
Abstract
Introduction
M
This is the first report of KATP channel-mediated mechanosignaling as a trigger for revascularization in vivo. We had reported earlier that KATP closure is an initial event with stop of flow; here, we show that the redox signaling events which follow were mediated partially by endothelial NADPH oxidase 2 (NOX2) activation that plays a large role in driving revascularization in vivo.
To study endothelial flow sensing and its attendant signaling, we have used the model of removal of shear on endothelial cells in vitro and in situ (11 –13, 44, 54, 65). This model would occur physiologically with vascular obstruction, resulting in ischemia. Using isolated intact lungs, isolated aorta, and flow adapted cells in vitro, we have shown that endothelial cells respond to acute loss of flow (ischemia) with a signaling cascade that is characterized by the generation of reactive oxygen species (ROS) (12, 13, 43, 54). We have also established that the mechanism for increased generation of ROS after stop of flow is the deactivation of a KATP channel which leads to endothelial membrane depolarization, resulting in NADPH oxidase assembly. ROS generation was prevented by pretreating cells with a KATP channel agonist to prevent endothelial cell membrane potential change (12, 13, 54, 65).
There have been extensive reports of NADPH oxidase 2 (NOX2)-derived ROS as a cell signaling molecule or in causing oxidative injury. The determining factors for oxidative damage or signal transduction seem to be based on the levels of ROS generated, its sources, and its cellular targets. We have reported earlier that the ischemia-induced superoxide generation (via activation of NOX2) during the initial 2 min of ischemia by pulmonary endothelial cells is ∼5.1 nmol/min per 106 cells (40). This superoxide is produced extracellularly and mostly dismutates into H2O2; our earlier reports detected ROS (produced on lung ischemia) with H2O2-sensitive dyes, Amplex red, 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), and carboxy-difluorodihydrofluorescein diacetate (H2DFFDA) (13, 40, 65).
In studies undertaken to understand the effect of ROS in signaling post ischemia, we showed that pulmonary endothelial cells, when subjected to loss of shear, exhibited increased cell proliferation (44 –46). The response to loss of shear required a preceding period of exposure to flow in order to reach a flow adapted state. We found that blocking ROS production or scavenging by either antioxidants or inhibitors (i.e., KATP channel agonists or NADPH inhibitors) or by using endothelial cells from gene-targeted mice (knockouts of KATP channel [KIR6.2−/−] or NOX2, i.e., cells that show diminished ROS generation with ischemia) markedly inhibited cell proliferation with ischemia (45, 46).
The aim of this study was to determine whether ischemia-induced ROS-mediated endothelial cell proliferation is sensitive to the KATP-dependent mode of NOX2 activation, and whether it has relevance to vascular remodeling. For this, we used an in vitro flow adapted endothelial cell system and the in vivo femoral artery ligation model; knockout mice and agonists/inhibitors were used to evaluate the role of KIR6.2 channel-dependent NOX2-derived ROS in angiogenesis and vascular remodeling associated with ischemia. This murine model of ischemia is particularly relevant to peripheral artery disease that is characterized by stopped flow in the systemic vasculature; the subsequent remodeling process is also clinically relevant in the context of ischemia in the systemic circulation.
Vascular remodeling is a complex phenomenon and involves myriad signaling pathways; indeed, it spans the scale from cells to budding vessels and, eventually, an established vascular network. All this involves the interplay of multiple interacting cells and signaling molecules. The particular focus in this study is ROS generated by NOX2 in endothelial cells. In addition to endothelial cells, NOX2 is also expressed in neutrophils and is a major contributor to the overall ROS production in the ischemic hind limb (59, 60). Thus, to address the role of endothelial NOX2-derived ROS (as compared with ROS from other sources) in remodeling post ischemia, we generated transgenic mice with endothelial-targeted overexpression of NOX2 (endoNOX2Tg). Proliferative signaling and expression of vascular remodeling markers, hypoxia inducible factor-1 alpha (HIF-1α) and vascular endothelial growth factor (VEGF), were also measured in ischemic tissue to determine whether their induction is driven by ROS produced by ischemia.
Based on our results, we conclude that mechanotransduction via the KATP channel drives vascular remodeling via ROS generation by endothelial and other cells, which subsequently increases VEGF and HIF-1α production. Thus, the biochemical response to altered flow generates a signal for growth of collateral vessels and represents an attempt at revascularization to restore the impeded blood flow.
Results
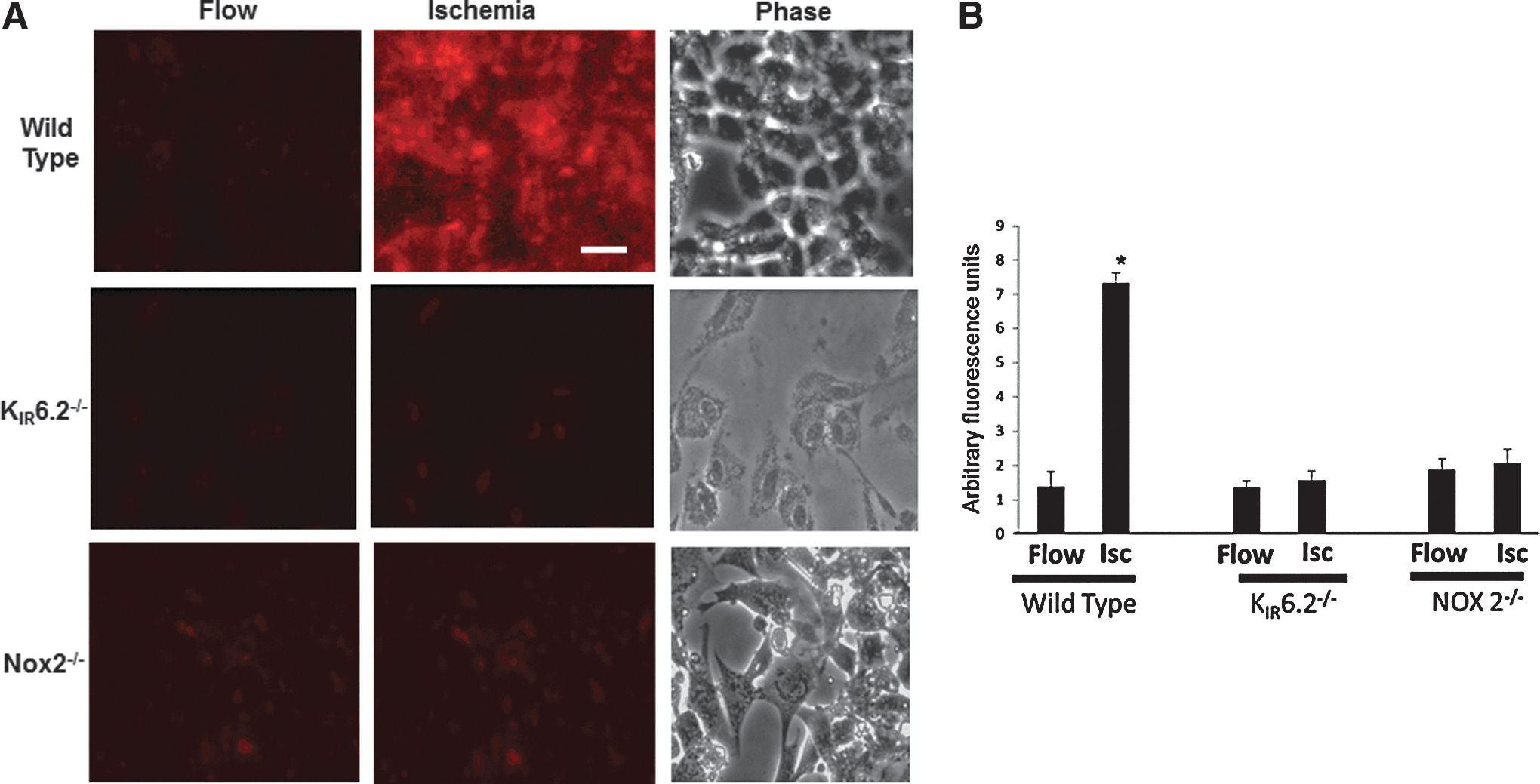
ROS production with ischemia in vitro
Mouse pulmonary microvascular endothelial cells (PMVEC) isolated from wild-type (WT), KIR6.2, and NOX2 null lungs (13) were kept in a parallel plate flow chamber (Warner, Inc.) and flow adapted for 24 h. Endothelial cells were labeled with dihydroethidium (DHE) and imaged for superoxide production in real time with stop of flow. Superoxide production was evidenced by increased fluorescence on stop of flow in WT cells. This was significantly lower in cells derived from KIR6.2−/− and NOX2−/− lungs (Fig. 1A, B).
