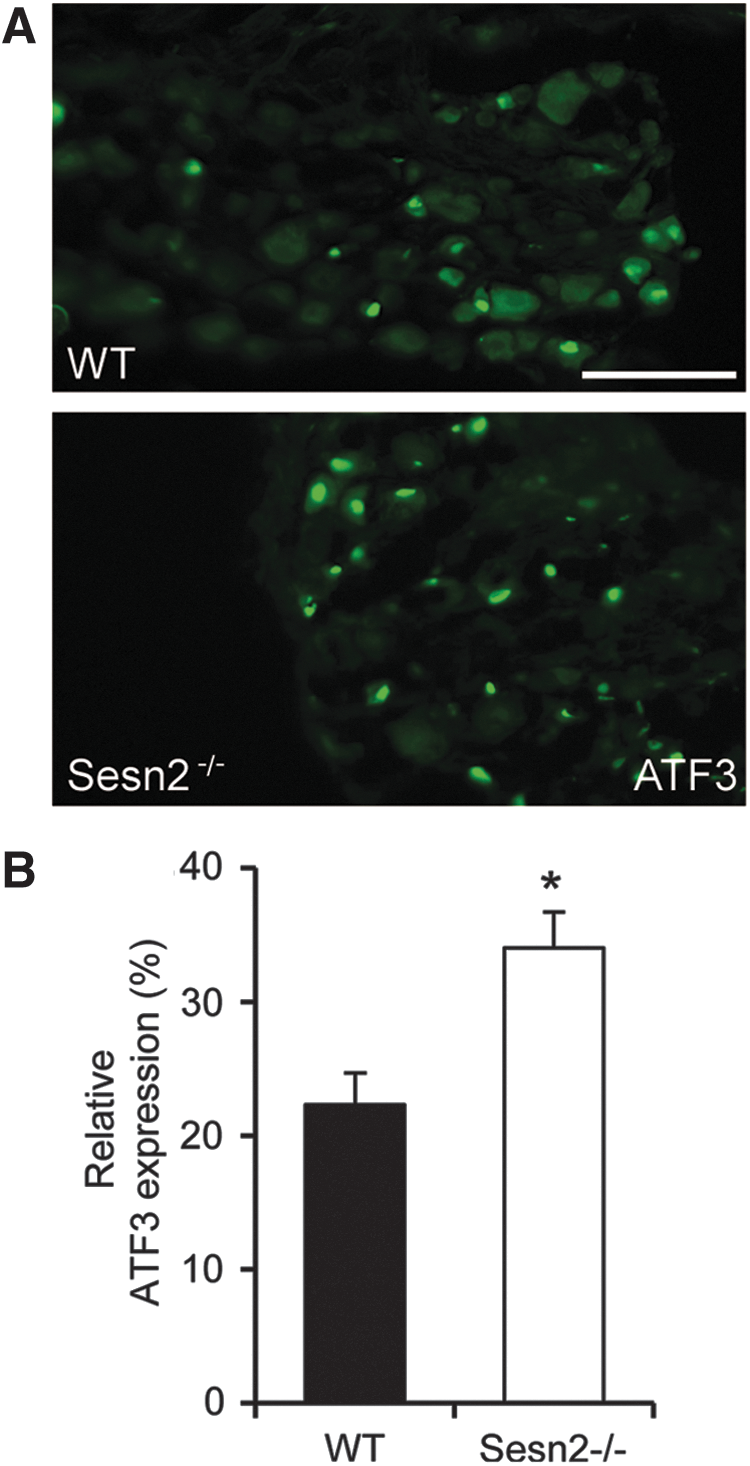
Peripheral nerve injury and neuropathic pain are accompanied by an induction of activating transcription factor 3 (ATF3) in sensory neurons (1, 34, 39). ATF3 is a sensitive cellular marker of nerve injury (39), and a recent study revealed that ATF3 can be induced in an ROS-dependent manner (9). To investigate whether the increased injury-induced ROS production and neuropathic pain behavior in Sesn2−/− mice is accompanied by an increased ATF3 induction, we stained lumbar DRGs of Sesn2−/− and WT mice 21 days after SNI using an anti-ATF3 antibody (Fig. 5A). In WT mice, ATF3 was induced in about 22% of all DRG neurons. In Sesn2−/− mice, however, 34% of DRG neurons displayed an induction of ATF3 expression (Fig. 5B). This finding suggests that Sesn2 controls SNI-induced ATF3 induction in a population of sensory neurons.
