
Abstract
Introduction
GSH is commonly used as the reference standard when calculating thiol redox potentials, but different reports use different values of Eo’ (GSH/GSSG) . Herein, all values have been corrected using of Eo’ (GSH/GSSG) =−240 mV.
ND=not yet been determined.
OSH pK a has been estimated based on that measured for the model compound 1,5-dimethyl-4-thioimidazole.
pKs =microscopic acid dissociation constant of H3N+-Cys-SH. pKns =microscopic acid dissociation constant of H2N-Cys-SH.
This surprisingly low value was determined by a recently developed mass spectrometry procedure. However, it is worth noting that the same procedure provided an unusually low value for Cys (−283 mV).
GSH, glutathione; MSH, mycothiol; BSH, bacillithiol; ESH, ergothioneine; OSH
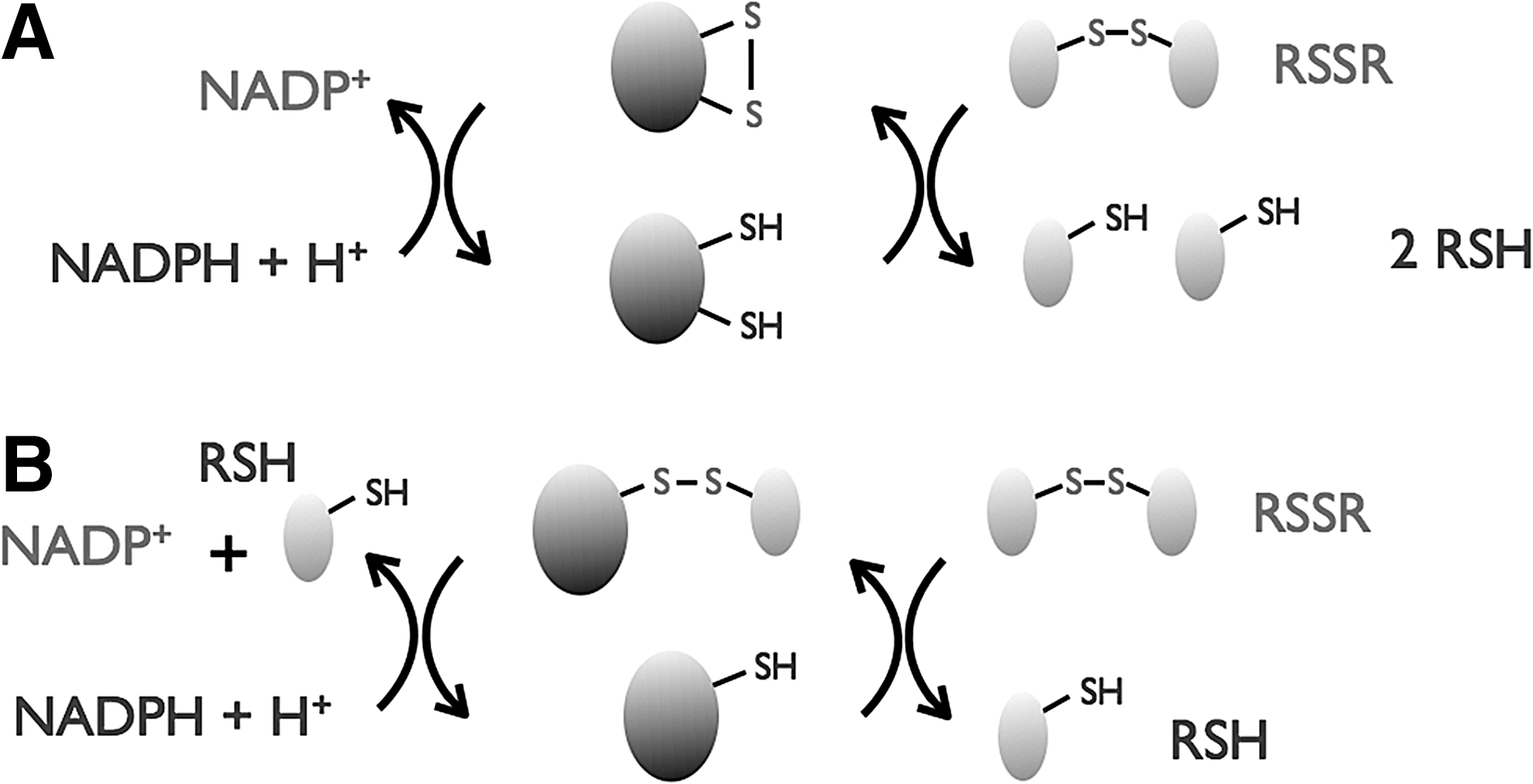
In the cell, the thiol and disulfide forms of these LMW thiols are not in equilibrium, but they are likely to be under kinetic control. Thiol–disulfide exchange mechanisms keep the steady-state equilibrium of LMW thiol ratio in balance. Also, the steady-state thiol modifications (e.g., glutathionylation, mycothiolation, cysteinylation, and bacillithiolation) are modulated by the changing redox potential of the cellular compartment and protect against oxidative stress (Fig. 2). These changes in the redox state are not only limited to the intracellular compartments, but also alter the redox state of the extracellular compartment, and are correlated with cellular processes, such as proliferation, differentiation, and death (12).
