
Abstract
Introduction
Mammalian cells are endowed with six peroxiredoxins; among these, only peroxiredoxin 4 (PRDX4) is localized in the endoplasmic reticulum (ER) (34).
This essay will focus on recent advances concerning the role of PRDX4, how its function of disposing of H2O2 is related to oxidative protein folding, and how impaired oxidative folding and H2O2 disposal can affect the metabolism of ascorbate. Finally, it will discuss the emerging cross-talk between the rate of oxidative folding, ER H2O2 content, ascorbate metabolism, and the organization of the extracellular matrix (ECM).
Concerted Protein Disulfide Bond Formation and H2O2 Production in the ER
The ER is the compartment in which the proteins destined for the secretory pathway and cell surface are folded: they enter in an unfolded state, and leave when they are correctly folded and assembled (18).
One feature of the proteins folded in the ER is the oxidation of free thiols on cysteines to disulfide bonds. This is done by means of a process known as oxidative folding, which is required for the stability and function of secreted and membrane-bound proteins. Oxidative folding is fundamental for cell fitness, as shown by the dramatic increase in unfolding protein response (UPR) signaling (a cell stress response, see (47) for a review) in cells treated with dithiothreitol (DTT), which reduces disulfide bonds (11).
In the early 1960s, Anfinsen et al. carried out pioneering in vitro studies of oxidative protein folding and showed that in the absence of catalysts, free thiols are oxidatively folded over a period of about a day (1).
In vitro oxidative folding largely depends on thermodynamic considerations, but in vivo folding is more complicated, because the transition from free thiols to disulfides is based on the dynamic and concerted formation, reduction, and reshuffling of disulfide bonds that favor the introduction of native (and therefore functional) disulfide bonds on new client proteins. In addition, the free thiols on proteins can be exposed to a competing H2O2-mediated oxidative process that produces oxygenated thiol derivatives. The derived sulfenylated proteins can be transient intermediates in the formation of more stable disulfides or precursors of higher oxidized sulfur oxides (sulfinic and sulfonic acids) or can react with GSH to produce S-glutathionylated proteins (27) (Fig. 1).

Given the complexity of oxidative folding and the multiple forms of H2O2-mediated cysteine modification, which can give rise to competing unproductive side reactions, it is necessary to consider kinetics in the case of in vivo folding. The intricate disulfide relay requires enzymatic assistance, and a specialized class of enzymes of the protein disulfide isomerase (PDI) family has evolved to promote the efficient formation of native disulfides in folding ER proteins. PDIs, through a redox-active thioredoxin-like catalytic domain (CXXC), directly introduce disulfide bonds into reduced client proteins and are finally reduced themselves (16).
Upstream of this redox reaction, endoplasmic oxidoreductin 1 (ERO1) reoxidizes and reactivates the thioredoxin-like domain of PDI for a new cycle of oxidative protein folding, and then by means of its cofactor flavin adenine dinucleotide, transfers electrons to molecular oxygen (the final electron acceptor in aerobic condition), thus producing H2O2 (44).
The final balance of oxidative folding is that one molecule of H2O2 is produced for every disulfide bond formed. Moreover, a related rough estimate indicates that ERO1-mediated oxidative folding accounts for 25% of the H2O2 produced during protein synthesis (45), thus raising the question as to how cells efficiently dispose of H2O2 in the ER.
In bacteria, the problem is solved by coupling oxidative folding and the respiratory chain (5), orchestrated in the form of a series of membrane electron transfer proteins that efficiently reduce molecular oxygen to water. However, as oxidative folding and respiration in eukaryotes take place in different cell compartments (respectively, the ER and mitochondria), ERO1-dependent oxidative folding is coupled with a rapid and efficient disposal of H2O2 to avoid detrimental effects on cell fitness.