ROS-induced nociceptive behaviors are increased in Sesn2−/− mice
Having established the functional role of Sesn2 in neuropathic pain processing, we next investigated whether Sesn2 also controls the immediate pain behavior induced by exogenous delivery of ROS donors. For that purpose, we first induced mechanical hypersensitivity by injecting the ROS donor tert-butyl hydroperoxide (TBHP) into the spinal cord of Sesn2−/− and WT mice (18). As shown in Figure 6A, an intrathecal (i.t.) injection of 1.1 μmol TBHP evoked transient mechanical hypersensitivity in both WT and knockout mice. However, at later stages (starting at 45 min after TBHP injection), hypersensitivity was significantly increased in Sesn2−/− mice as compared with WT mice (Fig. 6A). Mechanical hypersensitivity was not observed in both groups after i.t. administration of saline, which was used as a negative control (Fig. 6B).

We then injected TBHP (2.2 μmol) into a hindpaw and measured the evoked licking of the injected paw. While there was no difference in licking behavior between the WT and knockout mice during the first 15 min after the TBHP injection, Sesn2−/− mice continued licking for another 20 min (Fig. 6C), averaging a total licking time more than twice as long as the WT mice (Fig. 6D). No paw licking was observed in both genotypes after the injection of saline into a hindpaw (data not shown). Together, the increased ROS-induced nociceptive behavior at later stages after THBP administration further confirms that Sesn2 is neutralizing pronociceptive ROS in vivo. Moreover, these data suggest that the antinociceptive effect of Sesn2 is not restricted to the injured sciatic nerve, as observed in the SNI model. Sesn2 seems to generally contribute to the control of ROS levels in the nociceptive system at the site of ROS production or delivery.
Inflammatory pain behavior is normal in Sesn2−/− mice
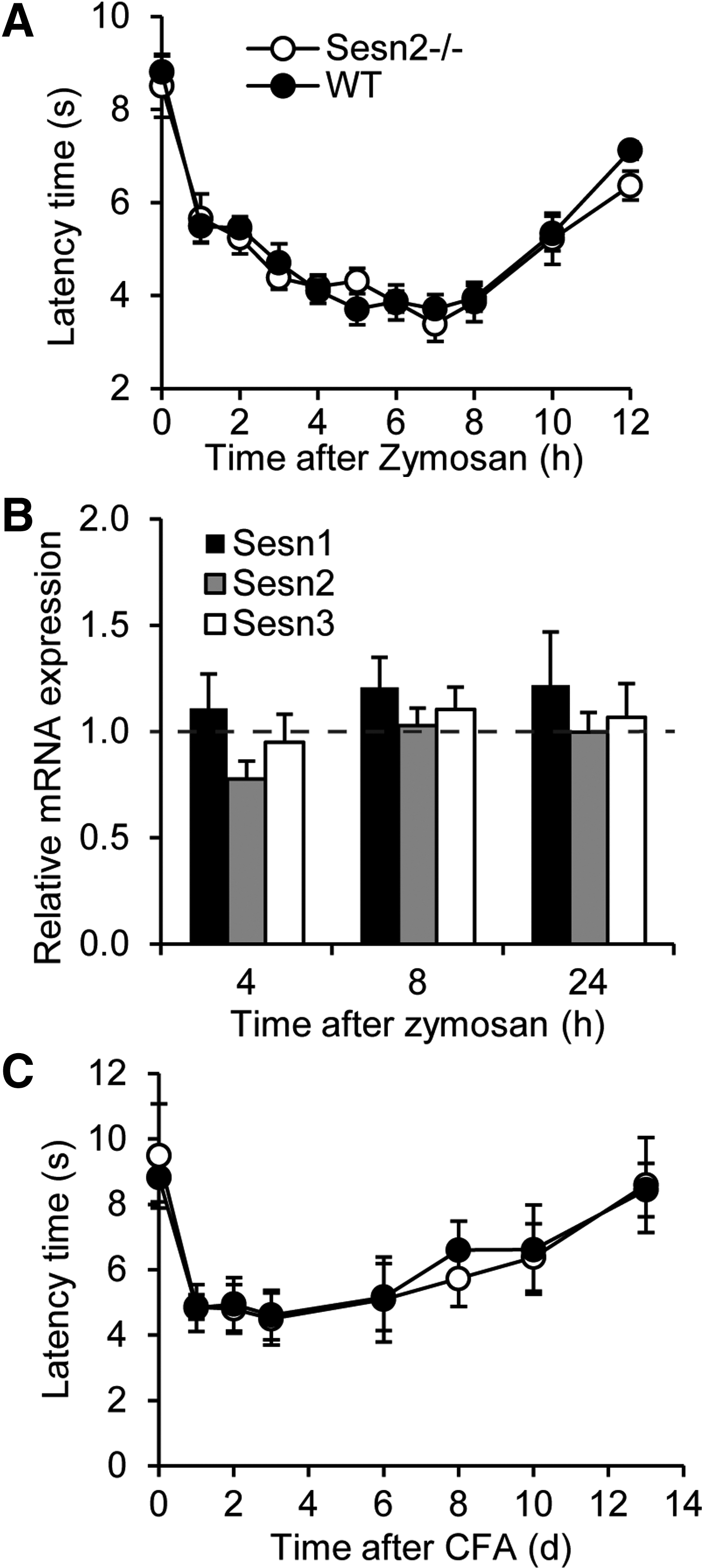
In addition to neuropathic pain, several studies showed that ROS also contribute to the processing of inflammatory pain (37, 38). However, ROS sources and effectors mediating inflammatory pain are believed to be different from those mediating neuropathic pain processing (10, 12, 17, 18), suggesting the involvement of different ROS susceptible signaling pathways. To investigate a potential involvement of Sesn2 in inflammatory pain processing, we characterized the inflammatory pain behavior of Sesn2−/− mice after a hindpaw injection of the proinflammatory yeast extract zymosan. As shown in Figure 7A, Sesn2−/− and WT mice developed mechanical hypersensitivity in the zymosan-injected hindpaws to a similar extent, indicating a limited role of Sesn2 in the processing of inflammatory pain. Moreover, unlike in