Tube formation in flow adapted, ischemic PMVEC is ROS dependent
To assess the role of ROS in tube formation in vitro, we compared cells that were flow adapted and cells cultured under static conditions (static cells). We have shown that stop of flow with flow-adapted cells results in bursts of ROS and cell signaling which were not observed in statically cultured cells (40, 44, 62). WT flow-adapted, ischemic PMVEC showed tube formation at 24 h, while statically cultured cells did not. NOX2 null PMVEC that were flow adapted also did not show tube formation. These cells lack the membrane subunit (NOX2) of NADPH oxidase, and, thus, ROS production in response to stop of flow was not detected. KATP −/− (KIR6.2−/−) PMVEC also did not show tube formation under the same conditions (Fig. 2). The latter cells do not depolarize with ischemia (13, 43, 65). Our previous studies have shown that cell membrane depolarization is necessary for activation of NADPH oxidase to produce ROS in response to ischemia.

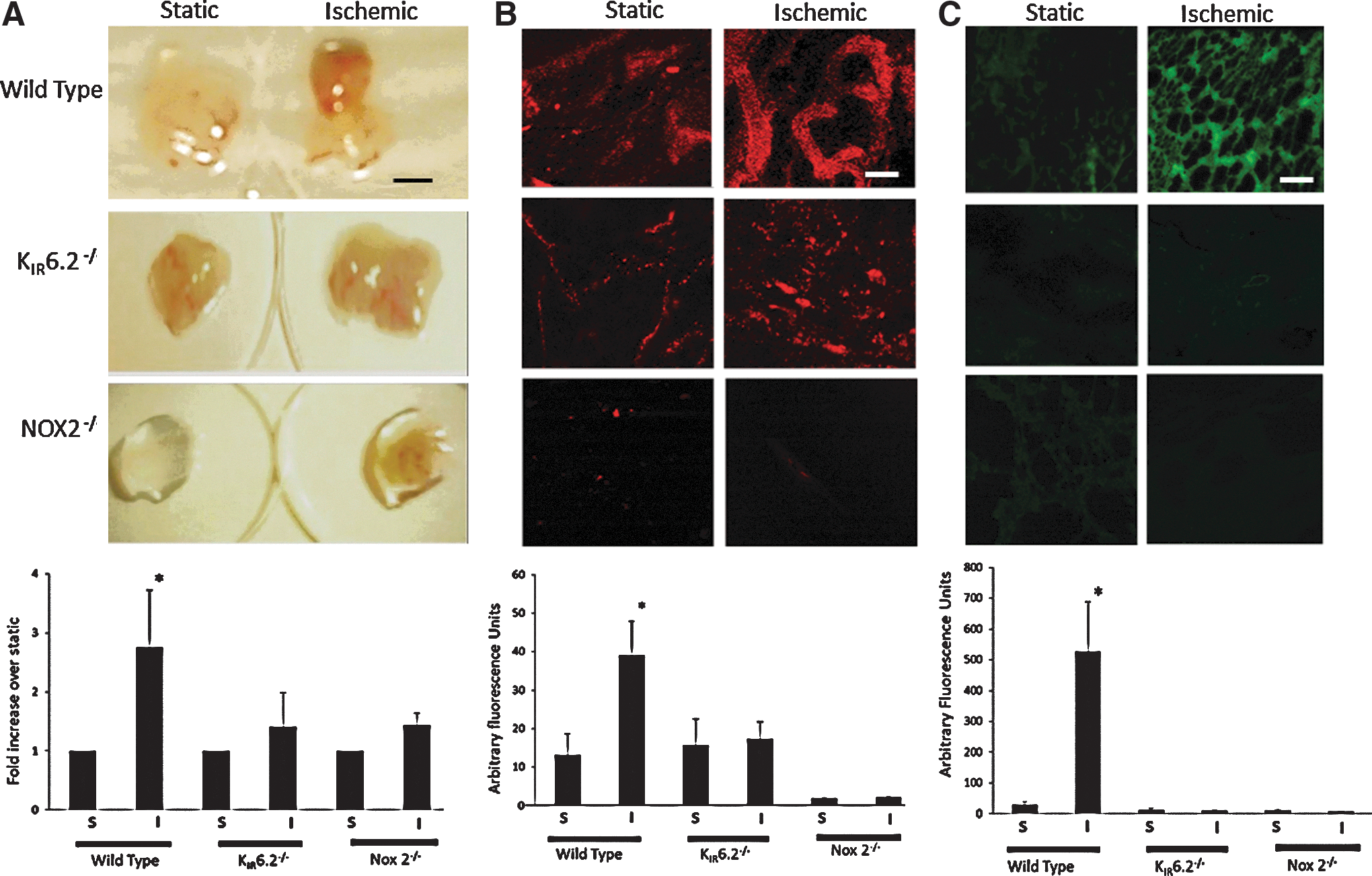
Vessel formation in vitro by flow-adapted, ischemic PMVEC is ROS dependent
To examine whether ROS produced post ischemia drives vascularization, flow-adapted, ischemic cells mixed in Matrigel were injected as subcutaneous plugs into nude mice. Plugs containing WT, ischemic cells showed increased hemoglobin content as compared with plugs that were seeded with static cells (Fig. 3A). These excised plugs were treated with the endothelial marker di–I acetylated low density lipoprotein (DiIAcLDL), and visible vessel formation was observed that was significantly greater in flow-adapted, ischemic cells as compared with cells cultured under static conditions (Fig. 3B). Cryosections of the plugs showed staining with anti-platelet endothelial cell adhesion molecule (PECAM), indicating endothelium within the plugs (Fig. 3C). The increased hemoglobin and endothelial expression of AcLDL and PECAM (by staining with DiIAcLDL and anti-PECAM respectively) indicated vascularization within the plugs containing WT, ischemic cells. However, DiIAcLDL staining was not observed in plugs containing static cells or ischemic KIR6.2 and NOX2 null cells that do not generate ROS with ischemia. In addition, PECAM staining that was observed in plugs with WT, ischemic cells was very low in plugs with ischemic knockout cells and statically cultured cells.