PRDX4 Couples Oxidative Folding and the Disposal of the H2O2 Produced by ERO1
The efficient disposal of H2O2 is done by means of ER peroxidases which, as their name suggests, are dedicated to degrading H2O2. The mammalian ER is endowed with three known (PDI) peroxidases (peroxidases capable of accepting electrons from PDIs): PRDX4 [in this essay, PRDX4 only refers to the ER isoform of the protein, but it has recently been shown that alternative mRNA splicing in the testis can give rise to a cytosolic isoform of PRDX4 (54)], and the two misnamed (since they do not efficiently accept electron from GSH) GSH peroxidases GPX7 and GPX8 (32, 39). PRDX4 is the oldest of these in evolutionary terms and has the highest tissue expression: it is found in the genome of Drosophila melanogaster and has been conserved in vertebrates (3, 54), whereas GPX7 and GPX8 appeared late during the evolution in Ascidian (3); furthermore, PRDX4 is more enriched in highly secretory tissues (22) than GPX7 and GPX8, which are also widely expressed, but at low transcriptional level (32). Moreover, its higher degree of reactivity with H2O2 (a second rate constant of 2.2×107 M −1·s−1) in comparison with the other ER peroxidases (49) also makes PRDX4 an efficient scavenger of low concentrations of H2O2.
The enzymatic mechanism by means of which PRDX4 reduces H2O2 to water is that of a typical 2-Cys peroxiredoxin (17). Structurally, PRDX4 is a pentamer of dimers (12, 49) whose catalytic mechanism is based on a conserved peroxidative cysteine (CP) and a resolving cysteine (CR). The catalytic cycle proceeds as follows: CP reacts with H2O2 to form cysteine sulfenic acid (CP-SOH) (peroxidation), which is attacked by the nucleophile-resolving cysteine CR of an adjoining PRDX4 protomer to produce a peroxiredoxin dimer (resolution); finally, this dimer is reduced by PDI to re-establish peroxidase activity (recycling) (40, 57).
An alternative redox fate of 2-Cys peroxiredoxin is the further oxidation of CP-SOH to sulfinic (CP-SO2H) and sulfonic (CP-SO3H) acids. To be reduced and reactivated, the oxidized sulfinic CP-SO2H species require the enzymatic assistance of sulfiredoxin, an ATP-dependent enzyme (8, 50), whereas the sulfonic CP-SO3H is irreversibly inactivated (Fig. 2).
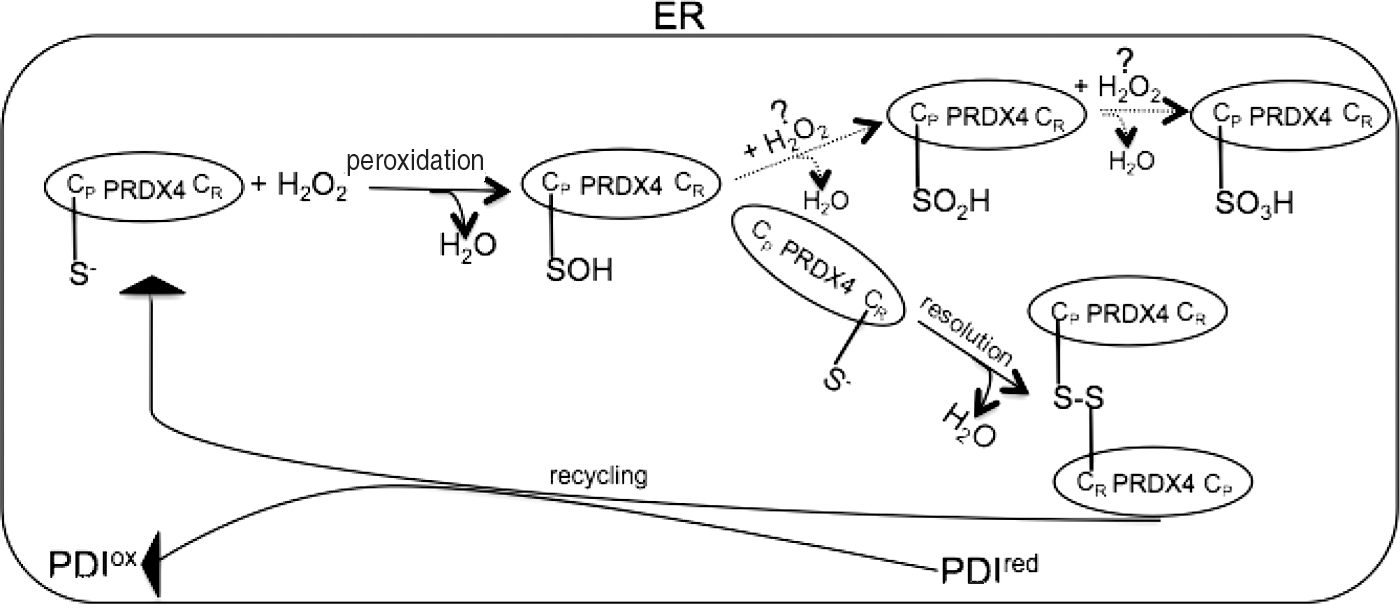
Various hypotheses have been proposed concerning the biological roles of hyperoxidized peroxiredoxins such as acting as a molecular chaperone and peroxide dosimeter regulating the cell cycle (26), but the lack of sulfiredoxin-like activity in the ER (12) and the unequivocal relationship between the content of H2O2 and the hyperoxidation of PRDX4 raises the question of the relevance of the H2O2-mediated terminal inactivation of PRDX4 (54).
Given the biological implications of PRDX4 loss of function, mechanisms (other than the H2O2-mediated mechanism) producing the terminal inactivation of PRDX4 are worth investigating.