DRGs of post-SNI mice, Sesn2 mRNA was not up-regulated in DRGs after zymosan injection (Fig. 7B), and zymosan had no effect on Sesn1 and Sesn3 expression in DRGs (Fig. 7B). Finally, we investigated the behavior of Sesn2−/− mice after the injection of complete Freund's adjuvant (CFA) into one hindpaw, a model of long-lasting inflammatory pain (5). The CFA-induced mechanical hypersensitivity, however, was not distinguishable between Sesn2−/− and WT mice (Fig. 7C). These data suggest a limited role of Sesn2 in the processing of inflammatory pain, in contrast to its function in peripheral nerve injury-induced neuropathic pain processing. The data further point to different ROS-dependent signaling pathways in inflammatory and neuropathic pain conditions.
Discussion
In the present study, we identified the antioxidant enzyme Sesn2 as a specific modulator of neuropathic pain processing after peripheral nerve injury. Analyzing the behavior of Sesn2-deficient mice points to an essential function of Sesn2 in degrading elevated ROS levels in peripheral nerves after injury, thereby reducing pronociceptive effects of ROS.
Sestrins are inducible proteins that protect cells against exaggerated ROS levels (2). Here, we show that expression of Sesn2, but not of Sesn1 or Sesn3, is induced in sensory neurons after peripheral nerve injury. It has been reported that Sesn2, in addition to its ROS-protective role, displays ROS-independent properties, including the control of target of rapamycin activity and stimulation of transforming growth factor beta-dependent pathways (2). However, here, we provide several lines of evidence that Sesn2 modulates neuropathic pain processing in an ROS-dependent manner. First, Sesn2−/− mice exhibited increased ROS levels in the sciatic nerve after injury as compared with WT. Since up-regulation of Sesn2 protein was also observed in the injured nerve, we assume that this is the main site of action of Sesn2 in injury-induced neuropathic pain processing. Second, the enhanced neuropathic pain behavior of Sesn2−/− mice was reversed by low doses of the ROS scavenger PBN, which did not demonstrate an antinociceptive effect in WT mice. The most likely explanation for this finding is that the exaggerated ROS levels in Sesn2−/− mice are scavenged by low-dose PBN, whereas higher PBN doses are necessary for scavenging injury-induced, but Sesn2-independent ROS elevations in both WT and Sesn2−/− mice. Finally, i.t. and intraplantar injection of the ROS donor TBHP evoked a prolonged nociceptive behavior in Sesn2−/− mice as compared with WT mice. Hence, Sesn2 obviously contributes to the neutralization of elevated ROS levels in the nociceptive system, thereby controlling pronociceptive effects of ROS.
