
Abstract
Introduction
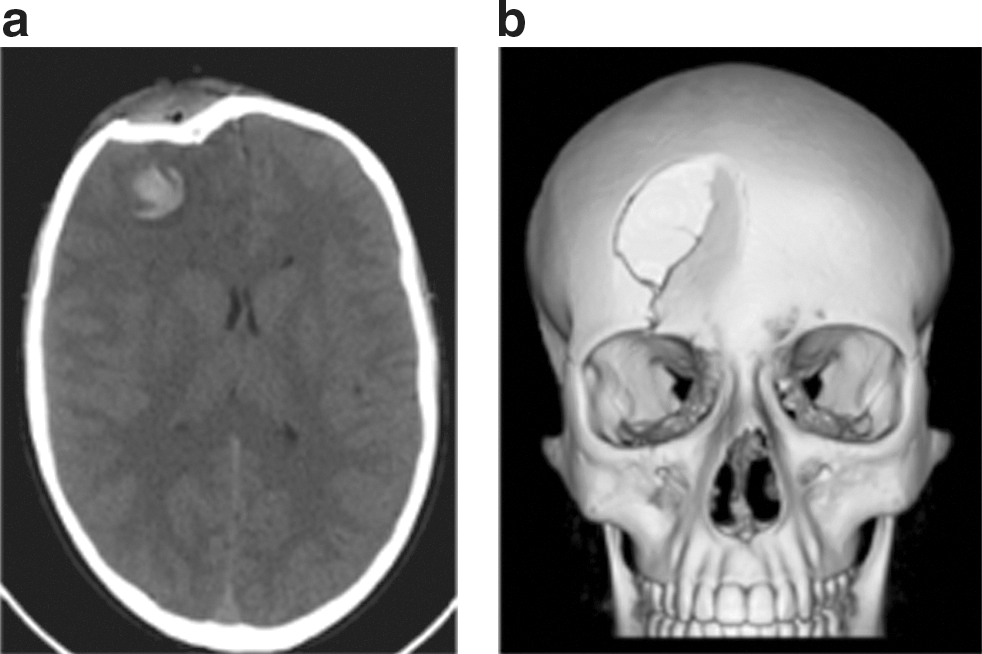
Some of the current imaging techniques used for diagnosis and treatment include computed tomography (CT) and magnetic resonance imaging (MRI). CT scan is useful in brain injury to detect hemorrhagic lesions. In Figure 1a and b, the CT scan showed the hemorrhagic lesion as a hyperdense area with surrounding hypodense area due to edema in the right frontal lobe; the CT scan demonstrated the soft-tissue involvement and depressed skull fracture; 3D volume rendering image allows to see the extension and the morphology of the depressed skull fracture for surgical planning (Fig. 1).
The common causes of TBI are car accidents; others include physical abuse, shaken baby syndrome, and the like (Fig. 2).

The following review represents the authors' effort to piece together the current concepts and the recent findings about the complex basic physiology underlying TBI. Further, we tried to clarify that the role of oxidative damage may be toxic to neuronal cells, examining the nature of the reactive species involved in TBI, and showing how different classes of antioxidant are implicated in the literature with beneficial effects on amelioration of damage after TBI.
TBI Classifications
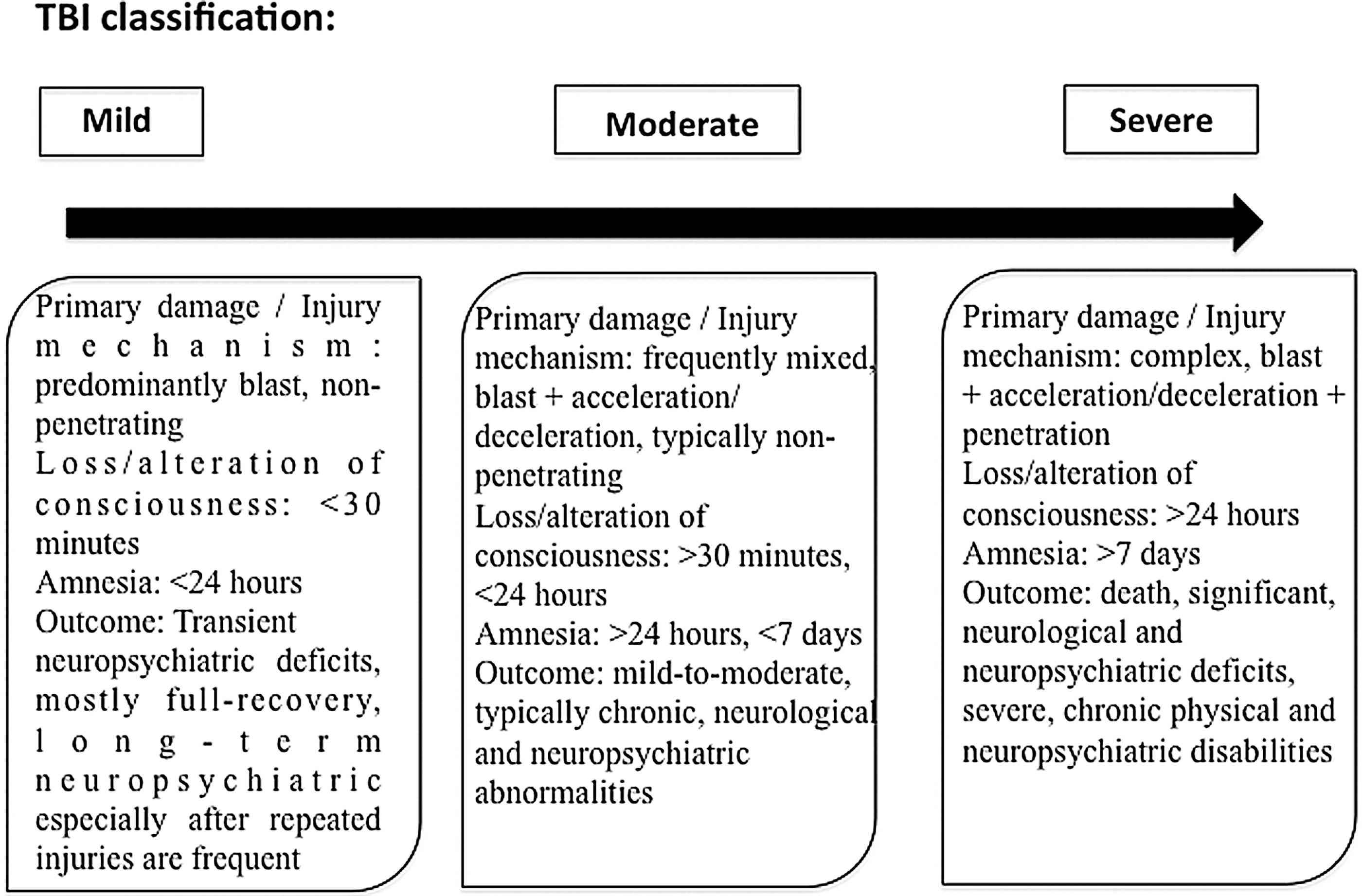
TBI is classified as mild, moderate, and severe (Fig. 3) (138). There are typically no penetrating injuries to the head or other organs, and neurological deficits are focal and transient in nature. The transient and mild neuropsychiatric deficits are followed by recovery. In the severe form, polytrauma, injuries to other parts of the body, frequently to the extremities, abdomen, and lungs can significantly contribute to and modify the pathology and outcome of brain injury.
Pathophysiology of TBI
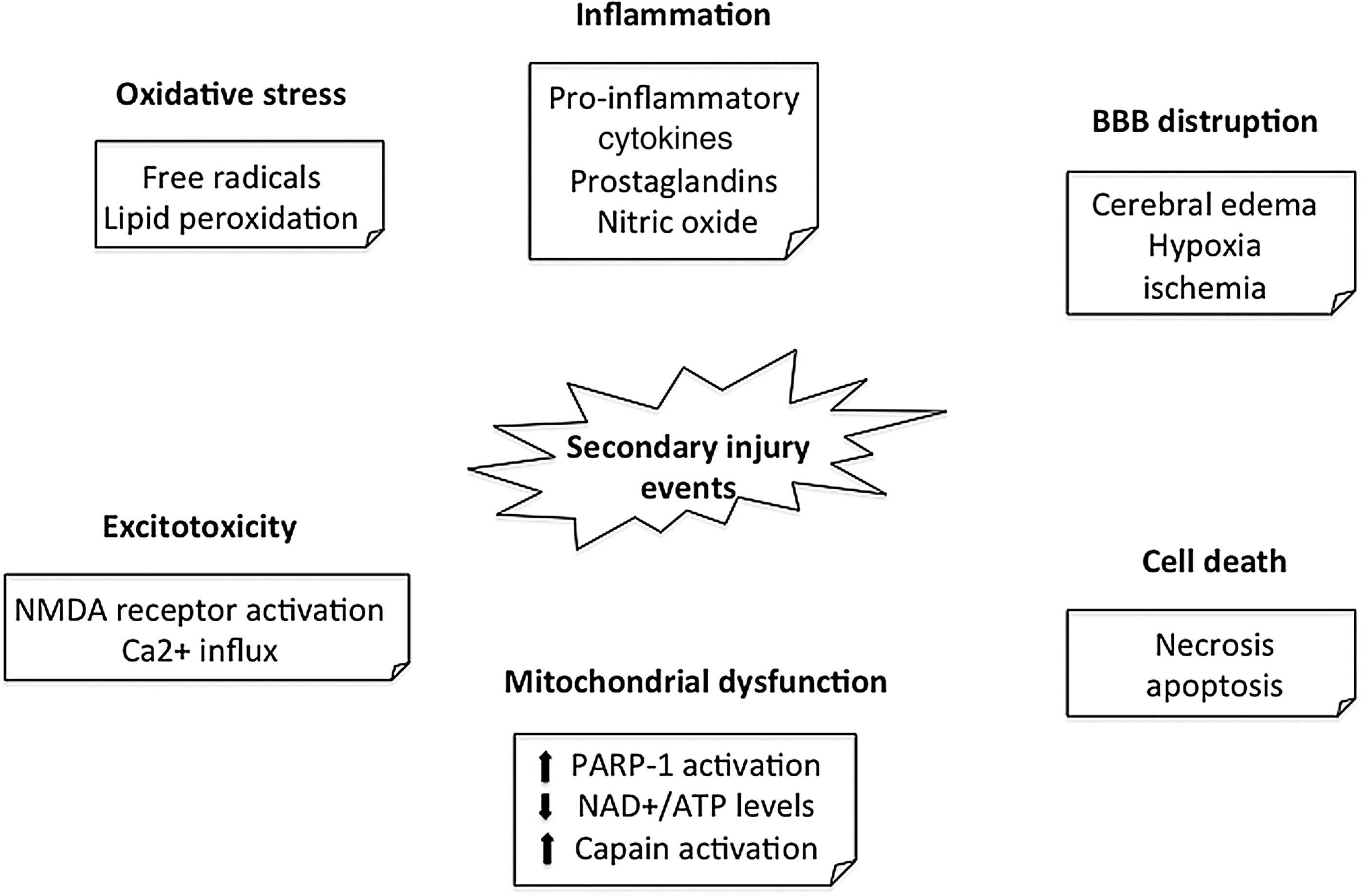