The reactivation of oxidized PRDX4 by PDI leads to the further insertion of disulfide bonds in new client proteins, which implies a catalytic cycle in which the H2O2 produced by ERO1-mediated oxidative folding works as the driving force of PRDX4-mediated oxidative folding (40, 57) (Fig. 3).

By coupling H2O2 catabolism with disulfide bond formation, PRDX4 is a part of an ingenious system for streamlining resources: the reduction of one molecule of oxygen leads to the introduction of two disulfide bonds in new client proteins (one via ERO1, and the other via PRDX4). In this respect, the massive upregulation of PRDX4 during the transition from B cells to highly secretory antibody-producing lymphocytes may be explained as a means of accommodating increased H2O2 and parallel immunoglobulin M synthesis (6).
Other Sources of H2O2 Fuel PRDX4-Mediated Oxidative Folding
ERO1 is the predominant disulfide oxidase in yeast, and is encoded by a gene that is essential for the viability of the organism (33).
Gene duplication events in vertebrates have generated two ERO1 isoforms: ERO1α and ERO1β. The expression of ERO1α and ERO1β overlaps in some tissues, but their tissue distribution pattern differs considerably. ERO1α is broadly expressed throughout the body, whereas ERO1β is enriched in highly secretory cells, such as pancreatic β-cells and antibody-producing lymphocytes (55). Genetically modified mice carrying a single mutation in ERO1α and ERO1β show a subtle and tissue-specific phenotype. Mice lacking ERO1α have an abnormal cardiac response to adrenergic stimulation, and mice lacking ERO1β develop a mild and nonprogressive form of diabetes associated with pancreatic cell dysfunction and a defective insulin biogenesis (13, 55). Hence, it is possible that the activity of one specific ERO1 isoform is crucial in some tissues, whereas ERO1 function in other tissues can be complemented by the activity of the paralog. Despite these differences between the two ERO1 isoforms, biochemical analyses leave no doubt that like yeast ERO1, both mammalian ERO1s function as disulfide oxidase accepting electrons from reduced PDI (9, 20, 48, 57).
However, the dogma that ERO1 is necessary for oxidative folding has been recently questioned by experiments in which fly ERO1 and mouse ERO1α and β activities were abolished (in the case of the fly) or greatly inhibited (in the case of the mouse) by means of genetic perturbation (43, 55).
Mice double mutant for both ERO1 are viable and display only a modest delay in terms of oxidative folding. In this genetic context, PRDX4 backs up disulfide oxidase and drives oxidative folding (57).
Mice lacking PRDX4 are viable and fertile (22), which suggests that the enzyme is not essential for disulfide bond formation under physiological conditions, but becomes an important disulfide oxidase in cells with impaired ERO1 activity (57).