Cysteine
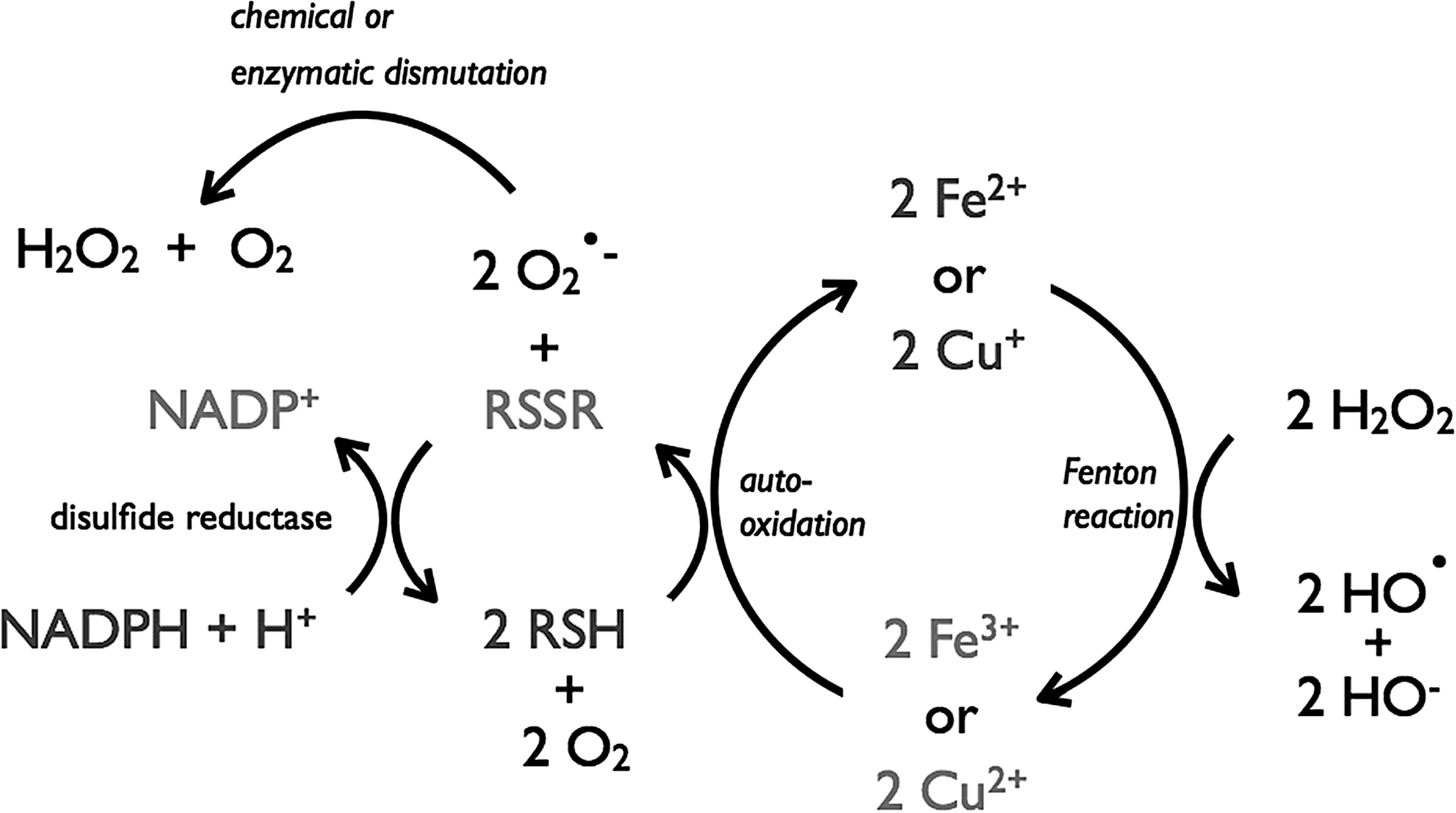
Cys is frequently found at functionally and structurally important sites in many proteins where it partakes in a wide variety of biological functions, such as catalysis, structure stabilization, signal transduction, metal binding, protein splicing, post-translational modifications, and others (81). Cys's broad functionality can be explained by its reactive thiol side chain, which can be deprotonated to a thiolate anion, enhancing its nucleophilicity (81). The intrinsic pK a value for the free Cys thiol–thiolate equilibrium in aqueous solution is ∼8.6 (97). Upon oxidation, two Cys molecules can react with each other via their sulfur atoms and form a cystine. In the cell, Cys is predominately present as Cys. The concentration of Cys and cystine is low in the cell, and the Cys/cystine couple is the most oxidized at −160 mV in proliferating cells and −125 mV in differentiated cells, but is unresponsive to Cys deficiency (49).
In normal human plasma, concentrations of free Cys (8–10 μM) and free cystine (40–50 μM) (50) are considerably higher than those of GSH (2.8 μM) and GSSG (0.14 μM) (48). These correspond to redox potentials of −80 mV for the Cys/cystine couple and −140 mV for the GSH/GSSG couple. Growing E. coli cells maintain an intracellular concentration of free Cys in the range of 0.1–0.2 mM (77). Human HT29 cells maintain an intracellular free-Cys concentration of 125 μM and 31 μM cystine (49). Such low Cys concentrations are a sufficient feedstock for protein biosynthesis and are below its toxicity threshold. High intracellular Cys concentrations undergo auto-oxidation in the presence of transition metal ions (such as Fe3+). During this reaction, O2 generally acts as oxidant generating hydroxyl radicals (HO•) via the Fenton reaction (45). Copper (Cu+ and Cu2+) and iron (Fe2+ and Fe3+) are the most efficient, biologically relevant, catalysts of Cys auto-oxidation (104). Cu+ and Cu2+ substantially increase the oxidation rate of Cys at submicromolar concentrations. A concentration of 0.2 μM Cu2+ or Cu+ can oxidize half of the intracellular Cys concentration in 60 min. Albumin has a strong binding site for Cu2+ that could drive the oxidation of Cys to cystine in the plasma and explain the higher abundance of cystine in the plasma (35,50). The auto-oxidation of Cys by Fe2+ and Fe3+ is slower than that of Cu+ and Cu2+. While there is no difference in the rate of auto-oxidation with Cu+ or Cu2+, Fe2+ is a faster oxidant of Cys than Fe3+ at concentrations > 10 μM. The soluble Fe3+ complex, hemin, is more efficient in catalyzing Cys auto-oxidation than Fe2+ and Fe3+, but still much less efficient than Cu+ or Cu2+ (104).
As such, most organisms therefore employ other, less-reactive, LMW thiols as their intracellular redox buffer. The parasites Giardia duodenalis, Entamoeba histolytica, and Trischomonas vaginalis are exceptions (106). They produce up to 0.6 mM free Cys as a major redox buffer. The microaerophilic lifestyle of these pathogens might facilitate their tolerance against these high levels of intracellular Cys (106).
Perturbances in the Cys/cystine redox balance have been associated with cardiovascular diseases and age-related diseases, although the GSH (GSH/GSSG) redox balance is here also modified. A redox cycle over the plasma membrane is maintained in mammalian cells by the oxidative-stress-induced cystine/glutamate exchange system, system
The GSH-dependent cystine reductase activity of the C-terminal glutaredoxin-like domain of 5-adenylyl sulfate reductase (13) has a reported KM of 1.1 mM and a Vmax of 54.3 μM·min−1·mg−1 [for GSH in the presence of saturating (0.35 mM) cystine], which corresponds to a kcat value of ∼48 s−1 and a kcat /KM value of ∼40,000 M −1·s−1. This would be lower at physiological cystine levels. Furthermore, a decrease in GSH of 90% showed no effect on the rate of reduction of extracellular cystine, which indicates a GSH-independent mechanism (3). In this respect, it should be noted that in the absence of intracellular GSH, the cytosolic thioredoxin reductase system seems to be able to function as a backup reducing system of cystine (63). Altogether, the cellular reactions are not clear, and so far no enzyme is known that reduces cystine in mammalian tissues.