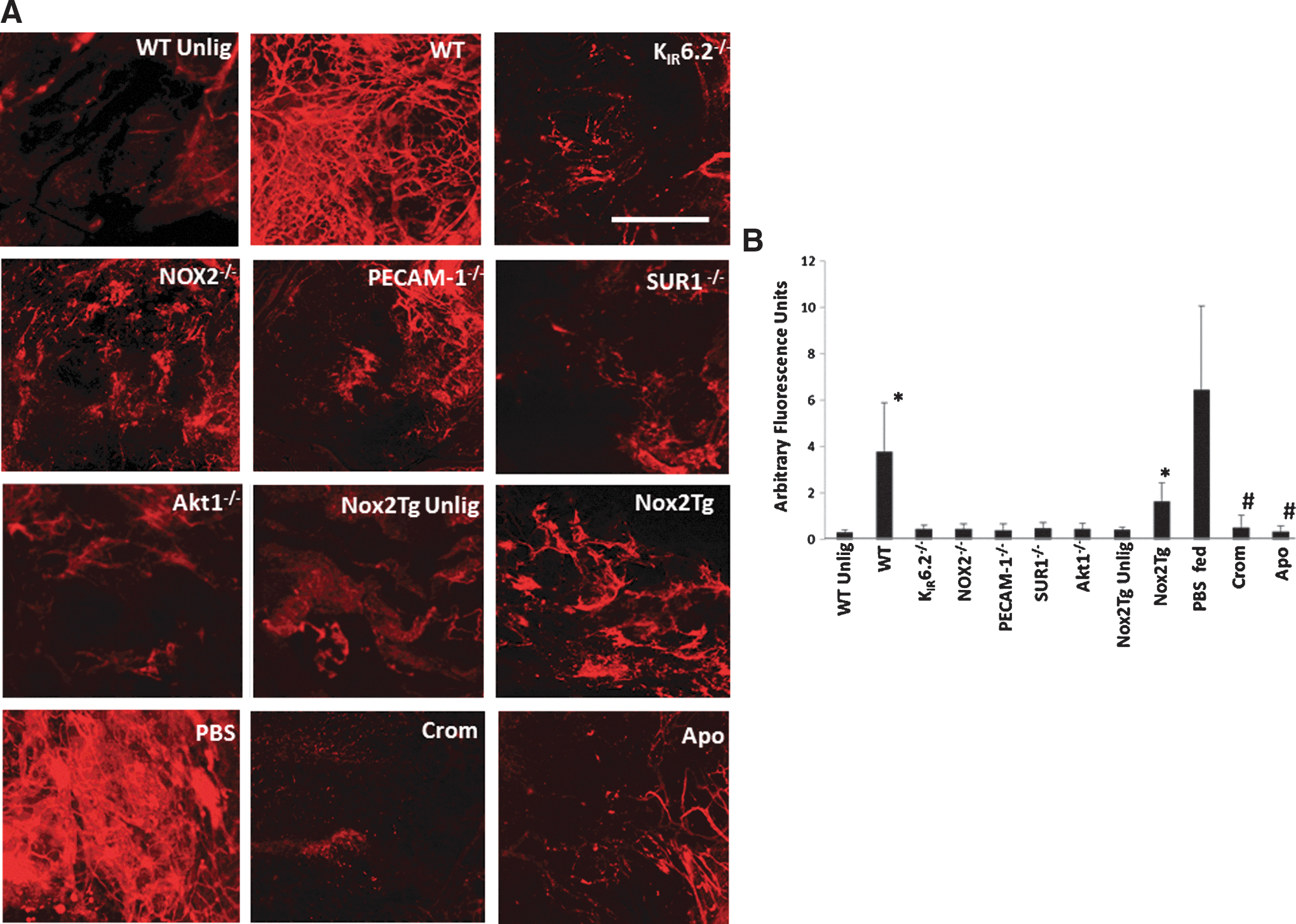
Vascular remodeling (neovessel formation) in hind limb ischemia is dependent on the KIR6.2 channel and NOX2-induced ROS
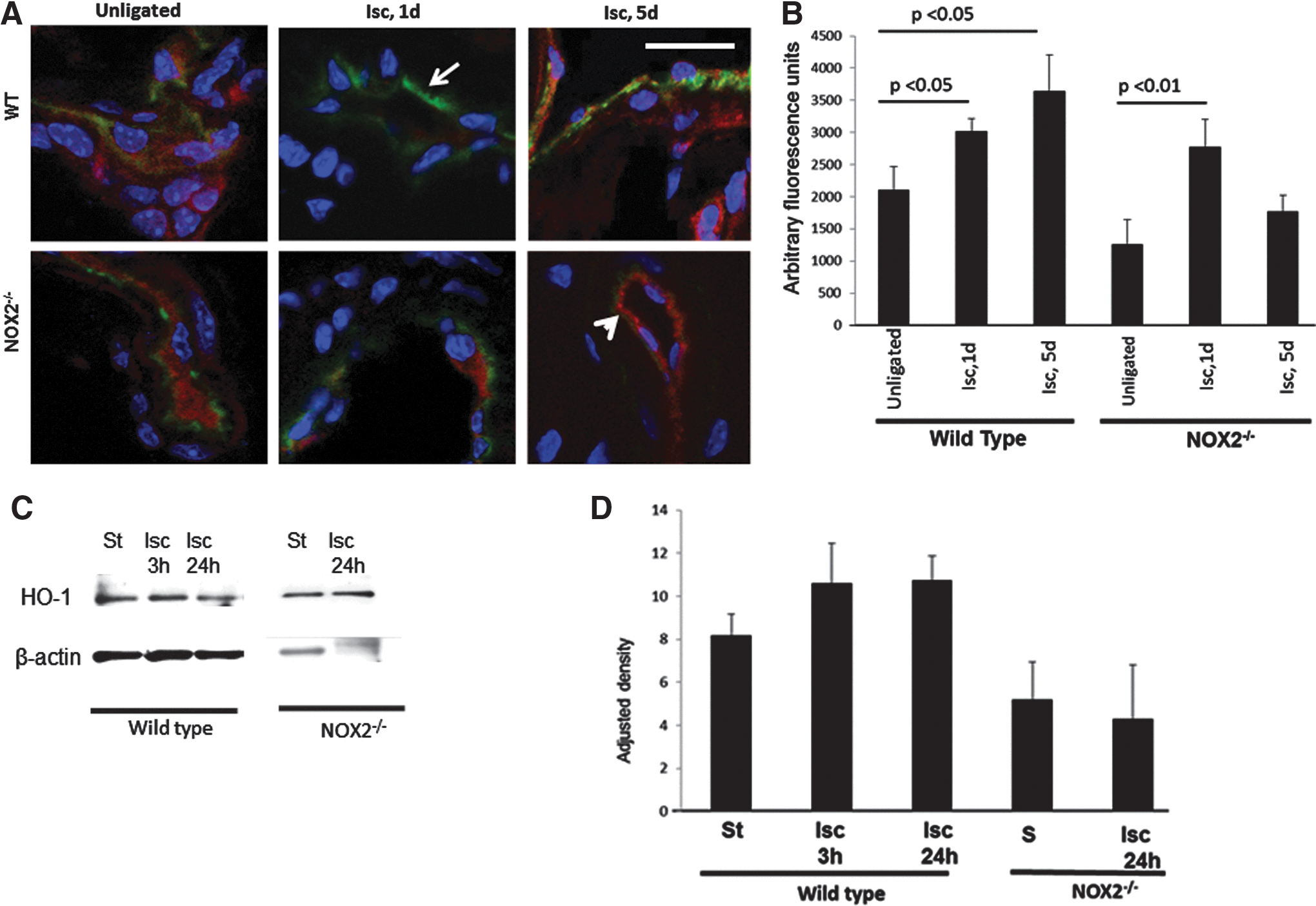
We evaluated the formation of new vessels in our model of limb ischemia in which the vessel is only tied, not excised. Our initial inspection, in WT mice, of the ischemic region, that is, the area between the sutures immediately on ligation and 5 days later, revealed the appearance of a fragile reddish area between the ligatures. We sought to obtain more accurate methods to quantify the extent of vascularization in the various mice used by using a quantitative fluorescence assay based on entrapment of 40 nm beads inside neovessels. The fluorescent signal that emanates from the beads entrapped inside vessels <40 nm is indicative of revascularization (Fig. 4A). Ischemic tissue from WT mice when imaged by confocal microscopy showed the entrapped fluorescent beads, while unligated hind limb controls had almost no bead entrapment. Mice with knockout of KIR6.2 and NOX2 also showed less revascularization post ischemia as compared with WT mice. Revascularization post ischemia was also compromised in mice fed with the KATP agonist cromakalim or the NADPH oxidase inhibitor apocynin, as well as in mice in which the channel function was compromised (SUR1−/−). Deletion of the SUR subunit is reported to compromise KATP channel function (25, 42). PECAM-1−/− and Akt1−/− mice that do not produce ROS with ischemia (12) showed less neovascularization