ERO1 contributes to the ER pool of H2O2 under normal circumstances, but the source driving PRDX4-dependent disulfide bond formation in cells lacking ERO1 is clearly different. It can be speculated that there are some extra-ER sources of H2O2, because the diffusion of H2O2 through cell compartments may be modulated by changes in the membrane permeability, or as a result of transport through the transmembrane aquaporins (24).
Consequently, the mitochondrion-associated membrane junctions between mitochondrial and ER membranes could channel H2O2 into the ER for PRDX4-mediated oxidative protein folding, which would explain previous findings of the mitochondrial respiration-dependent production of disulfides in mammalian cells (53).
As mitochondria could theoretically be an important source of H2O2, it is tempting to speculate that in a highly secretory cell system, such as that of pancreatic β-cells, the close coupling of mitochondrial respiration and nutrient availability may provide a mechanism for the accelerated oxidative folding of proinsulin, which is translationally activated by nutrients (21). This may also explain an interesting peculiarity of β-cells, which are endowed with superoxide dismutase (which converts oxygen radicals to H2O2), but lack catalase and the GSH peroxidase required to breakdown H2O2 (42). This is something that may have evolved to enable β-cells to harness H2O2 efficiently for the disulfide bond formation required during proinsulin folding.
However, at an experimental level, ero1-1 yeast (yeast with the highly penetrant temperature-sensitive ero1-1 mutation) with impaired mitochondrial respiration due to the deletion of coq3 is nonetheless rescued by PRDX4, thus indicating that an intact respiratory chain is not an exclusive requisite for PRDX4-mediated oxidation in yeast (57). Similarly, ERO1-deficient mouse embryonic fibroblasts (MEFs) are not conspicuously hypersensitive to impaired mitochondrial gene expression in a culture medium containing ethidium bromide (E. Zito and D. Ron, unpublished data).
In brief, although experimental observations do not completely exclude the possibility that mitochondria are a source of the H2O2 that fuels PRDX4-mediated oxidation, they argue against the fact that this is the only alternative source of H2O2 in the ER.
By coupling the β-oxidation of long-chain fatty acids with oxidative protein folding, peroxisomes are another potential source of H2O2 for PRDX4-mediated disulfide bond formation (15). However, there are no experimental data that consider this source of H2O2 for this purpose.
H2O2 can also be produced by the NADPH oxidase (Nox), a family of integral membrane proteins: Nox generates superoxide by transferring one electron from NADPH to O2, and the superoxide then undergoes dismutation to H2O2.
In this regard, it is worth mentioning that by channeling the H2O2 produced by Nox, cytosolic peroxiredoxins regulate the activity of signalling molecules such as oxidation-sensitive phosphatases (51). A similar diffusion of H2O2 into the lumen from ER-localized NADPH oxidases (Nox4) may therefore couple extracellular signaling and PRDX4-mediated oxidative folding.
Experimentally, ERO1-deficient MEFs are not hypersensitive to diphenylene iodonium, an inhibitor of Nox (E. Zito and D. Ron, unpublished data), and so the experimental data suggest a redundancy in the H2O2 sources driving PRDX4-mediated oxidative folding, and this is also reflected by the apparent lack of effect of the ERO1 genotype on the rate of endogenous PRDX4 reoxidation after a pulse of DTT (57).
Finally, it has been shown that most cell types elicit a small oxidative burst that generates low concentrations of H2O2 when they are stimulated by cytokines, growth factors, and hormones, for example, interleukin 6, transforming growth factor-1 (TGF-1), granulocyte–macrophage colony-stimulating factor, and fibroblast growth factor-2 (41). This again leads to the speculation that various cellular cues may lead to the regulated buildup of H2O2 and consequent oxidative folding (Fig. 3).
