
Abstract
Introduction

The Discovery of DsbD
Although it is now clear that the bacterial cell envelope harbors a large array of redox enzymes, the presence of redox reactions in the bacterial periplasm was thought to be rather limited. It is the discovery of DsbA in 1991 that opened the way to the study of the redox processes taking place in the envelope (2). The early studies in the field focused on DsbA and DsbB, the two proteins that function in the disulfide formation pathway. Then, in 1994, two different groups independently reported the identification of an additional periplasmic oxidoreductase, which they called DsbC (41, 59). When DsbC was discovered, the protein was first proposed to catalyze disulfide bond formation in the periplasm. However, it is now clear that DsbC rather functions as a protein disulfide isomerase (Fig. 1) correcting the non-native disulfides that can be introduced by DsbA in proteins with multiple cysteine residues (see below).
DsbC is a V-shaped soluble homodimeric protein in which each subunit presents both a catalytic domain and a dimerization domain linked together by an α-helix (39, 67). The catalytic domain adopts a thioredoxin fold and possesses a CXXC motif that is kept reduced in the periplasm (25, 52). This enables DsbC to catalyze the isomerization of incorrect disulfides: the reaction starts by a nucleophilic attack of the first cysteine of the CXXC motif on the non-native disulfide, which results in the formation of a mixed-disulfide between DsbC and the substrate protein. Then, the mixed disulfide undergoes a nucleophilic attack either by another reduced cysteine in the substrate or by the second cysteine of the CXXC motif. In the first scenario, DsbC is released in the reduced state and acts as a true isomerase. In the second scenario, DsbC is released in the oxidized state and acts as a reductase, while the non-native disulfide of the substrate is reduced. The substrate then needs one or more rounds of oxidation by DsbA to acquire its native disulfides. This second scenario is supported by a study showing that expression of a thioredoxin-like protein that only exhibits a reductase activity is able to complement an E. coli dsbC mutant (60).
To allow DsbC to react with non-native disulfides, it is important to maintain the CXXC motif of this protein in the reduced state. A search for mutations that restore disulfide bond formation in a dsbA-null strain led to the identification of DsbD, the protein that provides reducing equivalents to DsbC (42, 51).
Properties of
E. coli
DsbD
DsbD is a 59-kDa monomeric protein, which is located in the cytoplasmic membrane. DsbD has three distinct domains: an N-terminal periplasmic domain (DsbDα) with an immunoglobulin fold, followed by a membrane-embedded domain (DsbDß) with eight transmembrane segments (TM), and finally, a second periplasmic domain (DsbDγ), with a thioredoxin fold. Each domain of DsbD possesses a pair of redox-active cysteines that are essential for the activity of the protein (Fig. 2) (10, 62). These cysteines form a relay that shuttle electrons from cytoplasmic thioredoxin to periplasmic oxidoreductases via a cascade of thiol–disulfide exchange reactions: electrons are first transferred from the reduced Cys33 and Cys36 residues of the catalytic motif of thioredoxin 1 (Trx1) to the cysteine residues of DsbDß and then successively to the cysteines of DsbDγ and DsbDα. DsbDα then reduces substrate proteins, such as DsbC (28). After donating its electrons to DsbD, Trx1 is released in the oxidized state. It is then converted back to its reduced state by thioredoxin reductase at the expense of nicotinamide adenine dinucleotide phosphate (NADPH). The reaction catalyzed by DsbD is thermodynamically driven as electrons flow from the more reducing protein, Trx1 (E°′=−270 mV), to DsbDß (−246 mV), then to DsbDγ (−241 mV) and DsbDα (−239 mV), and finally to oxidoreductases with significantly more oxidizing redox potentials (DsbC has a redox potential of −130 mV) (11, 55, 67).

The reaction between DsbDγ and DsbDα has been thoroughly characterized using a combination of biochemical and biophysical techniques (36 –38, 54, 56). Moreover, the structures of the two periplasmic domains, either as independent entities or in complex, have been solved (19, 22, 31, 56). In contrast with the abundance of details regarding the interaction between DsbDγ and DsbDα, we only have a partial understanding of the mechanism used by DsbDß to transfer electrons across the membrane. The current working model for the structure of DsbDβ was built by Beckwith and his coworkers (7, 9, 29). The model was constructed by probing the accessibility of selected membrane-embedded residues to membrane-impermeable alkylating reagents. These experiments revealed that the catalytic cysteine residues of DsbDβ (Cys163 and Cys285), which are located in TM1 and TM4, respectively, are exposed to both sides of the membrane. The residues located at the C-terminus of TM1 and TM4 are exposed to the aqueous environment, whereas the residues located at the N-terminus are not (Fig. 3). Moreover, analysis of the amino acid sequence of DsbDß suggests that TM1–3 and TM4–6 present an antiparallel architecture (32). These results, together with the fact that Cys163 and Cys285 can form a mixed disulfide complex with Trx1 (28) and DsbDγ (9), respectively, provide support for a model in which DsbDβ adopts an hourglass structure (9). In this structure, the catalytic cysteine residues are located at the juncture of the two cavities where they are exposed to both thioredoxin in the cytoplasm and DsbDγ in the periplasm (Fig. 3). Interestingly, the water accessibility of the TM residues was shown to be independent of the redox state of the protein, which suggests that the conformation of DsbDß is largely the same in its oxidized and reduced forms (7).