as compared with WT mice. Quantitation of the fluorescence signal emitted by the entrapped beads was achieved by integrating the intensity of fluorescence of the images obtained by confocal microscopy of the ischemic region using Metamorph Software. Revascularization based on this assay was several fold lower in mice that do not have ROS generation with ischemia as compared with WT mice (Fig. 4B). Transgenic mice with endothelial targeted expression of NOX2 (endoNOX2Tg) were subjected to hind limb ischemia; these also showed revascularization that was somewhat intermediate between WT and NOX2 and KATP null mice, indicating a role for endothelial NOX2 in revascularization.
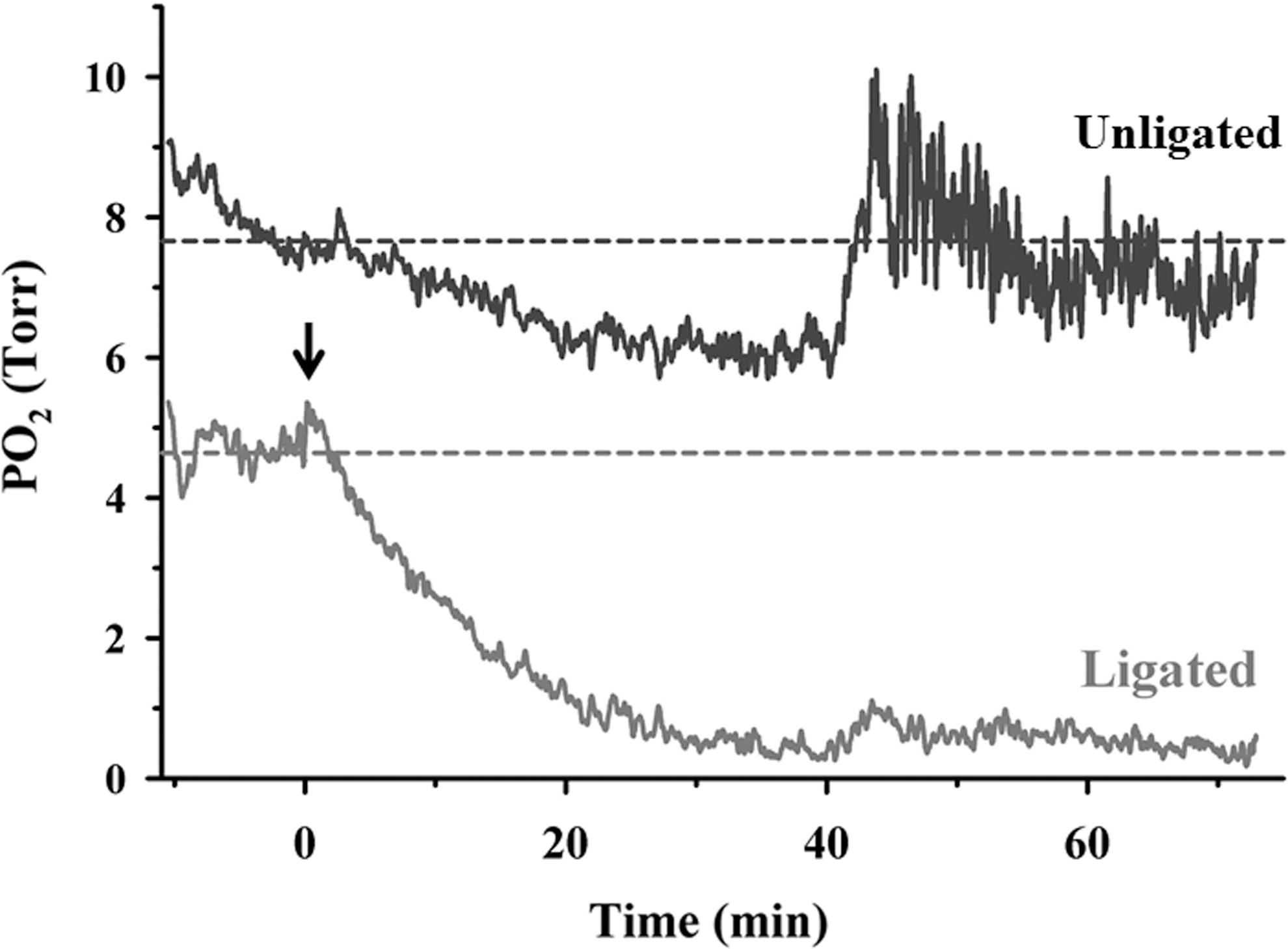
Hypoxia in hind limb tissue post ischemia
To ascertain the effects of stopped flow on the oxygen delivery in the ischemic limb, we measured tissue pO2 levels using an oxygen-sensitive microelectrode (10, 57, 61). Tissue pO2 was measured before ligation and immediately after ligation, and measurement continued till 1 h post ligation. For the unligated hind limb recordings, similar conditions were used except that the ligatures were not tied; that is, the skin was cut open, the femoral vessel was exposed, and the oxygen-sensitive electrode was kept in contact with the tissue. We observed that within 20 min after ligation, tissue pO2 values fell by ∼80% (Fig. 5). This did not occur if no ligation was performed, indicating that the reduced oxygen supply within a few minutes was due to the stop of blood flow.