The fact that ROS levels are increased after peripheral nerve injury has been demonstrated in earlier studies. Important ROS sources are the NADPH oxidases Nox4 and Nox2, which in response to peripheral nerve injury produce ROS in primary afferent neurons and spinal cord microglial cells, respectively (12, 17). In addition, mitochondria in primary afferent neurons and spinal cord dorsal horn neurons increase their ROS production after peripheral nerve injury (5, 23). In contrast to the historical view that ROS are purely harmful, it is now clear that ROS can function as specific regulators of intracellular signaling pathways, in large part through covalent modifications of redox-sensitive cysteine residues within target proteins [for review, see (6, 22)]. Notably, there is much evidence that specific ROS signaling occurs in persistent pain states. For example, superoxides derived from Nox2 induce microglia activation in the spinal cord (12), while H2O2 derived from Nox4 affects myelination properties of injured peripheral nerves (17). Moreover, Nox4−/− mice demonstrated a reduced neuropathic pain behavior, while their behavior was normal in models of acute nociceptive and inflammatory pain (17). In addition, administration of a combination of the antioxidant molecules vitamin C and E specifically inhibited neuropathic pain behavior without displaying any effect on persistent inflammatory pain behavior (18). While these data imply pronociceptive functions of ROS, other studies demonstrated that ROS can also exert antinociceptive effects. For example, redox-reactive compounds such as 5,5′-dithio-bis-(2-nitrobenzoic acid) or α-lipoic acid have been shown to inhibit T-type calcium channel currents in DRG neurons by oxidizing specific thiol residues on the extracellular face of the channel, thereby decreasing cellular excitability of DRG neurons and nociceptive behavior (17, 37, 38). Thus, different ROS sources and species as well as unique localization of target proteins obviously affect distinct pronociceptive and antinociceptive signaling pathways during pain processing. Our observation that Sesn2−/− mice only demonstrated an exaggerated neuropathic but a normal acute and inflammatory pain behavior further confirms the hypothesis that ROS serve as specific messenger molecules during pain processing. Another important conclusion is that antioxidant proteins such as Sesn2 are not merely passive disposers of intracellular oxidants but rather active participants in redox signaling (22).
The antibody we used for western blot analysis failed to detect a specific Sesn2 signal in immunohistochemical stainings, probably due to additional proteins that are recognized by this antibody (Supplementary Fig. S1). Furthermore, we could not detect a specific Sesn2 protein signal using three other Sesn2 antibodies (data not shown). We, therefore, investigated the cellular localization by in situ hybridization and found Sesn2 mRNA to be expressed in DRG neurons. After SNI, the hybridization signal was enhanced and the number of Sesn2 mRNA-positive DRG neurons was increased, confirming the up-regulation of Sesn2 mRNA detected by quantitative real-time RT-PCR. In contrast to these findings, Sesn2 protein was increased only in the injured peripheral nerve after SNI. The most likely explanation for this finding is an axonal transport of the Sesn2 protein synthesized in DRG neurons to the site of injury. However, one should consider that Sesn2 is also expressed in non-neuronal cells [e.g., (4)], and that after peripheral nerve injury, various cells infiltrate to the site of injury (22, 30, 35, 41). We, therefore, cannot exclude the possibility that Sesn2-positive non-neuronal cells contribute to the increased Sesn2 protein levels at the site of injury. Even though the lack of Sesn2 mRNA detection in the injured sciatic nerve using in situ hybridization speaks against this possibility, further studies using specific tools to detect Sesn2 protein will be needed to elucidate the origin of Sesn2 in the injured nerve.
Our observation that the SNI-induced ATF3 induction in DRG neurons is increased in Sesn2−/− mice as compared with WT mice provides further evidence for a neuronal localization of Sesn2 in DRGs. ATF3 is a transcription factor that is induced by neuronal stress such as peripheral nerve damage and is considered a marker of neuronal injury (39). It belongs to a family of basic-region leucine zipper transcription factors regulating multiple cellular processes. Depending on the interaction with other proteins, ATF3 can function as an activator or repressor of transcription (36). The exact functions of ATF3 induction after peripheral nerve injury are poorly understood, but it is presumably associated with regenerative properties of injured peripheral nerves (15, 25, 31). Interestingly, only recently it has been demonstrated that glutathione depletion induces ATF3 expression in human monocytes, suggesting an ROS-dependent induction of ATF3 (9). Thus, it is likely that the elevated ROS levels in Sesn2−/− mice lead to increased ATF3 induction in DRG neurons, resulting in an exaggerated stress response after peripheral nerve injury.