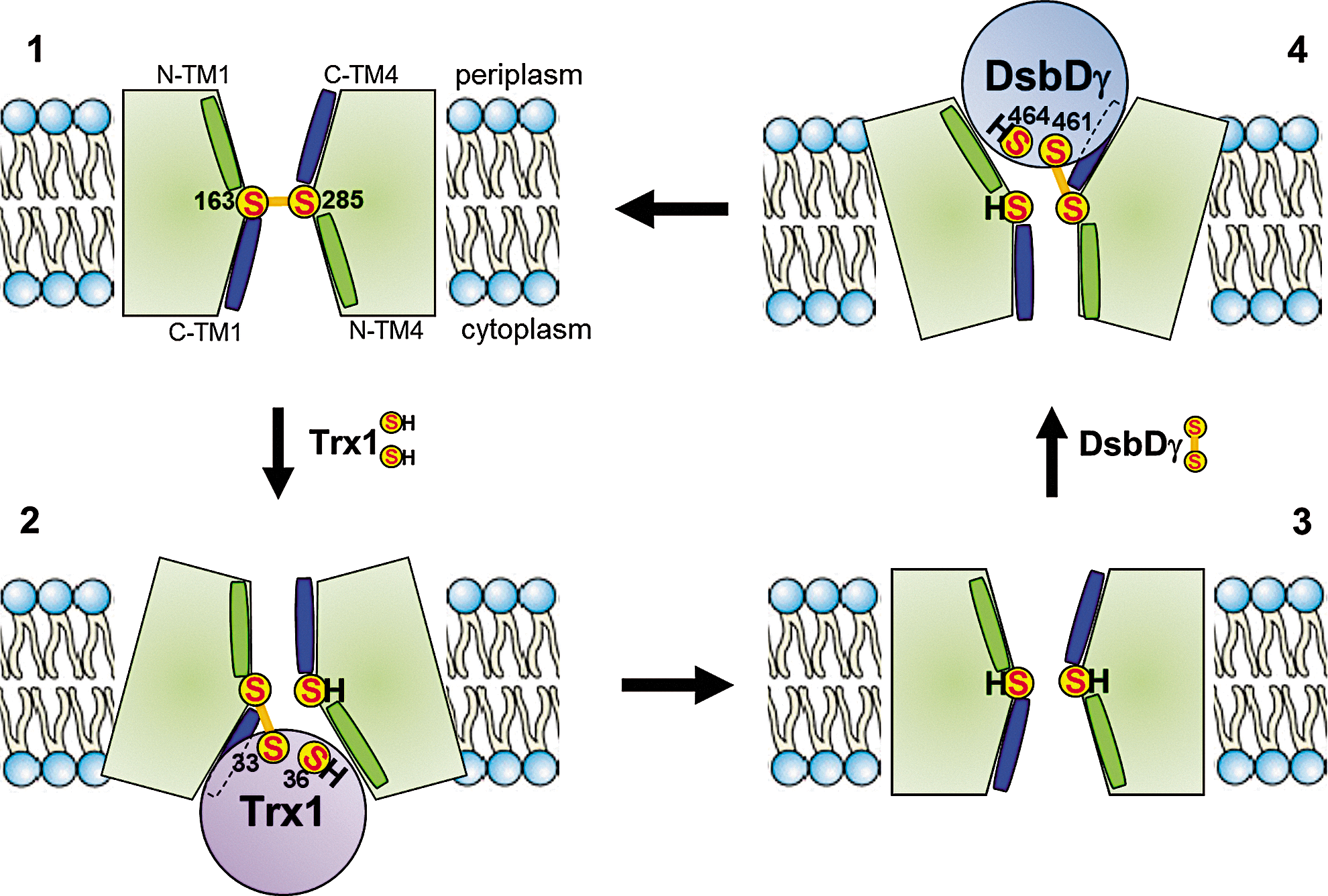
The properties exhibited by DsbDß are reminiscent of those of channel-like transporters, such as aquaporins and the protein-conducting channel SecYEG complex (43, 64). Channels usually adopt an hourglass-like structure whose both sides remain steadily open while the central pore is maintained small enough to allow the passage of specific ligands (note that in the case of SecY, there is a removable plug in the central pore). In contrast, many pump-like membrane transporters alternate between an open and a closed conformation depending on the binding of ligands (18).