PRDX4 Protects Newly Nascent Proteins Against an Alternative Oxidative Fate
An unexpected level of complexity in the reaction of oxidative folding has been revealed by phenotype studies of compound Ero1 (α and β) and Prdx4 mutant mice. First of all, the recovery of the triple mutant mice (although with a frequency of 18% in comparison with the expected 100% in the Mendelian ratio) reinforces an emerging theme in the redox field: the plasticity and redundancy of the mammalian oxidative folding pathway (24). In this regard, it has recently been shown that Vitamin K epoxide reductase fulfills oxidative folding in the absence of ERO1 and PRDX4 (36).
Furthermore, it was thought that professional secretory cells, such as pancreatic β-cells and antibody-producing lymphocytes, would be particularly susceptible to the loss of disulfide oxidases, because a cell's level of ER stress is related to the protein load passing through its ER. However, these cells in compound (Ero1 and Prdx4) mutant mice are surprisingly little affected in comparison with the conspicuous phenotype at the level of collagenous tissue. The underlying mechanism of this unexpected phenotype unveils an intricate redox scenario in the ER.
In a native cell environment, kinetic delay in oxidative protein folding after the ERO1 mutations exposes the free thiols of new client proteins to competing oxidative conversion into sulfenic acid (triggered by the H2O2 coming from alternative sources), and this process is further enhanced in cells that are devoid of PRDX4, which are even more impaired in terms of oxidative folding and their ability to metabolize H2O2. This implies that another physiological oxidative reaction competes with oxidative folding, and therefore that the folding depends on a regulated equilibrium between the concentration of H2O2 in the ER and the fast kinetics underlying the formation of disulfide bonds. Impairment in this chemical equilibrium causes an excess of sulfenylated proteins at the expense of disulfide-bonded proteins (Fig. 4).
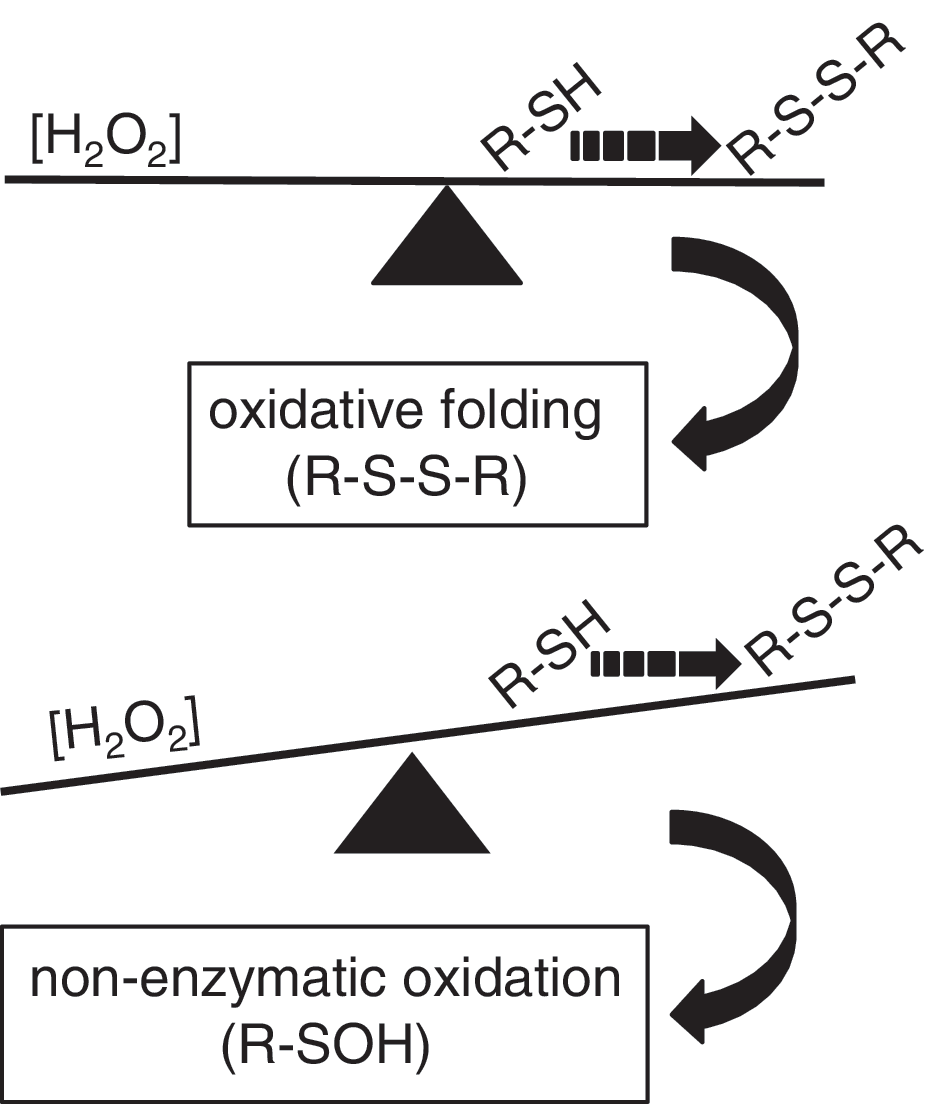
In theory, the formation of cysteinyl sulfenic acid on one thiol followed by its resolution by a second thiol creates a disulfide. In the presence of reduced PDI, the disulfide can isomerize to a correctly placed protein disulfide or be reduced back to the dithiol, and thus initiating a PDI-mediated disulfide relay (25). However, both processes are slow, and cysteinyl sulfenic acid is probably subject to alternative fates.