Glutathione
GSH (
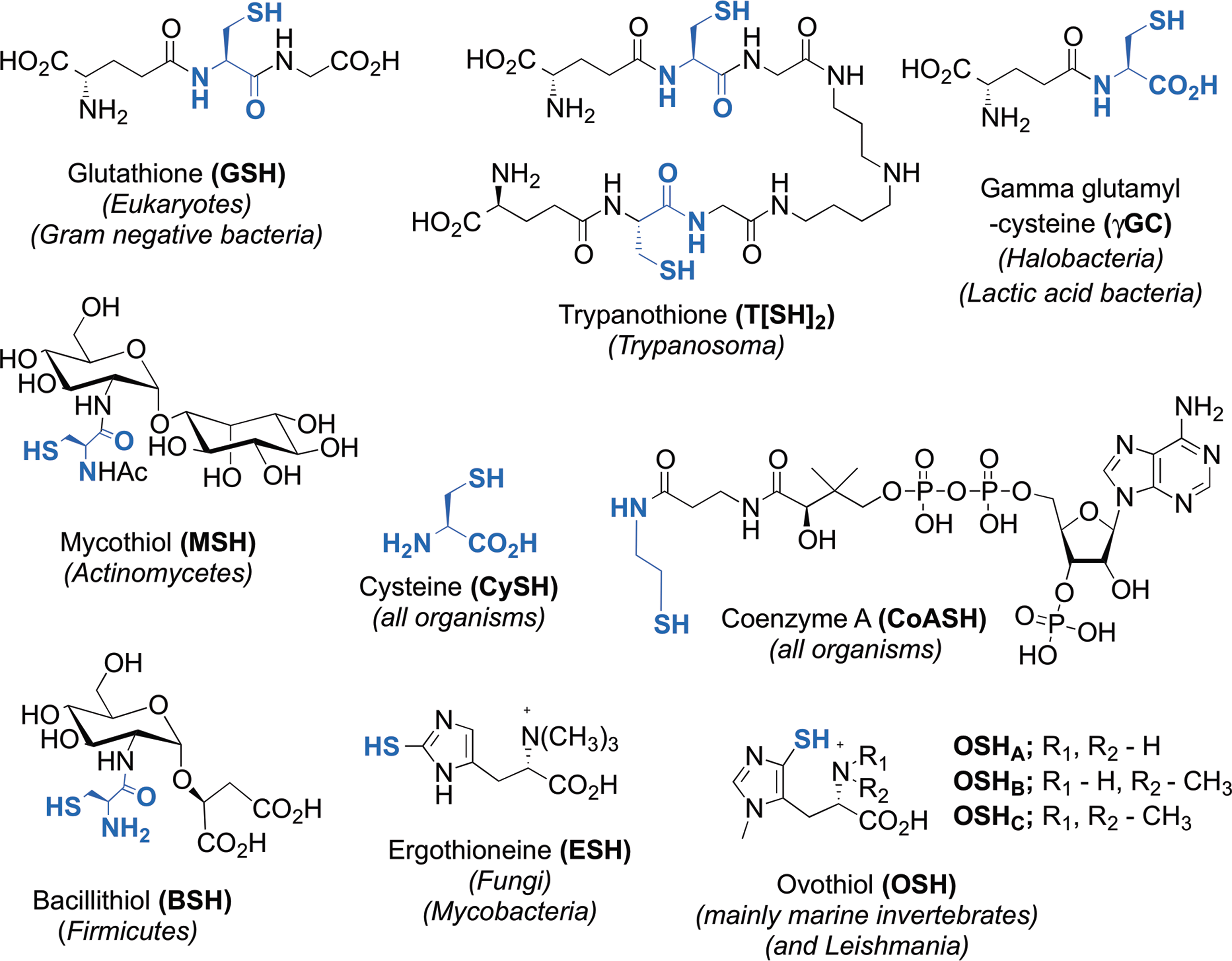
Thiols with a low pK a are more prone to oxidation to sulfenic acids by ROS (82) (Fig. 2). Although due to their high reactivity, a sulfenic acid is often only an intermediate in a reaction mechanism. For example, in 2-cys peroxidases, the presence of another Cys residue in close proximity to the sulfenic acid will lead to the formation of a disulfide between both Cys as final oxidation product (40). Most probably, only a minority of sulfenic acids are formed as final oxidation product without catalysis.
The relatively high pK a of GSH (∼9) helps protect it from facile oxidation to its sulfenic acid (GSOH). To perform a nucleophilic attack, the thiol pK a of GSH needs to be decreased, for the thiolate form to predominate at physiological pH. Glutathione S-transferases decrease the pK a of GSH and enhance the nucleophilicity of GSH, facilitating the conjugation of GSH to electrophilic substrates (6) (Fig. 3B). In the enzyme–GSH complex, hydrogen bonding and electrostatic effects lower the pK a of the GSH thiol from ∼9 to ∼6 (18,59), so that it is predominantly present as thiolate at physiological pH.
GSH is indirectly involved in peroxide scavenging by participating in the recycling of ascorbate, an electron donor to ascorbate peroxidases (84), and by supplying selenocysteine glutathione peroxidases (Fig. 4) (32,98).

GSH can also covalently modify protein Cys residues in a process called S-glutathionylation (Fig. 2). Although <0.1% of the total protein Cys are glutathionylated in nonstressed cells, this increases to >15% during disulfide (i.e., diamide) stress (41). S-glutathionylation is reversible and is assumed either to protect Cys residues from irreversible oxidation or to regulate proteins whose Cys residues are essential for activity or folding. In general, S-glutathionylation is reversed by the action of Grxs that almost specifically reduce mixed disulfide adducts formed between protein thiols and GSH. Mechanisms for S-glutathionylation can be schematically classified as ROS dependent or ROS independent (37). Although the final product is the same, the rates of the reactions may be very different. The ROS-independent mechanism occurs via thiol–disulfide exchange between GSSG and the proteins. However, under physiological conditions, the low concentration of GSSG in the cytosol is unlikely to favor this mechanism for the formation of mixed disulfides with protein thiols in this compartment. Moreover, the thermodynamic barrier limits the oxidation of proteins by GSSG via the nucleophilic attack of a protein thiolate on GSSG, as the mixed disulfide formed must have a redox potential higher than that of the GSH/GSSG couple, which is theoretically possible, but extremely unlikely (34). In contrast, in the endoplasmic reticulum, where the GSH/GSSG ratio is lower, the formation of protein–GSH-mixed disulfides is more likely to occur via the direct reaction of a protein thiolate on GSSG (34). Under oxidative stress conditions, when high concentrations of hydrogen peroxide (H2O2) accumulate by the inactivation of 2-cys peroxiredoxins (108), the Cys residues are first oxidized to an unstable sulfenic acid that can be irreversibly oxidized to sulfinic and sulfonic acids (82) (Fig. 2). H2O2 acts also as a signaling molecule by oxidizing critical thiol groups on redox-regulated target proteins. In this case, the oxidation to sulfenic acid might be catalyzed by H2O2-scavenging peroxidases (39).