A particularly intriguing question regarding DsbD is how DsbDß alternatively interacts with Trx1 and DsbDγ. An attractive hypothesis is that Trx1 may interact first with some membrane-embedded residues of DsbDß, which would trigger major conformational changes within DsbD and allow the subsequent formation of the mixed-disulfide complex between these two proteins. As shown in Figure 3, DsbDγ and DsbDβ would interact in the same way. The crystal structure of the SecA–SecYEG complex provides support to this hypothesis (68): the binding of SecA, a soluble cytoplasmic protein, to the SecYEG complex leads indeed to conformational changes within the membrane domain of SecY. This subsequently allows specific helices from SecA to displace structural elements within SecY, allowing protein translocation.
The DsbD Family Is Diverse
A recent bioinformatic analysis of bacterial genomes has shown that homologs of E. coli DsbD are found in many bacteria, and that they can be divided into three classes: the DsbD-like proteins, the CcdA-like proteins, and the ScsB-like proteins (Fig. 4) (8). As E. coli DsbD is the prototype of the first class, this subgroup will not be further discussed here.
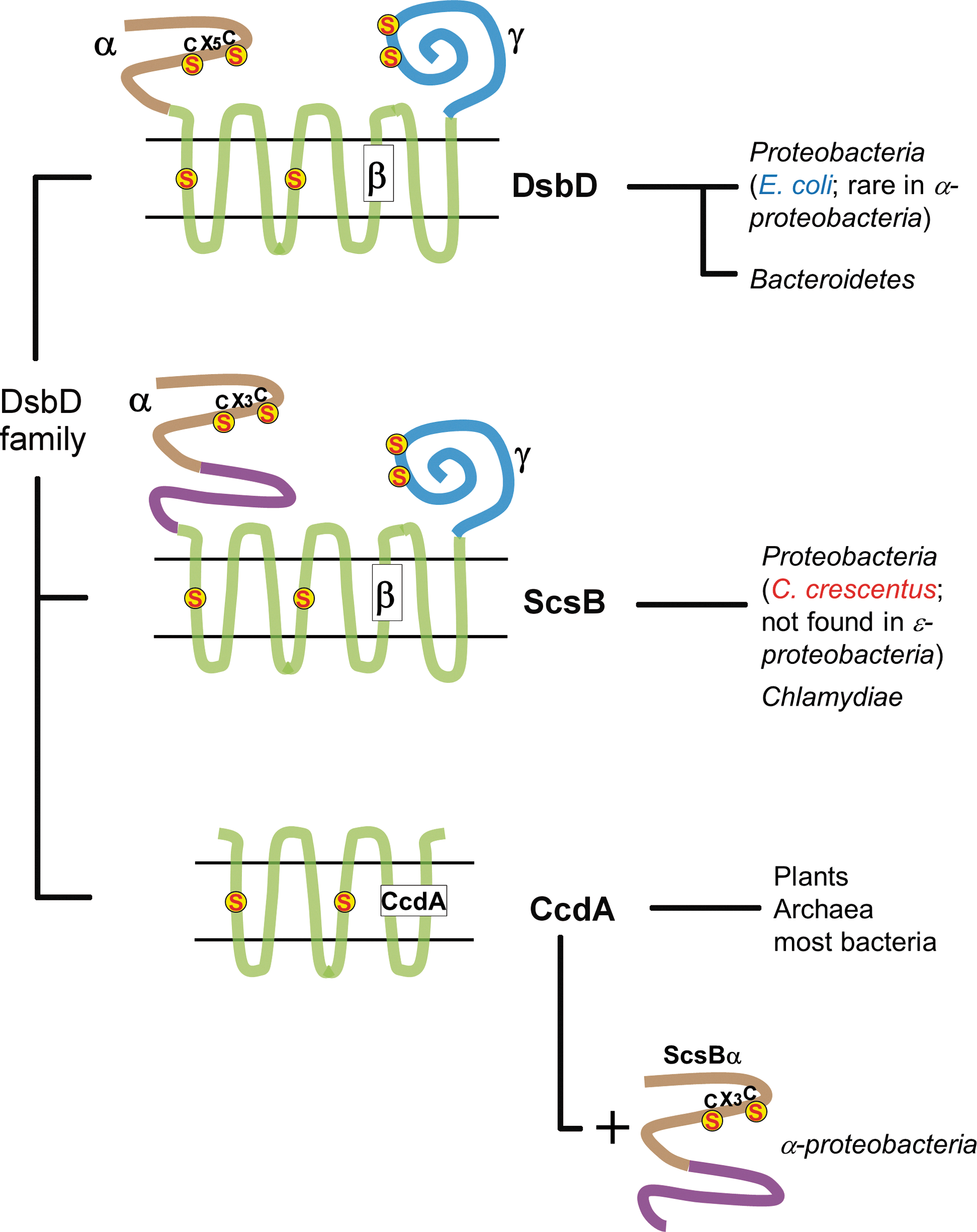
Some bacteria, such as Rhodobacter capsulatus, contain a homolog of DsbD, which is called CcdA (15). CcdA is also found in plants and archaea, and appears to have more broadly evolved compared to the other DsbD family members (8, 30, 45). CcdA is a stripped-down version of DsbD, which does not possess the two periplasmic domains (30) and only comprises six TMs instead of the eight found in E. coli DsbD (Fig. 4). It has been shown that R. capsulatus CcdA can provide electrons to separately encoded thioredoxin-like proteins such as CcmG (details in DsbD provide electrons to a variety of redox periplasmic pathways section). In contrast, the E. coli homolog of CcmG receives electrons from the periplasmic soluble domain of DsbD (DsbDα). Therefore, it can be postulated that DsbD evolved from the fusion of a primitive CcdA-like protein with two soluble periplasmic proteins to facilitate electron transfer (30).