Because of their highly reactive properties, sulfenylated proteins are transient intermediates of more stable, hyperoxidized (and therefore inactive) sulfinic and sulfonic proteins (27). Thus, a unique process takes place to protect redox homeostasis in the ER and re-establish the initial reduced redox state of the cysteines: the abundant and well-placed ER-resident ascorbate [which reaches millimolar concentrations in most tissues (30)] is recruited to reduce the sulfenylated proteins (56) (Fig. 5).

The two-electron reduction from sulfenic acid back to free thiol converts ascorbate to its unstable oxidized dehydroascorbate derivative (31).
Dehydroascorbate could theoretically later reoxidize thiols to disulfides, and thus contribute to the ER disulfide relay, but this process is too slow to recycle them back to ascorbate effectively (37) in comparison with the rapid hydrolytic process that produces the 2,3-diketo-l-gulonate (2,3-DKG) derivative (10).
Finally, other abundant intracellular reductants such as GSH could take part in the reduction of sulfenylated proteins. However, as it is believed that the concentration of ascorbate in the ER is proportional to its requirement (29), and that collagenous tissues largely rely on ascorbate (as discussed later), a hypothetically higher ascorbate/GSH ratio in the ER of the collagenous tissues would lead to a preferential engagement of ascorbate as a reductant of the oxygenated thiol derivatives.
Moreover, it has been recently shown by three different laboratories that cells deficient in disulfide oxidases have a higher level of oxidized GSH (2, 56, 36). These observations imply that mutant cells may have less-reduced GSH in the ER, thus explaining why the sulfenic species might selectively exploit ascorbate as their reductant.
Pathophysiological Implications of Defective PRDX4-Mediated Oxidative Folding in an ERO1 Mutant Background
The probable conversion of ascorbate to the hydrolyzed, and therefore dead-end product 2,3-DKG in the process described above starves the triple-mutant (Ero1 α and β and Prdx4) ER of an important cofactor. This impairs the activity of ER-localized proline 4-hydroxylase, proline 3-hydroxylase, and lysyl hydroxylase, which are all enzymes dedicated to collagen biogenesis that require ascorbate as cofactor (23). The result is a delay in collagen maturation, the retention in the ER of an unfolded (i.e., not structured as triple-helix) procollagen that is structurally incompetent for secretion, and a consequently collagen-defective ECM that shares some aspects with the consequences of a nutritional deficiency in Vitamin C (scurvy) (Fig. 6A).