Altogether, our results demonstrate that the antioxidant protein Sesn2 controls persistent neuropathic pain processing after peripheral nerve injury. Hence, specific activation or induction of Sesn2 in the nociceptive system might be a novel strategy to inhibit neuropathic pain.
Materials and Methods
Animals
Experiments employed C57BL6J WT and C57BL6J Sesn2−/− mice (RRJ141/Sesn2Gt(RRJ141)Byg ). RRJ141 (C57BL6J/129P2) mice were rederived at UC-Davis/USA from frozen embryos distributed by the UC-Davis branch of the Mutant Mouse Regional Resource Centers (MMRC) and back-crossed to C57BL6J mice for at least 10 generations before use. Genotyping was performed by genomic PCR using mouse tail DNA and primers complementary to sequences flanking the gene trap insertion site. All primers are available on request. Behavioral experiments were performed in 6- to 12-week-old animals of either sex. In addition, male C57BL6N mice (Harlan) were used for RT-PCR and western blot analyses. Animals were housed on a 12/12 h light/dark cycle with free access to water and food. Experiments were approved by the local Ethics Committee for Animal Research and were performed in accordance with the International Association for the Study of Pain.
Real-time RT-PCR
Total RNA of the entire lumbar spinal cords (L4-L5) and DRGs (L4-L5) was extracted under RNase-free conditions using an RNA isolation kit (for spinal cord: RNeasy Lipid Tissue Mini Kit, Qiagen; for DRGs: RNAqueous Micro Kit, Ambion), according to the manufacturer's instructions. To minimize genomic DNA, contamination samples were treated with DNase for 15 min and quantified using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies). Reverse transcription was performed with a Verso Kit (Thermo Scientific) according to the manufacturer's instructions. Quantitative real-time RT-PCR was performed with an ABI Prism 7500 Sequence Detection System (Applied Biosystems) using TaqMan Gene Expression Assays for murine Sesn1 (Mm01185732_m1), Sesn2 (Mm00460686_m1), Sesn3 (Mm01171504_m1), β-actin (Mm00607939_s1), and GAPDH (Mm99999915_g1), purchased from Applied Biosystems. Reactions (total volume, 10 μl) were performed in duplicate or triplicate by incubating for 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Water controls were included to ensure specificity. Relative gene expression levels were calculated using the comparative 2-ΔΔCt method and normalized to GAPDH. The Ct value represents the cycle number at which the signal of the PCR product crosses a defined threshold that is set within the exponential phase of the PCR. Control experiments revealed similar results if β-actin was used for normalization.
In situ hybridization
Probe generation and in situ hybridization was performed as previously described (8). Briefly, cDNA synthesis was performed with gene specific primers 5′-gga gtg tct atc gcc gta ga-3′ and 5′-taa gaa cac tgg tga cgg gc-3′ corresponding to the nucleotides 140–664 of Sesn2 mRNA (NCBI accession number NM_144907.1). Control experiments were performed with a second fragment corresponding to nucleotides 1665–2198 of Sesn2 mRNA designed with the primers 5′- tgt gaa ctt gct gct cct tg-3′ and 5′- gaa tga gga agg agg gga gg-3′. PCR products were cloned into the pCR4-TOPO plasmid vector (Invitrogen), amplified in One Shot TOP10 Chemically Competent Escherichia coli cells (Invitrogen), and plasmid isolation was performed using the Qiaprep Spin Miniprep Kit. After sequence verification (LGC Genomics), restriction digestions were performed using PmeI and NotI, and sense and antisense probes were transcribed using T3 or T7 RNA polymerases (Roche Diagnostics).