In a recent study, we identified a third distinct class of DsbD-like homologs (8) (Fig. 4) found in proteobacteria and Chlamydiae. The prototype of this new class is Salmonella typhimurium ScsB (suppression of copper sensitivity), a protein that has been shown to confer copper tolerance to E. coli copper-sensitive mutants (21). Like DsbD, the ScsB class has a three-domain structure, each domain, including a pair of cysteines, active in the transfer of electrons. The transmembrane domain (ScsBβ) has eight TMs, and the C-terminal domain (ScsBγ) has a thioredoxin fold. However, the N-terminal domain (ScsBα), which is presumably the final electron donor for substrate proteins, differs significantly from DsbDα (Fig. 4) and acts on a different array of substrate proteins from those already known for DsbD (see below). One interesting feature of ScsBβ is that its N-terminal part (N-subdomain), which harbors the two catalytic cysteines, is relatively well conserved, in contrast to the C-terminal part of ScsBβ (C-subdomain), which is not (Fig. 4). Some organisms do not even have the C-subdomain of ScsBα, which suggests that the acquisition of this subdomain is related to a different substrate specificity. Interestingly, many α-proteobacteria do not express a full-length ScsB, but rather two independent proteins corresponding to ScsBα and either CcdA or ScsBβγ. The co-occurrence of these proteins suggests that they cooperate in electron transfer and supports the gene fusion hypothesis advanced above (Fig. 4).
DsbD Provides Electrons to a Variety of Redox Periplasmic Pathways
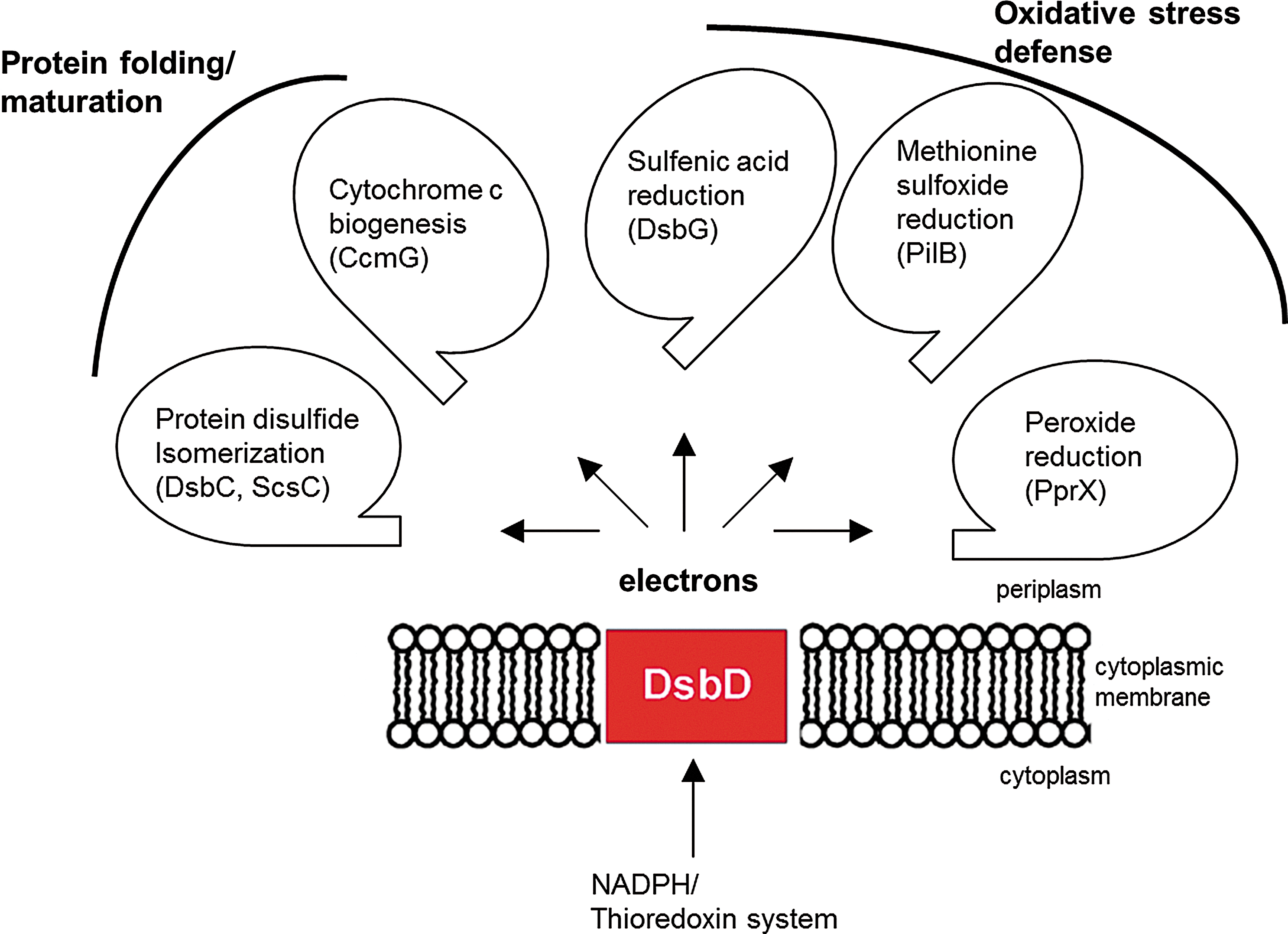