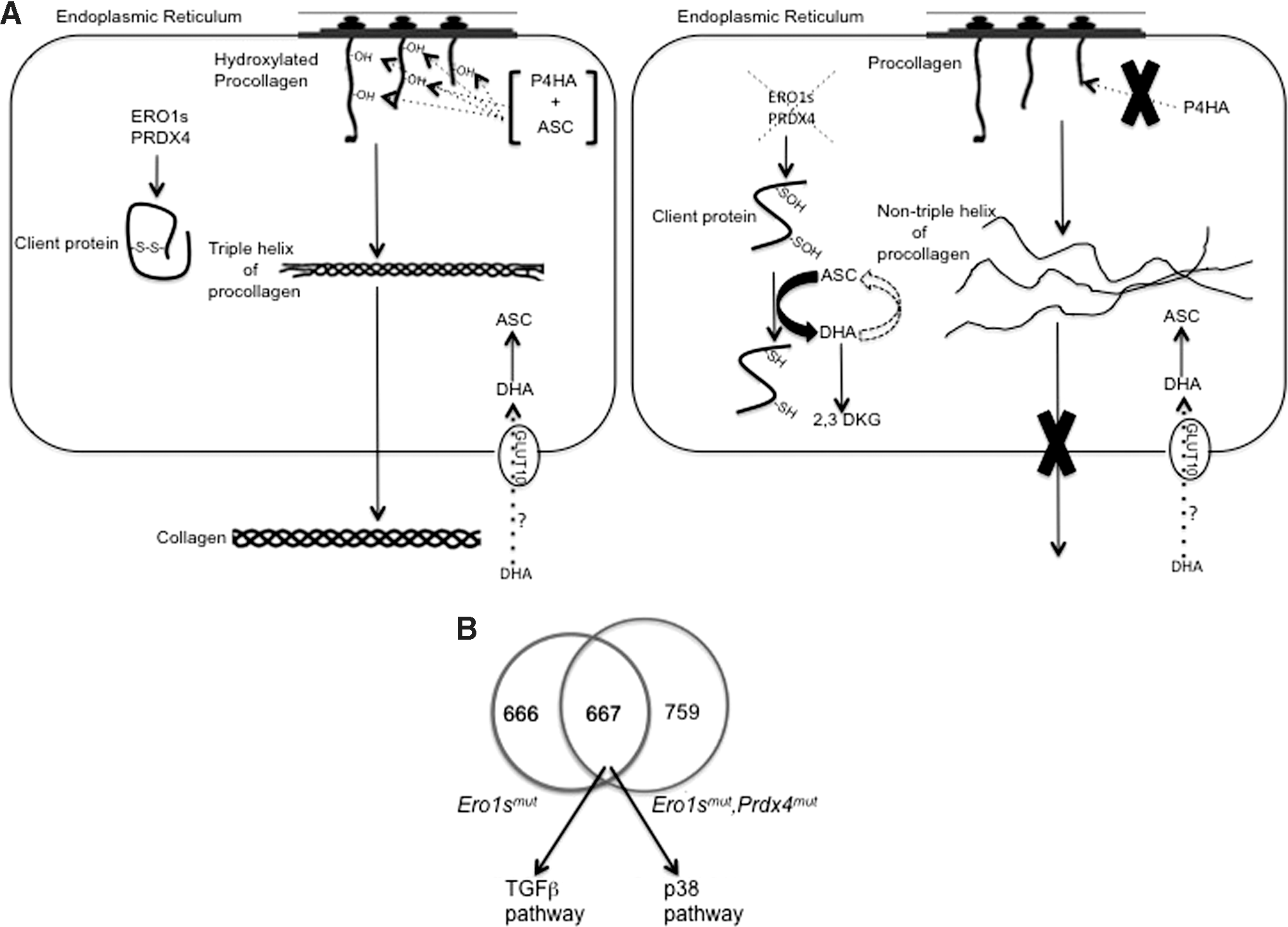
The ECM acts as a reservoir of various cytokines and growth factors, thus creating a microenvironment that commits cells to a specific metabolic fate such as senescence or differentiation. Alterations in the architecture of the ECM trigger the deregulated release of ECM-bound cytokines and signaling molecules (such as TGFβ) that can directly or indirectly regulate cell proliferation (28).
Furthermore, it has been reported that the p38 pathway is activated downstream of TGFβ, and that this is sufficient to trigger a senescent cell phenotype and a senescence-associated secretory phenotype (SASP), a condition that aids tissue repair as a result of the secretion of growth factors (35).