GSH will react with the more electrophilic sulfenic acid to form a GSH–protein-mixed disulfide, which effectively protects those Cys residues from irreversible oxidation (Fig. 2). For example, the α-glutamyl transpeptidase is protected from oxidative damage by S-glutathionylation (25). S-glutathionylation can also regulate protein function (21). For instance, the enzyme α-ketoglutarate dehydrogenase is reversibly inactivated by S-glutathionylation in response to alterations in GSH levels in mitochondria (64). Altogether, the reversible S-glutathionylation of specific proteins has implications for the regulation of cellular homeostasis in health and disease. Changes in the S-glutathionylation state of specific proteins play important roles in diabetes, cardiovascular, lung, and neurodegenerative diseases (21,64). Finally and depending on the organisms, the essential function of GSH might not be in thiol-redox control, but rather in iron homeostasis, as recently revealed by work done in the yeast Saccharomyces cerevisiae (58).
Trypanothione
In the Kinetoplastida, which are primitive eukaryotes that parasitize animals and plants, most of the GSH is found in the form of N1,N8-bis(glutathionyl)spermidine. This molecule, better known as trypanothione [T(SH)2], is composed of two GSH molecules connected via a spermidine linker (Fig. 1) (29). TSG is maintained in its reduced state through reduction of its oxidized intramolecular disulfide TS2 by the FAD disulfide oxidoreductase TSG reductase (TR) (33). The cellular T(SH)2 concentrations range from 0.2 to 1.5 mM, and the TS2/T(SH)2 couple determines the redox potential in the cell. The standard redox potential of T(SH)2 is almost similar to that of GSH (Table 1) (30). On the other hand, T(SH)2 is more reactive than GSH in thiol–disulfide exchange reactions under physiological conditions, due to the lower pK a value of its thiol group (∼7.4) (68), which provides optimal conditions for thiol–disulfide exchange (38). The dithiol character of T(SH)2 favors formation of an intramolecular disulfide in comparison to the intermolecular disulfide formed upon oxidation of GSH.
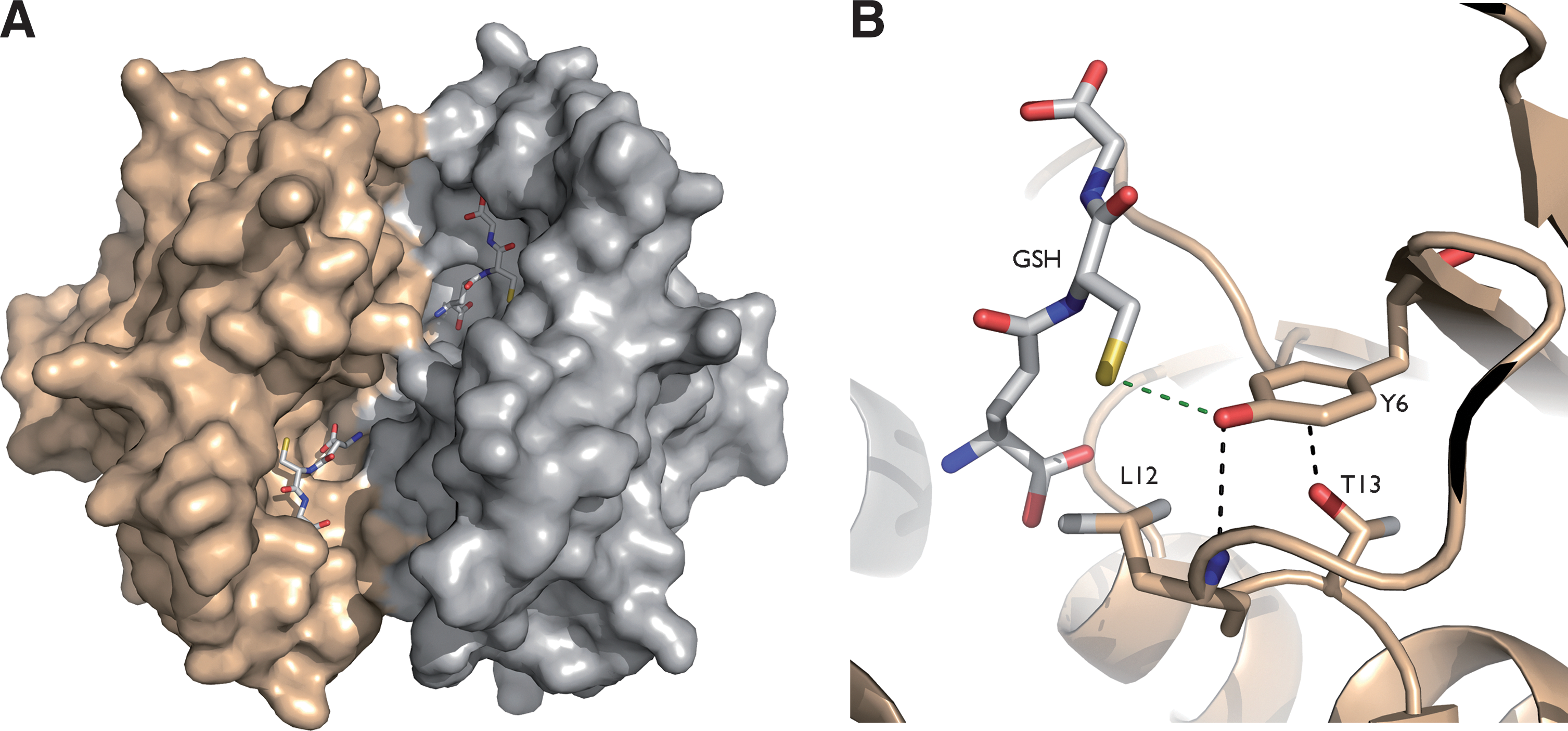
Hydroperoxide detoxification in trypanosomatids relies on a sophisticated cascade of reactions in which TSG, TR, and tryparedoxin (TXN) play central roles as the carriers of reducing equivalents from NADPH onto two types of peroxidases [2-Cys-peroxiredoxin (Prx) and glutathione peroxidase type (Gpx)] (Fig. 5A). T(SH)2 functions also as the donor of reducing equivalents to several enzymes of the parasite, including thioredoxin (85), TXN (61,75), monothiol glutaredoxin-1 (31), and ribonucleotide reductase (26). T(SH)2 can also spontaneously reduce protein sulfenic acids in the model protein glyceraldehyde-3-phosphate-dehydrogenase (31). This makes that the disulfide-exchange TR/T(SH)2/TXN system is essential and responsible for maintaining the cytosolic redox homeostasis.