In the previous sections, we focused on the main properties of DsbD and on the diversity of the DsbD family. We will now review recent data that highlight the function of DsbD as an electron hub dispatching reducing equivalents to various redox pathways present in the cell envelope.
DsbD provides electrons for protein folding and maturation
The best-documented function of DsbD proteins is to provide electrons to periplasmic protein disulfide isomerases, such as E. coli DsbC. Envelope proteins are synthesized in the cytoplasm as unfolded polypeptides and are then transported in an unfolded state across the inner membrane. It has been shown that DsbA preferentially introduces disulfides in a vectorial manner in polypeptides entering the periplasm (26). Thus, if disulfides need to be formed between cysteine residues that are not consecutive in the sequence, for example, between Cys1 and Cys3 and between Cys2 and Cys4, DsbA will likely introduce non-native disulfides (Cys1–Cys2 and Cys3–Cys4) in the secreted protein. The function of DsbC and of other periplasmic protein disulfide isomerases, such as the recently described Caulobacter crescentus ScsC (8), is to correct these non-native disulfides using the catalytic mechanism described in The Discovery of DsbD section.
While about half of the E. coli envelope proteins are known or predicted to be DsbA substrates, the number of DsbC substrates is more limited. The list of DsbC substrates includes a few soluble periplasmic enzymes with multiple cysteine residues, such as ribonuclease I (RNase I; 8 cysteines, 1 nonconsecutive disulfide), endonuclease 1 (End1; 8 cysteines, 1 nonconsecutive disulfide), a murein endopeptidase (MepA; 6 cysteines, 3 nonconsecutive disulfides), and a phytase (AppA; 8 cysteines, 1 nonconsecutive disulfide) (3, 23, 35, 40, 65). Noteworthy, a new artificial DsbC substrate has been developed, which confers a semiquantification genetic selection of DsbC or DsbD mutants simply by correlating bacteria ampicillin resistance to DsbC and DsbD activity (50).