HIF-1α and VEGF in ischemic tissue is ROS dependent
The ischemic tissue was assessed for HIF-1α activation, as ROS-induced HIF translocation has been reported in other studies (9). HIF-1α activation as determined by its increased expression in the nuclear fraction (Fig. 6A) of ligated tissue was significantly higher in WT mice as compared with KIR6.2 and NOX2 knockouts, that is, mice that do not produce endothelial ROS post ischemia (Fig. 6B).

Blocking HIF-1α in vitro abrogates tube formation
We next checked the role of HIF-1α in driving tube formation with ischemia in vitro. When flow-adapted PMVEC were pretreated with 30 μM CAY-10585, an inhibitor of HIF-1α accumulation and transcription (5, 34), before ischemia, we observed complete abrogation of tube formation under similar plating density conditions on Matrigel dishes (Fig. 6C). Instead of forming sprouts and eventual tubes, these cells flattened out and grew as clusters. This indicates that the HIF-1α pathway plays a role in transducing the ischemic signal (ROS) into an angiogenic response.
Expression of hemeoxygenase-1 and VEGF
Hemeoxygenase-1 (HO-1), the inducible isoform of HO, catalyzes heme oxidation and via the products biliverdin, CO, and free ferrous iron, plays a role in vascular homeostasis (19, 36, 38, 47). HO-1 is expressed in response oxidative stress and inflammation, and is reportedly induced by HIF-1α activation. HO-1 has also been reported to drive angiogenesis via VEGF expression (36, 47). We monitored HO-1 expression at 0, 1, and 5 days post ischemia in vivo by immunostaining sectioned tissue around the revascularized region. HO-1 expression increased with ischemia and was highest at 5 days post ischemia. However, in sections obtained from NOX2 null mice, there was an initial increase in HO-1 expression followed by a decrease at day 5 (Fig. 7A, 7B). When mouse PMVEC were flow adapted and subjected to ischemia in vitro, no significant change in HO-1 expression was observed in either WT or NOX2 null cells (Fig. 7C, 7D).
As is well established, VEGF drives angiogenesis. We monitored VEGF expression at 5 days post ischemia by immunostaining sectioned tissue around the revascularized regions (region between the ligatures). There was increased VEGF expression in sections derived from ischemic regions of WT mice as compared with the knockout models (Fig. 8A, B). In addition, we quantified VEGF expression by ELISA from the whole vascular bundle of the ischemic region. VEGF production is significantly higher in mice that generate ROS with ischemia as compared with mice that do not (Fig. 8C). We investigated whether the restoration of VEGF expression in the null mice would rescue the revascularization to WT levels. For this, we delivered VEGF protein to KIR6.2 null mice in vivo, post–hind limb ischemia (by Chariot delivery method, see Materials and Methods section) (4, 41). Introduction of VEGF in KIR6.2−/− mice restored revascularization to WT levels (Fig. 9A, B). The tissue sections from the ischemic region in the rescued mice showed VEGF expression (Fig. 9C).


Increased nitric oxide (NO) bio-availability may play a role in neovascularization. NO generated in our in vivo model post-ligation was assessed by monitoring the tissue levels of NOx (nitrate, nitrite, and S-NO) (28, 56) in homogenized hind limb. WT mice contained a slightly lower amount of NO products (mean±SE=1.9±0.2 NOx/mg protein) when compared with NOX2 null (2.5±0.2 nmol NOx/mg protein). These results for n=3 samples were not statistically different (p>0.05).
Discussion
We have been engaged in studying the response of endothelial cells to cessation of flow as would happen in the in vivo state with various obstructive pathologies (2, 3, 12, 13, 54, 65). In general, stop of flow involves (i) alteration of the mechanical component of blood flow, (ii) compromised delivery of oxygen and nutrients to tissue, and (iii) failure of removal of CO2 and other metabolites.
In order to understand endothelial mechanotransduction and subsequent endothelial signaling with loss of the mechanical component of flow, we established models in which stop of flow did not compromise oxygen delivery and nutrient supply. Using these models (i.e., intact perfused rat and mouse lungs and flow-adapted PMVEC adapted to shear values of 1–6 dynes/cm2 that typically represent the in vivo state), we showed that abrupt cessation of flow causes an endothelial cell membrane depolarization which leads to ROS production via activation of endothelial cell NADPH oxidase (3, 12, 13, 54, 65). Patch clamp of flow adapted cells in vitro revealed that stop of flow resulted in an immediate decrease of the inward rectifier K+ (KIR) current, indicating depolarization with abrupt loss of shear. Lungs and cells with “knock-out” of KIR6.2 or treated with KATP agonist showed almost no change in membrane potential and showed markedly reduced ROS production with ischemia (13, 64, 65), leading us to conclude that KIR6.2-induced depolarization and NADPH oxidase-induced ROS generation were the result of altered mechanotransduction.
Our experiments with ischemic cells in vitro showed that ROS production (as detected by oxidation of DHE) and cell proliferation post ischemia were dependent on the presence of KIR6.2 channels and NADPH oxidase, indicating that ROS drives cell proliferation (45, 46). DHE is reported to be reasonably specific for superoxide (8). To understand the implications of cell proliferation post-stopped flow in vivo, we investigated angiogenic potential of ischemic cells. Since the Matrigel matrix is similar to the natural environment of a basement membrane for endothelial cells, the formation of capillary-like structures that comprise rows of endothelial cells reminiscent of “beads on a string” can be considered an index of angiogenesis. Our data (Fig. 2) showed that cells which produce ROS on ischemia (as seen in Fig. 1) show angiogenesis in vitro. In the in vivo plug model, angiogenesis within the plug is induced by angiogenic signals emanating from endothelial cells within the plug. Nude mice, being immunocompromised, do not reject cells in Matrigel-injected in vivo and angiogenic signals from the injected endothelial cells recruit endothelial cells from the host animal and form blood vessels within the plug. An assessment of angiogenesis by measuring hemoglobin or by scoring selected regions of histological sections for vascular density also showed that ROS-generated post ischemia drives neovessel formation; cells that do not generate ROS with ischemia do not show extensive neovessel structures within the plug, although they stain positively for endothelial markers (Fig. 3).
Next, we extended this study into an in vivo model to understand whether stopped flow in blood vessels might drive vascular remodeling. Based on its pervasive vascularity, the lung is not a suitable model for studying angiogenesis. This led us to consider the hind limb ischemia model to test the effect of the stop of flow signaling cascade on revascularization.
We, thus, checked the effect of stop of flow signaling in vivo using mouse hind limb ischemia as a model of stopped flow and monitored revascularization in the ischemic region 5 days post ligation. We studied the link between membrane depolarization, ROS, and vascular remodeling in the hind limb with stopped flow by monitoring revascularization in mice that do not exhibit changes in membrane potential (KIR6.2 channel null mice) and/or do not generate ROS with stopped flow (KIR6.2−/−, NOX2−/−, Akt1−/−, SUR1−/−, PECAM-1−/− mice).