Mice were intracardially perfused with 4% paraformaldehyde (PFA) in 0.1 M phosphate-buffered saline (PBS), pH 7.4. DRGs and sciatic nerves were dissected, postfixed for 10 min and cryoprotected in 20% sucrose. Cryostat sections were cut at a thickness of 14–16 μm. Sections were incubated with 3% H2O2 for 5 min, postfixed in 4% PFA for 5 min, and prehybridized in hybridization buffer (50% formamid, 5×SSC, 5×Denhardt's solution, 500 μg/ml herring sperm DNA, and 250 μg/ml yeast tRNA in nuclease-free water) at 65°C for 2 h. Sesn2 sense or antisense probe was incubated over night at 65°C. Slides were washed in 0.2×SSC and PBS, blocked for 2 h in blocking buffer (0.12 M maleic acid, 0.15 M NaCl, pH 7.5, 1% Blocking Reagent, Roche Diagnostics), and incubated with alkaline phosphate-conjugated anti-digoxigenin antibody (Roche Diagnostic). Sections were equilibrated in alkaline phosphate buffer (0.1 M Tris-HCl, 0.1 M NaCl, 0.05 M MgCl2, pH 9.5, 0.2 mM levamisol, and 0.2% Tween) and incubated with BM-Purple (Roche Diagnosics). In situ hybridization was analyzed using an Axio Observer.Z1 microscope (Zeiss) equipped with a monochrome CCD camera (AxioCam Mrm; Zeiss). Images were taken using different filters, pseudocolored, and superimposed with the Zeiss AxioVision 4.7.2 software. Final adjustment of contrast and intensity was done using Adobe Photoshop CS software.
Western blot
For Western Blot analysis, entire lumbar spinal cords (L4-L5), DRGs (L4-L5), and sciatic nerves were rapidly dissected and snap frozen in liquid nitrogen. Samples were homogenized in Phosphosafe buffer (Novagen) mixed with a protease inhibitor cocktail (Complete Mini; Roche Diagnostics) and centrifuged at 14000g for 1 h to remove cellular debris. Protein extracts were separated by sodium dodecyl sulfate-polyacrylamid gel electrophoresis and transferred onto a nitrocellulose membrane. Ponceau red staining was used to confirm equal sample loading. Membranes were blocked in blocking buffer (Odyssey blocking buffer, LI-COR Bioscience; diluted 1:1 in PBS) for 1 h at room temperature (RT) and then incubated with rabbit anti-Sesn2 (1:200; Proteintech) dissolved in blocking buffer containing 0.1% Tween-20. After incubation with secondary antibodies, proteins were detected on an Odyssey Infrared Imaging System (LI-COR Bioscience). GAPDH (1:2000, Ambion) was used as a loading control. Densitometric analysis was performed using the NIH ImageJ software.
Immunohistochemistry
Immunofluorescence staining of spinal cord and DRG slides was performed as previously described (8, 17). Briefly, animals were intracardially perfused with 4% PFA, and dissected lumbar spinal cord (L4-L5) and DRGs (L4-L5) were cryostat sectioned at a thickness of 14–16 μm and stored at −80°C until use. Sections were permeabilized, blocked in 10% normal goat or donkey serum and 3% bovine serum albumin in PBS, and incubated with primary antibodies. The following antibodies were used: rabbit anti-calcitonin gene-related peptide (1:500; Abcam), rat anti-substance P (1:200; BD Biosciences), mouse anti-NF200 (clone N52; 1:1000; Sigma Aldrich), and rabbit anti-ATF3 (1:100; Santa Cruz). After rinsing in PBS, slides were incubated with secondary antibodies conjugated with Alexa Fluor 488 (Invitrogen) or Cy3 (Sigma-Aldrich). Alexa Fluor 488-conjugated Griffonia simplicifolia isolectin B4 (IB4; 3 μg/ml in PBS; Invitrogen) was incubated for 2 h at room temperature. To reduce lipofuscin-like autofluorescence, slides were incubated in 0.06% Sudan black B in 70% ethanol (27, 29), rinsed in PBS, and coverslipped in Flouromount G (Southern Biotech). Images were taken using an Axio Observer.Z1 microscope as described earlier.
Cell counting
Sections of L4-L5 DRGs were processed as described earlier. Cell counts were performed on randomly selected sections with at least 60 μm apart. The number of DRG neurons positive for Sesn2 mRNA, substance P, IB4, NF200 or ATF3, and the total number of DRG neurons were counted and the percentage was calculated.