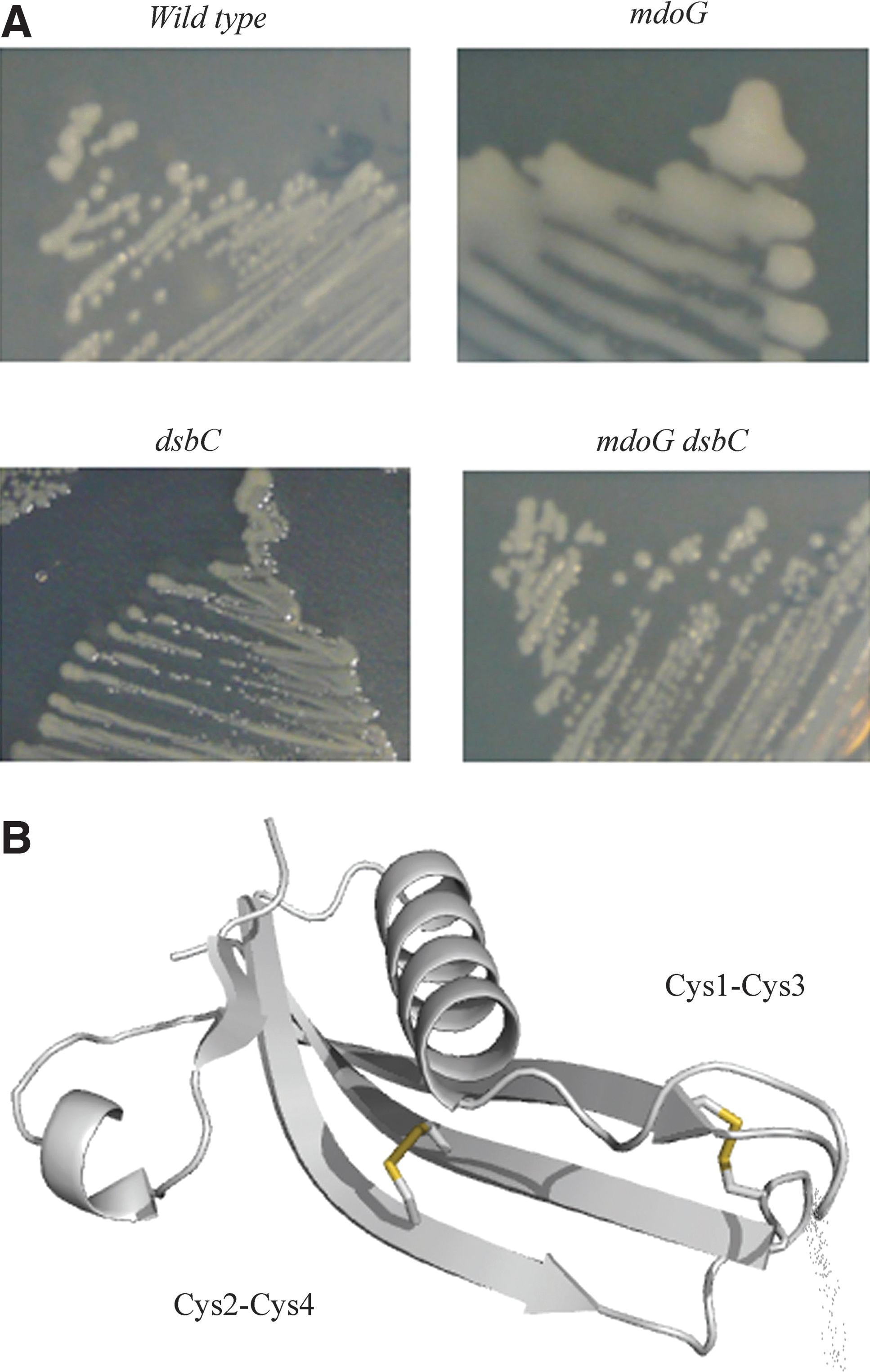
Recently, we showed that DsbC is also involved in the oxidative folding of two proteins that localize in the outer membrane (OM), the ß-barrel protein LptD and the lipoprotein RcsF (12, 34). These two OM substrates of DsbC play an important role in the cell: LptD is an essential protein that inserts lipopolysaccharides in the OM, while RcsF is a sensor that upon detection of damages occurring in the cell envelope activates a signaling cascade. The altered oxidative folding of these proteins in strains lacking dsbC leads to specific phenotypes that can be attributed to the lack of disulfide bond formation. This is particularly striking in the case of RcsF: the protein has two nonconsecutive disulfide bonds (Cys1–Cys3 and Cys2–Cys4) that are essential for folding and activity. Failure to form these two disulfides leads to the degradation of RcsF by periplasmic proteases, which renders the bacteria unable to detect and respond to certain envelope perturbations. For instance, deletion of mdoG, a gene coding for periplasmic glycans that play an osmoprotectant role, leads to the constitutive activation of the Rcs phosphorelay. This constitutive activation depends on RcsF and causes the production of colanic acid, an exopolysaccharide that accumulates at the cell surface, yielding a dramatic mucoid phenotype (Fig. 5). However, the decreased levels of properly folded RcsF in mdoG dsbC double-mutants do not allow activation of the Rcs phosphorelay, as illustrated by the nonmucoid phenotype of the double mutant (Fig. 5). Noteworthy, one of the two RcsF disulfide bonds bridges two cysteine residues that are present on two adjacent β-strands (Fig. 5), forming a so-called cross-strand disulfide (CSD). These CSD are described as notoriously unstable (66), which suggests that RcsF may be redox regulated.