Gene expression analyses show the selective and conspicuous upregulation of TGFβ and the related p38 mitogen-activated protein kinase (MAPK) pathway in mice with mutant ERO1 and PRDX4, which fits their senescent cell phenotype (Fig. 6B).
As the ER is transmitted during cytokinesis rather than being synthesized de novo, even in a simple organism such as yeast, the fate of the newborn cells is controlled at each mitotic division by a tight program that guarantees that only stress-free ER is transmitted to the offspring. This signaling program (the ER surveillance pathway) links ER stress and arrest of the cytokinesis (4), as a result of the activation of Hog1 MAPK (the yeast homolog of p38) (7) to preserve the integrity of the cell.
A parallel connection between ER redox stress and delayed cytokinesis is also found in mammals, although the relay of events is different, because the only evidence in mammals so far is that the biological signals involved in delaying the cytokines start from an altered ECM.
Thus, in mammals, an imbalance in ER redox, an altered ECM, the activation of the TGFβ and p38 pathways, and ultimately, senescence creates an imaginary bridge between the ER, the ECM, and the nucleus: the molecular events triggered by a redox-impaired ER and altered ECM lead to a delay in cytokinesis.
As this senescent cell phenotype is associated with the secretion of numerous growth factors, cytokines, and proteases that can ultimately promote tissue repair (35), it is possible to speculate that this relay of events helps cells to cope with the ongoing stress controlling cell cycle progression by imposing growth arrest (and thus a more suitable and less metabolically active phenotype) and the consequent secretion of the factors involved in tissue repair, while the UPR tries to re-establish the ER function (Fig. 7).

Concluding Remarks
What is remarkable about PRDX4-mediated oxidative folding is the coupling of H2O2 metabolism and disulfide bond formation in newly nascent proteins. The inactivation of PRDX4 has two deleterious consequences for oxidative folding: the slow transition from free thiols to disulfide-bonded proteins, and the build-up of H2O2. The combination of these two phenomena leads to an increase in sulfenylated proteins and the consequent depletion of the reductant ascorbate. The progressive deficit of ascorbate culminates in a defective ECM that triggers the TGFβ and p38 pathways involved in delayed cytokinesis and the senescent phenotype.
In a simple incarnation, this array of transduction signals linking the ER and cytokinesis may serve to put the cells in a dormant state while the UPR tries to resolve ER stress, and so further investigations of the cell mechanisms regulating the pathway linking ER redox imbalance and cytokinesis could pave the way to finding new therapeutic strategies.
The striking difference between mice and humans (the former produce their own ascorbate, whereas the latter depend on exogenous intake for survival) makes humans more sensitive to the enhanced intracellular clearance of ascorbate. This makes it possible to speculate that as they increase the cell content of sulfenylated proteins (and thus the clearance of ascorbate), mutations in ERO1 and PRDX4 are not compatible with human life.
However, combined ER and oxidative stress accompanies many pathophysiological states (19) that could have a similar, but less severe, underlying redox mechanism than that operating in Ero1/Prdx4 mutant mice. In the context of such diseases, it is worth studying the intracellular metabolism of ascorbate, because intracellular ascorbate depletion (due to oxidative stress) may contribute to the altered secretion of extracellular proteins and the development of tissue abnormalities. One correlate of this is the reduced Vitamin C levels observed in the cells of diabetes patients, whose disease is also largely associated with oxidative stress and defects in the ECM (46).
If the reduced intracellular ascorbate levels (due to enhanced clearance) in such diseases are associated with low serum vitamin levels, a treatment paradigm based on the administration of high-dose ascorbic acid might be evaluated.
Footnotes
Acknowledgments
I am grateful to Prof. David Ron for his generous advice and inspiring guidance during my stay in his laboratory. A special thanks also to Prof. Jean-Francois Collet for the critical reading of the manuscript.
EZ is a recipient of an EMBO long-term fellowship ALTF649-2008, a Marie Curie International Reintegration Grant (IRG), and a DTI (Dulbecco Telethon Institute) career award.