In most of the earlier studies that utilized hind limb ischemia, the artery and vein were tied, cauterized, and excised (48, 55, 63). We reasoned that the injury arising from the excision of a portion of the femoral artery would make it difficult to separate the effects of stopped flow from the complex signaling cascade and associated factors activated by inflammation and tissue damage. Thus, in our model of hind limb ischemia, the vessel was tied and neither cut nor excised. Ligation of the femoral artery has been reported earlier as causing neovascularization, which was observed to be NOX2 dependent (58). This latter study found that the NOX2 in bone marrow-derived cells was involved in the vascular remodeling; indeed, the authors followed this study by subsequent reports of NOX2-driven mobilization of progenitor and bone marrow-derived cells post ischemia that eventually led to revascularization in the ligated area (59, 60). Although the link between ischemia in the hind limb and the activation of NOX2 in the bone marrow environment is not clear, the authors observed an NOX2-dependent ROS production in bone marrow-derived cells post ischemia and, thus, concluded that ROS from these cells drives revascularization. To separate the effects of bone marrow cells from other cell types, these studies had used bone marrow chimeras that necessitated irradiation of the mice. However, the effects of irradiation on endothelium are not clear and were not considered. Thus, it was unclear whether endothelial NOX2 activity had a possible contribution toward revascularization in the ischemic region. We sought to address these questions by generating transgenic mice with endothelial-targeted overexpression of NOX2. Since NOX2 expression and activation in these mice was limited to the endothelium, their attenuated revascularization as compared with WT and significantly higher revascularization as compared with NOX2 null indicates that endothelial NOX2-driven ROS is important but does not play an exclusive role in vascular remodeling; it is possible that NOX2 and ROS from other cells (polymorphonuclear neutrophils, endothelial progenitor cells, bone marrow-derived cells) also play a role in revascularization with ischemia.
The ROS production post ischemia could arise from an altered mechanical component or a metabolic component or both. We have reported earlier that the mechanosignaling post ischemia is directly modulated by a KATP channel on the endothelium (13). Both KATP null and cromakalim-fed mice showed reduced revascularization post ischemia. In statically cultured cells, the KATP channel agonist, cromakalim, by keeping the channel open, keeps the cell membrane hyperpolarized (33). However, in endothelial cells under flow, the open-state probability of KIR channels is enhanced by shear stress and, in such a milieu, addition of cromakalim would not have any additional hyperpolarizing effect (49). Instead, with stop of flow, cromakalim would, by keeping KATP channels open, prevent depolarization (and thus halt the downstream signaling) that accompanies ischemia (13). Thus, reduced neovascularization post ischemia in cromakalim-fed mice (and in KATP channel nulls) points to a role for the mechanical component of flow, though it does not completely exclude contributions from metabolic changes induced by ligation.
Based on our earlier data, the link between KATP channel and NOX2 activations seems to be the activation of the PI3K/Akt1 pathway (12). In the context of hind limb ischemia, other studies too have shown a role for Akt1 in vascular remodeling (1, 30), although not via NOX2. However, these involved very severe models of femoral artery damage and may, therefore, have initiated a different signaling pathway. Using our model, we observed that Akt1 null mice showed significantly compromised revascularization. In light of our earlier studies on ischemia (12), where Akt1 null lungs showed intact depolarization but no ROS production, we conclude that in in vivo ischemia, NOX2 activation occurs via Akt1.
What is the initiating signal for angiogenesis post ischemia? Clearly, the process is ROS mediated in our model, but the mechanism by which ROS drives angiogenesis was not clear. Both HIF-1α and VEGF are known to regulate angiogenesis (23). HIF-1α, a transcription factor, is composed of an inducible alpha subunit (HIF-1α) and a constitutive β subunit. HIF-1α is stabilized at hypoxic conditions (and degraded under normoxia) and translocates to the nucleus, where it binds to hypoxia-response elements in the promoter of target genes (51), one of which is VEGF (52). Another pathway for HIF-1α activation independent of hypoxia is direct interaction with ROS (6, 21). Agonists such as angiotensin II, lipopolysaccharide, or thrombin have been shown to activate nuclear translocation of HIF-1α under normoxic conditions; inhibition of ROS in the presence of these agonists was found to decrease HIF-1α translocation (26). Monitoring the pO2 values in the ischemic tissues showed that the pO2 declined steadily post ligation, although there was no appreciable change for the first 1–2 min. Since our earlier reports show almost immediate depolarization followed by ROS generation within a minute, it is conceivable that ROS generation in the hind limb occurs before hypoxia sets in. In support of this is, our data in which blocking ROS by the use of NOX2 null and NOX2 inhibitor apocynin almost abolished revascularization. Revascularization has been reported to be both VEGF dependent and independent (27, 53); our data point distinctly to VEGF dependence, as (i) revascularization in the hind limb was seen only in those mouse models that showed increased VEGF expression, and (ii) revascularization could be restored in the null phenotype (KIR6.2 null) by delivery of VEGF into the ischemic region post ligation.
What drives the induction of VEGF? Although VEGF induction has been reported to be NOX2 independent in some cases (50), our model of ischemia shows NOX2 dependence; indeed NOX2 nulls showed very low VEGF induction. Our data show that HIF-1α participates in transducing the angiogenic signal, as blocking HIF-1α accumulation and transcription completely abrogates the ischemia-induced tube formation in WT cells in vitro. HIF-1α is also reported to drive angiogenesis via several growth factors and via HO-1, the inducible isoform of HO, which also induces VEGF expression and angiogenesis in ischemic heart (19, 36, 47). In comparison to other models of neovascularization in which a robust increase in HO-1 is observed (19), our data show a modest HO-1 induction even where extensive neovascularization (WT, 5d ischemia) occurs. Besides, the fact that HO-1 increased in NOX2 null but VEGF expression and neovascularization were still low indicates that unlike other models of neovascularization, in which HO-1 induces VEGF expression (via Sp1 activation), (37) our model of hind limb ischemia probably does not involve HO-1 or that this pathway is a minor contributor here. In addition, HIF-1α activation in our model does not correlate with HO-1 induction in NOX2 null mice. There are instances of HIF-1α dependent but HO-1 independent regulation of VEGF (38); so, it is possible that the modest HO-1 induction in our model does not drive VEGF expression and revascularization. In tumor models of angiogenesis, PI3K/Akt activation causes VEGF induction (32) via endothelial nitric oxide synthase (eNOS) activation. A diminished capacity to produce ROS may also have an impact on the bio-availability of NO. Increased NO bio-availability has often been linked to the regulation of pro-angiogenic factors. The slightly higher NO in NOX2 null mice post ligation was not statistically significant as compared with WT mice under the same conditions; yet, the NOX2 null mice have attenuated levels of revascularization, suggesting that in our model an increase of NO is not sufficient to drive vessel growth in the absence of ROS. Thus, in our model of hind limb ischemia, we postulate that the lack of revascularization in Akt1 null is due to the diminished ROS and not reduced NO signaling. Studies elsewhere (63) have shown a role for NO (specifically eNOS) in ischemic remodeling of the hind limb; however, since those models involved greater injury than observed in our studies, it is possible that the remodeling process relied on different signals.