DsbD plays an additional role in protein assembly by providing electrons to thioredoxin-like proteins, such as E. coli CcmG, which are involved in cytochrome c maturation in the periplasm (Fig. 6). Cytochromes c are covalently attached to heme groups via thioether bonds between the tetrapyrrole ring of these groups and two cysteine residues of a CXXCH motif of the apoprotein. During the folding of the apocytochromes c in the periplasm, the cysteine residues of the CXXCH motif are thought to be oxidized to a disulfide by DsbA, probably to protect the apocytochromes from degradation (57). Reduction of this disulfide is then required to allow the covalent attachment of heme to the protein. This reaction is carried out by CcmG, which is maintained reduced in the periplasm by DsbD (16, 44, 63).
The roles of DsbD in the defense mechanisms against oxidative stress
Oxidative stress occurs when cells are exposed to elevated levels of reactive oxygen species (ROS) such as superoxide (O2 −), hydrogen peroxide (H2O2), and alkyl hydroperoxides (ROOH) (53). Oxidative stress can cause DNA damage and mutations, disassembly of iron–sulfur clusters, and lipid peroxidation. It also affects proteins by oxidizing cysteine and methionine residues, which can lead to the inactivation of cellular proteins and cellular death. The presence of protection mechanisms against oxidative stress is therefore essential for cell survival. When DsbD was discovered, the function of the protein was thought to be restricted to providing electrons for disulfide isomerization and cytochrome c maturation. However, recent data obtained on E. coli, Neisseria meningitides, and C. crescentus, three gram-negative bacteria, have revealed that DsbD also plays an important role in the defense mechanisms against oxidative stress by providing electrons to envelope pathways that either reduce proteins that have been oxidatively damaged, or directly reduce harmful ROS in the cell envelope (6, 8, 13).
DsbD provides electrons for sulfenic acid reduction in the envelope
E. coli DsbG is a periplasmic protein that shares 26% sequence identity with DsbC. Like DsbC, it is also a V-shaped dimeric protein whose CXXC catalytic motif is kept reduced by DsbD (4). Because of the similarities between DsbC and DsbG, DsbG was proposed to function as a second protein disulfide isomerase in the E. coli periplasm. However, a search for DsbG substrates led us to find out that this is not the case. We found that the function of DsbG is to protect periplasmic proteins that contain single cysteine residues from oxidation (Fig. 6) (13). We identified three
DsbD provides electrons for methionine sulfoxide reduction in the envelope
The examples of reductive pathways we have talked about so far are largely the ones that are specific to the bacterial cell envelope. However, it has recently become clear that certain antioxidant enzymes that have previously been detected in the cytoplasm of cells have their counterparts in the bacterial periplasm. For example, methionine residues, like cysteine residues, are also sensitive to ROS and can be oxidized to methionine sulfoxides (MetO). Two diastereoisomers of MetO are generated, referred to as R and S, owing to the asymmetric position of the sulfur atom in the lateral chain. The oxidation of methionine residues in proteins leads, if unrepaired, to changes in hydrophobicity, alterations in protein conformation, and loss of biological activity. For this reason, most cells contain methionine sulfoxide reductases (Msr) that catalyze the reduction of MetO back to methionine [see the following review: (5)]. The bacterial cytoplasm contains MsrA and MsrB proteins that reduce the S and R form of MetO, respectively (5), the electrons being provided to them by the thioredoxin system.