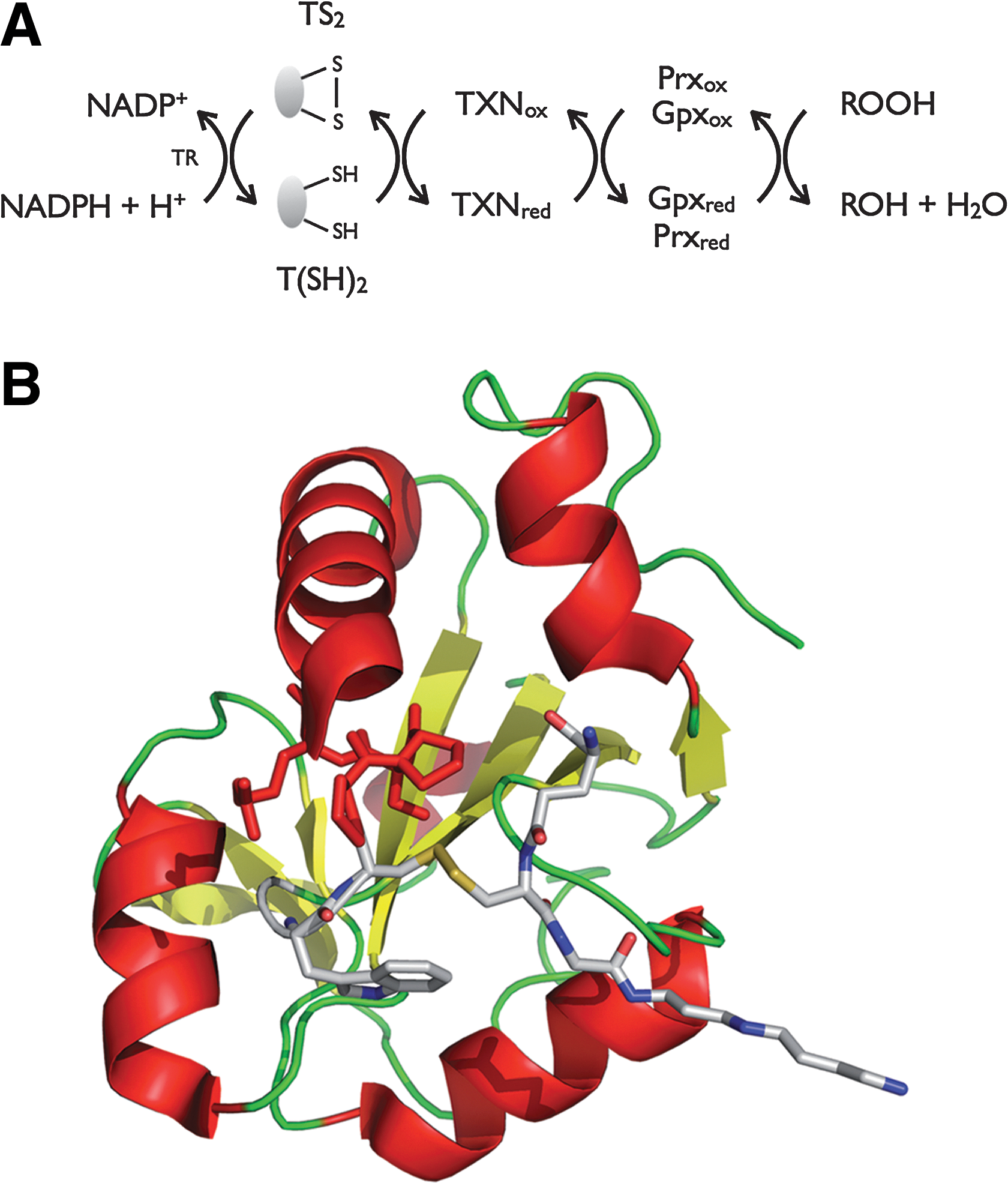
TXN is a distant relative of the thioredoxin superfamily (Fig. 5B), which has a WCPPCR active-site motif and is substantially larger (16 kDa) than most thioredoxins (56). As such, TXN is reduced by T(SH)2 (75) and can transfer reducing equivalents to a variety of protein targets, making the TXN/T(SH)2 couple the determining factor for the intracellular redox state of the parasite (80).
In addition to providing reducing equivalents to oxidoreductases, T(SH)2 can also efficiently scavenge H2O2, peroxynitrite, and radiation-induced radicals (10,16,96). T(SH)2 maintains the redox homeostasis by passing electrons to peroxidases via intermediate shuffle molecules, which can be TXN, ascorbate, or even GSH (17).
γ-Glutamylcysteine
γGC is a biosynthetic intermediate of GSH. Several halobacteria (72) and lactic acid bacteria (55) produce millimolar concentrations of γGC, but not GSH. An NADPH-dependent disulfide reductase has previously been purified from Halobacterium halobium that is able to reduce γGC disulfide, but not GSSG (93). This supports the notion that γGC serves as a redox buffer in these microorganisms. The free cysteinyl carboxylate of γGC makes it more prone to metal-catalyzed auto-oxidation, suggesting that it would be a poor substitute for GSH as a cellular redox buffer. However, in the presence of high salt concentrations (typical of halobacterial habitats), resistance of γGC to auto-oxidation is comparable to that of GSH (93).
Mycothiol
MSH (Fig. 1) is the major thiol in most Actinomycetes (e.g., Mycobacteria, Corynebacteria, and Streptomycetes) where it is produced at millimolar concentrations and serves as a GSH surrogate (51,71). It is comprised of N-acetylcysteine amide linked to a 1D-myo-Inosityl 2-acetamido-2-deoxy-α-
Like GSH, mycothiol exists in both a reduced (MSH) and disulfide (MSSM) state. However, the MSH thiol-redox potential is not yet known, probably due to its low availability. The chemical syntheses are convoluted (51,71) compared to its isolation from bacterial cell culture. This makes isolation from cell cultures still a more efficient protocol for MSH production (76).