Overall, blocking ROS generation by use of NOX2 null mice and the NOX2 inhibitor apocynin almost abolished neovascularization in the hind limb, pointing to a role for ROS. KIR6.2 channel null and the KATP channel agonist cromakalim (that prevents the channel closure and thus affects the role of KATP in mechanotransduction) also abolished the revascularization post ischemia.
We conclude that the KATP-NOX2 mechanosignaling axis plays a pivotal role in revascularization. The ROS produced by KATP-NOX2 signaling is transduced via HIF1α and VEGF into a physiological response. Our data indicate that HIF-1α transduces the angiogenic signal via VEGF; indeed, in our model VEGF is critical to the revascularization process. Thus, the KIR6.2-NOX2 machinery is a mechanosensing complex that drives regrowth and revascularization.
Materials and Methods
Chemicals
Apocynin (NADPH oxidase inhibitor) and Cromakalim (a KATP agonist) were from Sigma-Aldrich. Matrigel (growth factor reduced) was from BD Biosciences. Fluorospheres were from Invitrogen. Anti-VEGF was from Abcam. Anti-HIF1α, anti-PECAM, and anti-HO-1 were from SantaCruz Biotechnology. Pierce NE-Per extraction kit (ThermoScientific) was used to obtain nuclear extracts for quantitation of HIF-1α levels. VEGF-ELISA kit was a preconjugated 96-well plate from R&D Biosystems. DHE was from Invitrogen. A chemical inhibitor of HIF-1α, CAY-10585, was purchased from Cayman Chemical and used as directed. Chariot delivery system was from Active Motif. Recombinant mouse VEGF was from Life Technologies. The Chariot system was used in vivo as per the manufacturer's instructions. All in vitro experiments (flow adaptation, ischemia, and tube formation) were carried out at 37°C. Immunostaining of tissue sections and immunoblotting were carried out at room temperature.
Animals
Animal use was reviewed and approved by the University of Pennsylvania Institutional Animal Care and Use Committee. C57BL/6 and NOX2−/− mice weighing ∼20–25 g were obtained from Jackson Labs. Breeding pairs of mice deficient in KATP channels generated by targeted disruption of the KIR6.2 gene were obtained from Chiba University (26, 27) and bred in the University of Pennsylvania animal facilities. KIR6.2 is the pore-forming subunit of the KATP channel in lung endothelial cells (13, 14). KIR6.2−/− mice have been backcrossed for five generations to the C57BL/6 background. PECAM-1−/− mice were kindly gifted by Horace DeLisser's and Steven Albelda's laboratory at the University of Pennsylvania. Akt1−/− mice were a kind gift from the lab of Prof. Morris J. Birnbaum of the University of Pennsylvania (15). SUR1 is the regulatory subunit of the KATP channel in lung endothelial cells. SUR1−/− mice generation and genotyping have been previously described (28, 29). The SUR1−/− mice have been back crossed to the C57BL/6 background; they were bred and maintained at the Children's Hospital of Philadelphia animal facility. Both the KIR6.2−/− and SUR1−/− mice have white coat color because of the albino locus segregates with the KIR6.2 and SUR1 knockout alleles. Except for hair loss in patches on the skin of NOX2−/− mice, there were no obvious phenotypic differences between the WT and gene-targeted mice used in the experiments. Endothelial-specific NOX2-expressing mice were made by breeding male NOX2-overexpressing mice (human NOX2 under control of the endothelial specific Tie 2 promoter) from Keith M. Channon's laboratory at the University of Oxford (7) with NOX2 null female mice. Mice carrying the human NOX2 transgene were obtained by breeding with NOX2 null females. Genomic DNA analysis of tail tips was used to identify mice with the carrier transgene; polymerase chain reaction (PCR) showed the presence of the human NOX2 transgene (225 bp product). RNA from tail tip was extracted, and the cDNA was amplified by PCR using the following primers: for human NOX2 transgene, forward and reverse primers were CTGAGACTCA TCCCAGCCAGTGAGGTAG and GTCACACCCTTCGCA TCCATTCTCAAGTCAGT respectively. The expected band size is 225 bp (J.K. Bendall, pers. comm.). For WT (mouse NOX2) and mutant (NOX2 null) genes, the common forward primer was CTAAGAGAAACTCCTCTGCTGTGAAG and the reverse primers were CGCACTGGAACCCCTGAGAA AGG (for WT) and GTTCTAATTCCATCAGAAGCTTATCG (for NOX2 null). Information on primers was obtained from Jackson Labs Website, and WT primers were slightly modified. The WT allele showed a band at 240 bp; the mutant allele, at 195 bp; and the transgene, at 225 bp. PCR conditions were 95°C for 10 min followed by amplification at 95°C for 30 s, 55°C for 60 s, 72°C for 90 s, and 72°C for 10 s.
Cell culture
Isolation of PMVEC was carried out as previously described (13, 45). Briefly, lung tissue was trimmed at the periphery, digested and PECAM-positive cells were isolated by immunoselection using magnetic beads (22).
Flow adaptation of endothelial cells and ischemia in vitro
The artificial capillary system (FiberCell Systems, Inc.) for flow-adapting endothelial cells has been previously described (11, 45, 59). Cells were also adapted to flow for 24 h in parallel plate chambers (Warner, Inc.) at 5 dynes/cm2. Cells were labeled with the superoxide specific fluorophore DHE by adding the dye to the perfusate; the chamber was mounted on the stage of the microscope and imaged in real time with flow and stop of flow (λex=480 nm; λem=570 nm).
Tube formation
In order to evaluate angiogenic potential of these cells, both in vitro and in vivo tube formation assays were used.
(I) Tube formation in vitro, used to model the formation of three-dimensional vessels, involves growing endothelial cells in a gelled matrix. We used statically cultured and ischemic (flow adapted 72 h at 6 dynes/cm2) PMVEC and compared their capillary forming ability by plating each at a density of 2×104 cells in Matrigel (growth factor-reduced phenol red free)-coated wells. In separate experiments, flow-adapted PMVEC were pretreated with CAY-10585 (an inhibitor of HIF-1α accumulation and transcription) before ischemia. Matrigel gels at room temperature and forms a matrix. The cells were grown in regular culture medium at 37°C and imaged for capillary-like tube formation by phase contrast microscopy using a Nikon inverted microscope. For each condition, three to four randomly selected fields were imaged. Tube length was assessed by using Metamorph Software, drawing a line along each tube and measuring the length of the line.
(II) The Matrigel plug assay is used as a test for angiogenesis in vivo. In this assay, angiogenesis-inducing endothelial cells are introduced into cold liquid Matrigel, which after subcutaneous injection into nude mice solidifies and permits penetration of host endothelium to form new blood vessels inside the plug. PMVEC (1×106), either static or ischemic (72 h flow adaptation followed by 1 h ischemia), were mixed with 0.5 ml of growth factor-reduced Matrigel and injected subcutaneously into nude mice, using a B-D 26G1/2 needle so that the entire content can be delivered in one bolus to form a single plug. After 5 days, the animals were sacrificed; the plugs were extracted from under the skin, washed in cold phosphate-buffered saline (PBS) to remove blood and cells sticking to the plug surface, and photographed. The plugs were then analyzed for vessel formation by two methods. In one method, small pieces of the plug were stained with DiIAcLDL that labels vascular structures. For the other method, the plugs were cryosectioned and stained with anti-PECAM Ab to outline endothelial structures. The vascularization within the plug reflects the extent to which blood vessels from the host entered the plug.