The discovery of a periplasmic protein PilB involved in MetO reduction in Neisseria gonorrhoeae revealed that such antioxidant enzymes can be found in both the cytoplasm and the cell envelope (Fig. 6) (61). PilB is composed of three domains. The N-terminal domain has a thioredoxin fold and functions as a reductase, while the second and third domains are homologous to MsrA and MsrB, respectively (6). DsbD plays a role in periplasmic MetO reduction by providing electrons to the N-terminal domain of PilB. The latter then transfers the electrons to the MsrA and MsrB domains of the protein. Interestingly, deletion of dsbD was shown to increase the oxidative stress sensitivity of N. meningitidis mutants lacking the genes coding for two DsbA (dsbA1 and dsbA2). It has been proposed that the increased sensitivity may result from the lack of reducing equivalents delivered to the N. meningitidis PilB homolog (33).
Although the PilB protein is only found in a limited number of bacterial species, other bacteria contain gene clusters involving a CcdA-like protein and periplasmic homologs of thioredoxin, MsrA and MsrB. It seems likely that these proteins cooperate in periplasmic MetO reduction, illustrating the importance of this process in the cell envelope.
DsbD provides electrons for peroxide reduction in the envelope
In the cytoplasm, in addition to enzymes that reduce oxidatively damaged proteins, bacterial cells also express several catalases and thiol-dependent peroxidases, such as peroxiredoxins (Prxs), to directly scavenge harmful peroxides. Catalases deal with high concentrations of peroxides (mM), while Prxs are the major scavengers for peroxides generated at physiological concentrations (μM) (58).
Prx enzymes utilize a reactive cysteine residue to attack the O–O bond of the substrate, which leads to the formation of sulfenic acid. The latter is then attacked by a second cysteine residue, generating an intra- or intermolecular disulfide bond, depending on whether the resolving cysteine belongs to the same or to another Prx molecule. The Prx activity is then regenerated by reduction of the disulfide bond, a reaction that is usually catalyzed by proteins from the thioredoxin family [recently reviewed in (17, 47)]. While cytoplasmic peroxides scavengers are widespread in bacteria, none had been identified in the cell envelope. A recent search for substrates of C. crescentus ScsB (see above) leads to the identification of the first peroxide reduction pathway active in the bacterial periplasm (Fig. 6) (8). In this bacterium, ScsB was shown to deliver electrons to TlpA, a thioredoxin-like protein present in the periplasm. TlpA then uses the electrons to reduce a periplasmic peroxiredoxin, PprX (8). Characterization of PprX revealed that this enzyme is active against H2O2 and cumene hydroperoxide. Although the physiological importance of PprX remains to be determined, these results highlight the importance to directly scavenge peroxides in the cell envelope before they reach the cytoplasm.
Concluding Remarks
Since the discovery of DsbA in 1991, much of the attention given to the bacterial redox pathways was focused on the disulfide bond forming system involving DsbA and DsbB. The role of the DsbC/DsbG reducing pathway was thought to be mostly limited to the correction of non-native disulfides in a small number of periplasmic proteins. However, an abundance of data has recently revealed the unsuspected diversity of reducing pathways present in the bacterial cell envelope. As discussed above, envelope-reducing pathways provide electrons that are used not only proof read the oxidative folding process but also to protect envelope proteins from the harmful action of ROS. Many questions remain to be addressed. For instance, the physiological importance of the newly discovered redox pathways such as those involved in sulfenic acid reduction and peroxide scavenging needs to be established. Moreover, how the expression of DsbD and of its substrates is regulated depending on the conditions of the environment should also be investigated, as it is possible that in certain bacteria, these proteins are upregulated in response to oxidative stress. Thorough bioinformatic, biochemical, and genetic analyses, such as those that have led to the identification of the ScsB class of DsbD-like proteins and to the discovery of PprX, also need to be undertaken to fully understand the broad spectrum of the periplasmic reducing systems present in the hundreds of sequenced bacterial genomes, and also to discover novel reducing pathways. Finally, the electron transporter protein DsbD clearly plays a central role in these pathways by transferring electrons from the cytoplasm to the periplasm. Yet, the mechanism used by this protein to catalyze this reaction is still unknown. A detailed understanding of DsbD's mechanism will undoubtedly require solving the structure of the transmembrane domain of DsbD (DsbDß) or of its homologous protein, CcdA.
Footnotes
Acknowledgments
We thank Pauline Leverrier, Katleen Denoncin, and Alexandra Gennaris for critical reading of the manuscript and helpful comments. JFC is Chercheur Qualifié of the FRS-FNRS. This work was supported by the European Research Council (FP7/2007–2013) ERC independent researcher starting grant 282335–Sulfenic. We apologize to authors whose work was not cited directly due to reference limitations.