A key property of MSH is its high resistance to oxidation by molecular oxygen in the presence of redox metals. For instance, the copper-catalyzed auto-oxidation of MSH is about 30-fold slower than that of Cys and 7-fold slower than that of GSH (81), due to the N-acetyl and GlcN-Ins moieties blocking the amino and carboxyl groups of the Cys, respectively, thereby reducing its metal-ion coordination strength. In actinobacteria, MSH-disulfide reductase (Mtr), an NADPH-dependent flavoenzyme, reduces MSSM back to MSH to maintain the intracellular redox homeostasis required for the proper functioning of a variety of biological functions (79). In Corynebacterium glutamicum, mycoredoxin-1 (Mrx1), the glutaredoxin analog of the Actinomycetes, does not function with GSH, but has a strict specificity for MSH in a reaction coupled to Mtr and NADPH (76), suggesting that glutaredoxins and mycoredoxins have specific binding sites for GSH and MSH, respectively.
MSH S-conjugates derived from electrophiles and antibiotics are cleaved by MSH S-conjugate amidase Mca to release GlcN-Ins, which is used to resynthesize MSH, and a mercapturic acid, which is excreted from the cell (70). Mca can also cleave N-acetylcysteine from MSH (15). The constant Mca-catalyzed turnover of MSH serves to provide a controlled, but continuous, supply of Cys to serve other cellular needs while obviating the damaging effects of auto-oxidation. A mycothiol-dependent formaldehyde dehydrogenase MscR (66) was later identified as nitrosomycothiol reductase with a role in the protection against oxidative stress (102). Moreover, Ordóñez et al. have found that the activity of C. glutamicum arsenate reductases 1 and 2 depends on reducing equivalents transferred from MSH by the Mrx1 (76). Very recently, it has been shown that MSH provides the reducing power for Mycobacterium tuberculosis Mrx1 in reduction of synthetic mixed MSH disulfides (101). For the future, it will be important to identify the natural Mrx1 substrates (i.e., S-mycothiolated proteins) to unravel the roles of MSH in the redox regulation and protection of protein function in M. tuberculosis and other Actinomycetes.
Coenzyme A
CoA is an essential thiol cofactor in all living organisms that is widely implicated in central metabolic pathways, including the Krebs cycle and fatty acid metabolism. NAD(P)H-dependent CoA disulfide reductases (CoADR) have been isolated and biochemically characterized from Staphylococcus aureus (60), Bacillus anthracis (103), Borrelia burgdorferi (14), and Pyrococcus horikoshii (42), indicating that CoA has intracellular redox functions in many bacteria that do not produce GSH or MSH. Bioinformatic analyses indicate that CoADR is broadly distributed among bacterial and archaeal kingdoms (62). Unlike MSSM and GSSG reductases, CoADR is a group-3 pyridine nucleotide disulfide oxidoreductase with a single redox-active Cys that reduces CoA-disulfide via formation of a protein–SSCoA-mixed disulfide (Fig. 6) (60,62).
Some bacteria (e.g., B. anthracis and S. aureus) that utilize the CoADR/CoA redox couple also produce BSH in equivalent or greater quantities (73). Redundancy provided by these two thiol redox buffers may confer an advantage in virulence and/or survival of such pathogens when challenged by the oxidative burst of the host immune response (78). Little is yet known about the redox functions of CoA in a physiological setting. Its redox potential (−234 mV) (54) is comparable to that of GSH (Table 1), meaning that CoA could make a significant contribution toward the electrochemical potential inside the cell. However, a thiol pK a of 9.83 (53) means that CoA exists exclusively in its unreactive thiol form at physiological pH. It is reasonable to speculate that enzymes must be required to enhance the reactivity of CoA in cellular redox processes, although none have yet been discovered. In dormant spores of Bacillus megaterium, Bacillus subtilis, and Clostridium perfringens, ∼45% of cellular CoA is linked to proteins as mixed disulfides (88), and >75% of these are cleaved from these during germination. This clearly demonstrates a protein thiol-redox regulation and/or protection role for CoA similar to that of GSH. The mechanisms by which these CoASS proteins are reduced are not yet known. A CoADR null mutant of B. burgdorferi displays increased sensitivity to tertbutyl hydroperoxide, but not H2O2, supporting the role of the CoADR/CoA redox couple in defense against some forms of oxidative stress (27). In this instance, the lack of a H2O2 phenotype is probably due to the presence of peroxidases, which fulfill the H2O2 detoxification role. It will be interesting to see whether similar oxidative stress phenotypes occur in mutants of other CoADR-utilizing bacteria.
Bacillithiol
In 2009, BSH (Fig. 1) was identified as a LMW thiol among many low-G+C gram-positive bacteria (Firmicutes) (73), which do not produce GSH or MSH. These include many Bacilli and some, but not all, Staphylococci and Streptococci (73).
Structurally, BSH shares the same GlcN-Cys scaffold as MSH, but the inositol group is replaced by
Intracellular BSH is predominantly in its reduced form with BSH/BSSB redox ratios ranging from 40:1 (in B. anthracis) (78) to 400:1 (in B. subtilis) (73). This is presumably maintained by a BSSB reductase, although this has not yet been proven. Many BSH producers (e.g., B. anthracis and S. aureus) also produce CoADR, but the BSH/BSSB redox ratios remain unaltered in CoADR null mutants (78), indicating that CoADR is not also a BSSB reductase. BSH also forms mixed disulfides with protein thiols (bacillithiolation). In B. subtilis, the redox-sensitive peroxiredoxin transcription regulator (OhrR) is bacillithiolated during cumene hydroperoxide stress suggesting a function for BSH in redox sensing (90).
When exposed to sodium hypochlorite (NaOCl), a number of proteins in B. subtilis are reversibly bacillithiolated (19), including OhrR and two methionine synthases (MetE and YxjG). Bacilliredoxins (Brxs) must therefore exist to reverse protein bacillithiolation. Phylogenomic profiling has identified three candidate Brxs (YphP, YqiW, and YtxJ), which are widely conserved among BSH-producing Firmicutes (36). YtxJ is a Trx family protein with a conserved Cys residue in a TCPIS motif similar to what is found in the active site of many monothiol glutaredoxins. YqiW and YphP belong to the DUF1094 family of small, Trx-like proteins. However, the classical redox-active CXXC motif found in Trxs is replaced by an invariant CGC active-site motif. The reduction potential of YphP (−130 mV) (24) is also significantly higher than those typically observed for Trx and glutaredoxins (<−200 mV) (8). Under NaOCl stress, B. subtilis S-bacillithiolation of YphP is observed at the more solvent-exposed nucleophilic Cys53 residue of its redox-active CGC motif. This is a likely intermediate in the Brx catalytic cycle (19). Future investigations are required to confirm the Brx activity of these proteins.