Hind limb ischemia model
Hind limb ligation was performed with some modifications to the method (58). Briefly, mice were anesthetized, their hind limb hair was removed with depilatory cream, and the proximal portion of the femoral artery proximal to the popliteal bifurcation was ligated (with 6-0 silk sutures) at two locations 0.1 mm apart. Extreme care was taken not to injure the femoral vessel, as local bleeding leads to inflammation that can affect angiogenesis. The area surrounding the silk sutures was photographed immediately post ligation and 5 days later.
Microbead assay for quantitation of revascularization
A fluorescence bead assay was used for quantification of revascularization in the ischemic region (20). Ligated and unligated control mice were used 5 days later. Blood was removed from the tissue by perfusion with PBS and heparin (20,000 units). Fluorescent beads (0.04 μm FluoSpheres beads; Invitrogen) made in a solution of nitroglycerin and PBS were injected into the aorta. After 30 min, the tissue around the ligatures was harvested, and the animal was sacrificed. The harvested tissue was fixed in 4% paraformaldehyde in PBS for 24 h. Fixed tissue was then placed on a coverslip as a wet mount and imaged using confocal microscopy. Quantification of the fluorescence signaling emitted by the entrapped microbeads was assessed using Metamorph software. Images acquired under controlled acquisition conditions were evaluated using a masking algorithm for which the threshold was held constant at a level for optimizing vessel signal and minimizing background signal. For each tissue mount, three fields of view were acquired as representative images. The masked signal from each field was acquired and averaged. Tissue from three to four animals was analyzed for each condition.
Delivery of VEGF in vivo (into the hind limb)
We employed the Chariot™ transfection system that was designed to introduce proteins in vivo. Chariot has been reported to deliver biologically active proteins, peptides, and antibodies directly into cells in vitro and tissues in vivo (4, 39, 41). Our protocol involved incubation of the Chariot agent with protein (1 μg VEGF/6 μl chariot stock) for 30 min at room temperature followed by injecting it into the ischemic region (between the sutures) about 30 min post–hind limb ischemia and once each day (1 μg/day) thereafter for the next 5 days. Five days post ligation, the animal was sacrificed, and hind limb tissue was assessed for revascularization by monitoring the entrapment of fluorescence beads as described earlier. The tissue sections were also assayed for VEGF expression.
pO2 in hind limb tissue
Tissue pO2 was monitored using an oxygen-sensitive microelectrode (10, 57), ∼30 μm tip diameter with a 10-μm sensing region, calibrated at room temperature with saline. The anesthetized animal was placed on a firm surface, the left femoral artery/saphenous vein was exposed, and two sutures were positioned below the vessels. The electrode was inserted ∼1–5 mm into the tissue and ∼1 cm from the ligation site. A tubing containing saline was placed at the upper edge of the exposed tissue and saran wrap was placed over the exposed tissue enclosing saline drip to insulate tissue against evaporation. The electrode was allowed to equilibrate for ∼10 min to establish a baseline measurement. The tip of the electrode was placed close to the femoral artery presurgery, and recording continued while the suture was tightened. The pO2 measurements were recorded by an electrometer. Contact of the electrode with the tissue was maintained throughout preligation, ligation, and post-ligation periods.
Assay for HIF-1α
Anesthetized mice were perfused with PBS to wash out excess blood, and both hind limb muscle tissue and vascular tissue from the site of ligation to the knee was dissected and stored at−80°C. For the HIF-1α assay, nuclear extracts were prepared from vascular tissue and evaluated by immunoblotting using anti-HIF1α Ab (Abcam). Immunoblotting was performed by the Odyssey (Li-Cor) western blot analysis technique. Secondary antibody was IRDye™ 800 goat anti-rabbit for the green channel. Blots were run separately for each set (WT or knockout) of ligated and unligated tissue and scanned by placing the membrane on the Odyssey two-color scanner, and the scanned images were converted to grayscale. All manipulations of contrast were done for the entire gel. The individual bands were quantified using Odyssey software.
HO-1 induction in vivo and in vitro
Hind limb muscle tissue and vascular tissue from the site of ligation was excised, fixed, sectioned, and immunostained for HO-1 (anti-HO-1, 1:250; Santacruz Biotechnology). Anti-PECAM Ab (1:50; SantaCruz Biotechnology) was used to stain for endothelium. Secondary Abs used were Alexa 488-anti-mouse IgG and Texas Red–goat anti rabbit IgG (Invitrogen). Sections were imaged by confocal microscopy. For in vitro experiments, mouse PMVEC were adapted to flow in artificial capillary chambers and subjected to 3 or 24 h ischemia; cells were removed, lysed, and immunoblotted for HO-1 (1:1500, anti-HO-1; SantaCruz Biotechnology). β-Actin was used as a loading control.
Staining tissue sections with VEGF
In order to evaluate levels of VEGF expressed in the tissue surrounding the ligation, tissue in this region was excised and fixed in 4% paraformaldehyde. This was followed by dehydration by sequential sucrose treatment (10%, 20%, and 30% sucrose). The tissue was embedded in OCT blocks, and longitudinal sections were stained for VEGF and PECAM-1 using anti-VEGF165 antibody and PECAM-1 antibody, respectively. Sections were imaged using confocal microscopy.
Quantitation of VEGF in hind limb homogenates by ELISA
For the VEGF assay, the vessel and thigh muscle were dissected, snap frozen in liquid nitrogen, and stored at −80°C until they were ready to be used. VEGF protein levels were assayed from tissue homogenates from the ischemic region. Homogenates from four different animals were added to 96-well plates preconjugated with the anti-VEGF. The secondary signal was measured at 450 and 560 nm in a plate reader (Fisher Scientific Multiskan; ThermoScientific). The VEGF levels were normalized to protein concentrations of each sample.
Quantitation and statistical analysis
All fluorescence images were quantified by Metamorph Imaging Software (Molecular Devices). Immunoblots were quantitated by densitometric analysis, and data were expressed as adjusted density (i.e., relative to loading controls). Data were expressed as mean±SD and were evaluated for statistical significance by one-way ANOVA. Pairwise analysis was performed using the Bonferroni method. In the case of the VEGF expression, significance was determined with post hoc Student–Newman–Keuls analysis.
Footnotes
Acknowledgments
The authors thank Prof. Morris Birnbaum (University of Pennsylvania) and Prof. Keith Channon (University of Oxford) for providing them with Akt1 null and NOX2 overexpressing mice. This work was supported by National Institutes of Health Grant R01HL-075587, McCabe Foundation Award, and NIH-T32-HL07748-17. This work was presented in part at the 2011 Experimental Biology meeting in Washington, DC, 2012 and at the Experimental Biology meeting in San Diego, CA.
Author Disclosure Statement
None of the authors have any competing financial interests.