In TBI, we recognized two damages: the primary and the secondary. Primary damage (mechanical), occurring at the time of injury, is responsible for “focal” anatomical lesions, such as laceration, contusion, intracranial hemorrhage, and diffuse axonal injury; in treatment terms, this type of injury is exclusively sensitive to preventive but not therapeutic measures. The secondary damage (nonmechanical, delayed), produced by complex processes, is initiated at the moment of injury, but no clinical alterations are observed for a period of hours to days. In response to the typically short-lasting primary injury mechanism, there is a second wave of long-lasting pathological changes called secondary injury mechanism. These pathologies include metabolic changes, neuroinflammation, axonal injury, vascular abnormalities, and neuronal and glial cell death (58). Metabolic changes include abnormal levels of oxygenation, altered cell metabolism disrupted energy levels and systemic hormonal secretion, and an upregulation of inflammatory activity (Fig. 4) (48). Inflammation occurs in response to damaging stimuli, triggering the release and activation of cytokines and chemokines, and the activation and proliferation of microglia and astroglia in the central nervous system (CNS). Vascular abnormalities are marked by aberrations in the water content of the brain parenchyma, dysregulation of water channels, and a compromised blood-brain barrier (BBB). These primary and secondary injury processes may lead to a range of neuropsychiatric symptoms, including various forms of memory and learning deficits, anxiety, and depression (4). The first stages of cerebral injury after TBI are characterized by direct tissue damage and impaired regulation of cerebral blood flow (CBF) and metabolism. This “ischemia-like” pattern