In gram-negative bacteria, typical intracellular concentrations of GSH are ∼5 mM (69). BSH levels in the Firmicutes are significantly lower (i.e., ∼400 μM) and more comparable to those of Cys (73). The significance of this is not yet known. Perhaps, BSH is a more powerful reductant than GSH, and/or maybe BSH redox enzymes have enhanced catalytic efficiency compared to their GSH counterparts? The reported synthesis of BSH (89) and its commercial availability mean that it is now accessible in sufficient amounts to facilitate further investigations to address these fundamental questions.
Ergothioneine
ESH is a 2-thiolhistidine (Fig. 1) that has been detected (in millimolar concentrations) in bacteria, fungi, plants, animals, and humans. ESH is only biosynthesized in fungi (46) and Actinomycetes (86), whereas animals obtain it from dietary sources, and plants are believed to obtain ESH from the soil. ESH serves as an important antioxidant, which in higher organisms is primarily stored in tissues that are more often exposed to oxidative stresses.
ESH has a very high thiol-redox potential (−60 mV) (Table 1); hence, it is very difficult to oxidize to its less thermodynamically stable disulfide (ESSE). This is likely attributed to the more-stable thione form of ESH (Fig. 7A). The nucleophilic reactivity of the predominant aminothione form of ESH at physiological pH is attributed to the mesomeric release of the lone pair electrons on the nitrogen atom of the amino group (Fig. 7B) (52,65).
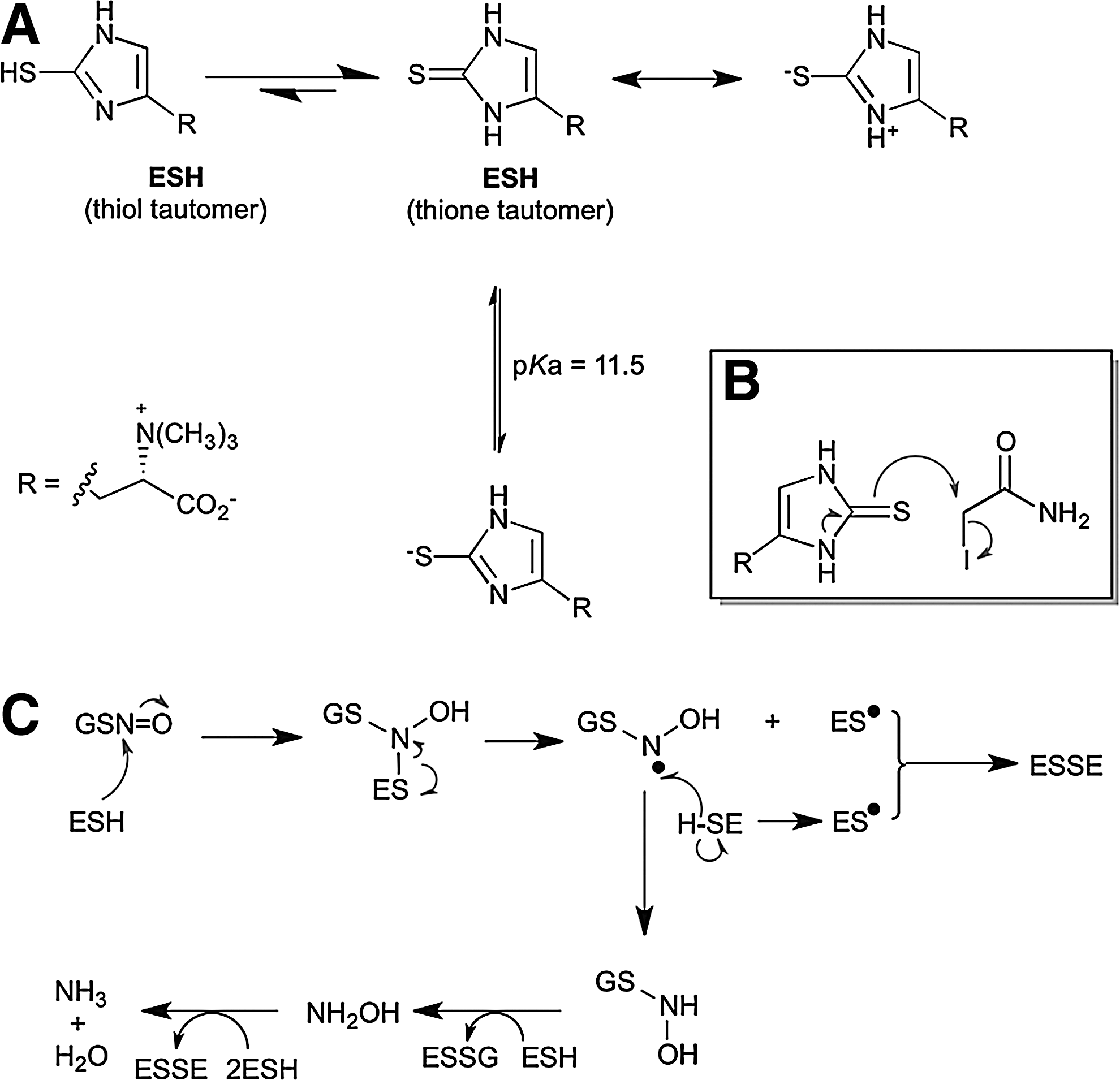
ESH strongly influences the GSNO decomposition, which can enhance its rate of decomposition 20-fold faster than GSH (Fig. 7C) (65). Even in the physiological presence of GSH, the preferential ESH-mediated pathway predominates and can be attributed to the nucleophilic character of ESH and the homolytic cleavage step that is favored by the stability of the ES• radical due to aromatic conjugation. ESH is a powerful scavenger of HO• (1,9), HOCl (1), singlet oxygen (1O2) (95), and peroxynitrite (47) at biologically significant rates, suggesting that it can play a relevant role in the detoxification of these oxidants. The relative formation constants of ESH–metal complexes are Cu2+>Hg2+>Zn2+>Cd2+>Co2+ ≥Ni2+ with ESH behaving as a unidentate ligand that coordinates the metal through the sulfur atom (67). In aerobic organisms (Fig. 8), cellular thiols such as Cys, GSH, and MSH are susceptible to metal- (e.g., Cu2+) catalyzed auto-oxidation (2,107) to disulfides with concomitant production of lower-oxidation-state metals (Cu+) that drive the Fenton reaction. However, at physiological pH, ESH forms a stable complex with Cu2+ in a nonredox active form, which prevents auto-oxidation and hydroxy radical formation (1).