H2O2 detection
Relative H2O2 levels were measured using Amplex Red (Invitrogen). The assay is based on a reaction of the colorless Amplex Red reagent with H2O2 to produce fluorescent resorufin. Lumbar spinal cords (L4-L5), DRGs (L4-L5), and sciatic nerves of control and SNI operated animals were rapidly dissected, weighted, and homogenized in ice-cold 500 mM phosphate buffer containing 10 mM sodium azide, incubated for 5 min at RT, and centrifuged at 10,000g for 10 min at 4°C. The supernatant was transferred into a 96-well plate (μCLEAR, BLACK, Greiner Bio One), and samples and H2O2 standards were incubated with 100 μM Amplex Red and 0.2 U/ml horseradish peroxidase for 30 min at RT. Fluorescence intensities were acquired at an excitation of 540 nm and an emission of 595 nm.
Behavioral tests
Behavioral studies were done by a blinded observer. Littermate mice of either sex were used in all investigations. Similar numbers of male and female mice per genotype were used, because sex differences in pain perception have been observed (7, 23, 24).
Rotarod test
Motor coordination was assessed with a Rotarod treadmill for mice (Ugo Basile) at a constant rotating speed of 32 rpm. After several training sessions, fall-off latencies from 3–5 tests were averaged, using a cutoff time of 120 s.
Hot- and cold-plate test
Mice were placed on a heated or cooled metal plate surrounded by a Plexiglas cylinder (Hot/Cold Plate; Ugo Basile). In the hot-plate test, the time between placing and the first nociceptive reaction (shaking or licking) of a hindpaw was recorded. Temperatures of 50°C, 52°C, and 54°C were used with cutoff times of 60, 40, and 20 s, respectively, to prevent tissue damage. For the cold-plate test, the number of paw withdrawal responses, licking the hindpaw, or jumping during 5 min at a temperature of 0°C was counted (16).
Dynamic-plantar test
Mechanical sensitivity of the hindpaw was measured using a Dynamic Plantar Aesthesiometer (Ugo Basile). This device pushes a thin steel rod against the plantar surface of the paw from beneath, and automatically stops and records the latency time at which the animal withdraws the paw. The force was increased constantly from 0 to 5 g in 10 s (ramp 0.5 g/s) and remained at 5 g for an additional 10 s (8, 28). Paw withdrawal latency was calculated as the mean of 4–6 measurements with at least 20 s in between.
Neuropathic pain
The SNI model (3) was used to investigate neuropathic pain behavior. Surgery was carried out under isoflurane anesthesia. Two branches of the sciatic nerve were ligated and cut distally, leaving the sural nerve intact, which leads to a hypersensitivity of the lateral surface of the hindpaw (sural nerve skin area). Paw withdrawal latencies were determined using the dynamic-plantar test. Phenyl N-tert-butyl-α-phenylnitrone (PBN, Sigma Aldrich) dissolved in saline was administered i.p. 22 days after SNI, and paw withdrawal latencies were measured 1–24 h after drug injection. PBN doses were selected on the basis of previous reports (14, 33).
TBHP-induced hypersensitivity
Intrathecal drug delivery was performed by direct lumbar puncture in awake, conscious mice as described earlier (18). TBHP (Sigma Aldrich) dissolved in saline was i.t. injected in a volume of 5 μl, and paw withdrawal latency times were measured using the dynamic-plantar test every 15 min over a period of 2 h. For peripheral drug delivery, TBHP dissolved in saline (20 μl) was injected subcutaneously into the dorsal surface of a hindpaw, and the time spent licking the TBHP-injected paw was measured every 5 min over a period of 45 min after drug injection. The TBHP doses were selected on the basis of previous studies (18) and our preliminary work.
Inflammatory pain
A zymosan A suspension (15 μl, 5 mg/ml in 0.1 M PBS, pH 7.4; Sigma-Aldrich) or 20 μl of CFA (containing 1 mg/ml heat-killed Mycobacterium tuberculosis in 85% paraffin oil and 15% mannide monooleate; Sigma-Aldrich) was injected subcutaneously into the plantar side of a hindpaw (20). Paw withdrawal latencies were determined using the dynamic-plantar test.
Statistical analysis
Statistical analysis was performed with SPSS software. For paired comparisons, the Student's t-test was used; multiple comparisons were analyzed using one-way ANOVA followed by a Fisher post hoc test. The Mann–Whitney U-test was used to compare rotarod fall-off latencies, and these data are presented as median and interquartile range. Other data are expressed as the mean±standard error of the mean. For all tests, a probability value p<0.05 was accepted as statistically significant.
Footnotes
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (SFB815-A14). The authors thank Karin Schilling, Christine Manderscheid, and Judith Fuchs for excellent technical assistance.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.