ESH can reduce ferrylmyoglobin (a higher oxidation state of metmyoglobin that can be formed under oxidative stress) back to metmyoglobin while itself being oxidized to ESSE. GSH then rapidly regenerates ESH via thiol–disulfide exchange. This may function as an important redox cycle to prevent ferrylmyoglobin-mediated peroxidation of membrane lipids and fatty acids (4).
To date, most studies of ESH function have been performed in eukaryotes, but a recent study of MSH-deficient mutants of Mycobacterium smegmatis showed a 20-fold to 30-fold increase in ESH production compared with the wild type, indicating that it may compensate toward the loss of MSH in these mutants (94). In the extensive in vitro and in vivo studies that have been reported, ESH appears to serve an important role as a biochemical antioxidant. So far, no enzymes have been identified that use ESH as a cofactor.
Ovothiol
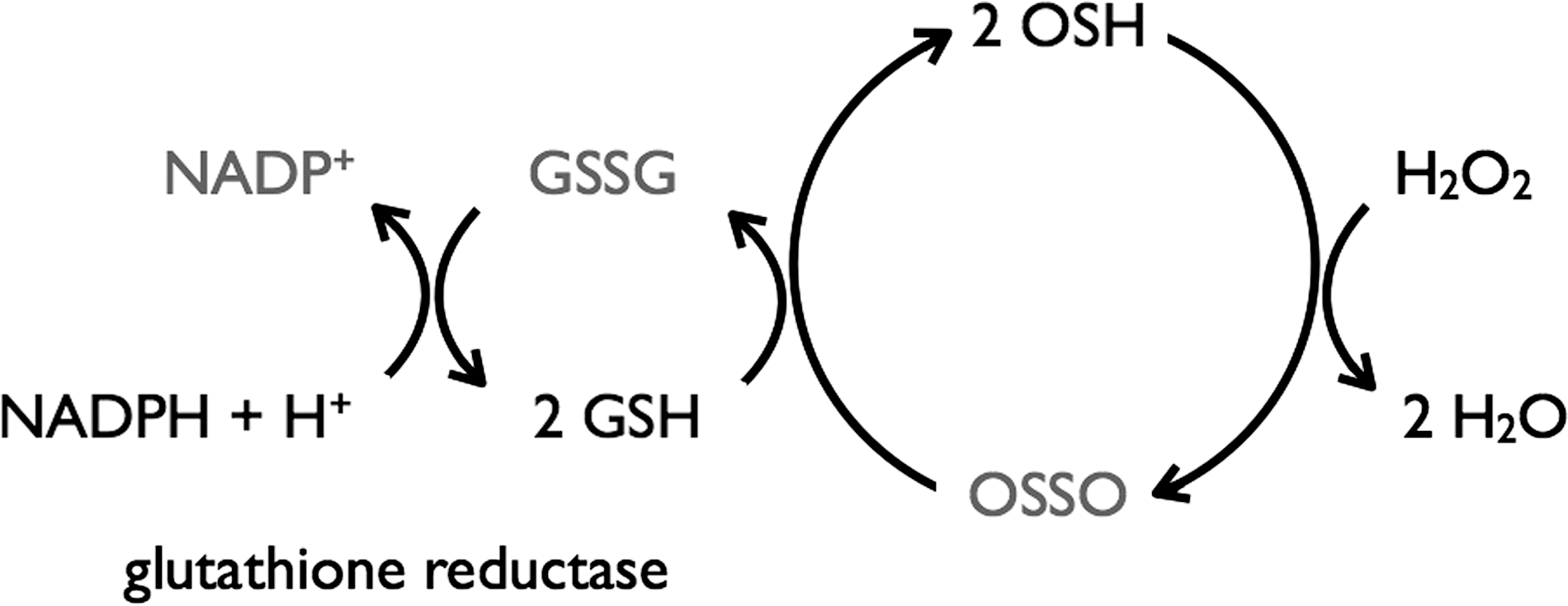
The ovothiols (OSHA–C) are a group of structurally related 1-methyl-4-thiol-histidines with differing degrees of N-methylation (Fig. 1). They are found alongside GSH in Echinoderms (100) and a variety of other marine invertebrates (83), some halotolerant blue-green algae (87), as well as the T(SH)2-producing parasites Crithidia fasciculate (92), Trypanosoma cruzi, and Leshmania spp. (5,91), and the eggs of rainbow trout and salmon (99). Ovothiols play an important role in resistance to oxidative stress. In fertilized sea urchin eggs, an oxidative burst produces large quantities of H2O2 to feed ovoperoxidase-catalyzed formation of the fertilization envelope. To prevent oxidative damage from excess H2O2, ∼5 mM OSHC is produced, which functions as an effective chemical equivalent of GSH peroxidase (Fig. 9), (99) and is even more effective than catalase in the detoxification of the H2O2 produced during egg fertilization. The redox potential of OSH is very high (105) (Table 1), and hence the disulfide is very unstable, and OSH is swiftly regenerated via thiol–disulfide exchange with other LMW thiols [e.g., GSH and T(SH)2]. Coupled with the acidity of the histidine thiol (pK a ∼2.3), this means that under physiological conditions, OSH exclusively exists in its thiolate anion.
Future Challenges
In many microorganisms, the LMW thiol-redox buffers have not yet been identified, and many elements of BSH and MSH redox biochemistry remain to be explored. More attention should also go to the function of mixed disulfides between two different LMW thiols. The future of scientific challenges in the LMW thiol biology (thiology) arena looks bright, and many new and promising paths will lead to essential discoveries.
Footnotes
Acknowledgments
Financial support was provided by the following institutions: (1) agentschap voor innovatie door Wetenschap en technologie (IWT), (2) Vlaams Instituut voor Biotechnologie (VIB), (3) the HOA project of the Vrije Universiteit Brussel (VUB), and (![]() ) the Biotechnology and Biological Sciences Research Council (BB/H013504/1). J.M. is group leader Redox Biology of the VIB. We also acknowledge the BMBS COST action BM1203 (EU-ROS).
) the Biotechnology and Biological Sciences Research Council (BB/H013504/1). J.M. is group leader Redox Biology of the VIB. We also acknowledge the BMBS COST action BM1203 (EU-ROS).