leads to accumulation of lactic acid due to anaerobic glycolysis, increased membrane permeability, and consecutive edema formation. The second stage of the pathophysiological cascade is characterized by terminal membrane depolarization along with excessive release of excitatory neurotransmitters, activation of N-methyl-D-aspartate, alfa-amino-3-hydroxy-5- methyl-4-isoxazolpropionate, and voltage-dependent Ca2+- and Na+-channels. Ca2+ activates lipid peroxidases, proteases, and phospholipases, which in turn increase the intracellular concentration of free fatty acids and free radicals. Activation of caspases, translocases, and endonucleases initiates progressive structural changes of biological membranes and the nucleosomal DNA. These events lead to membrane degradation of vascular and cellular structures and ultimately necrotic or programmed cell death.
Oxidative Damage in TBI
Free radicals are atoms or groups of atoms with an unpaired number of electrons and can be formed when oxygen interacts with certain molecules. Once formed, these reactive radicals can start a chain reaction, like dominoes. To prevent free radical damage the body has a defense system of antioxidants.
Antioxidants are molecules that can interact with free radicals and terminate the chain reaction before vital molecules are damaged. Several oxidants and their derivatives are generated after stroke, including superoxide anions (O2•−), hydrogen peroxide (H2O2), and hydroxyl radicals (•OH). Superoxide anions are formed when oxygen acquires and additional electron. O2•− are formed within the mitochondria, and 2%–5% of electron flow is employed to produce them. Enhanced production of reactive oxygen/nitrogen species (ROS/RNS) cause oxidative/nitrosative stress (83) leading to damage in lipids, proteins, and nucleic acids (133). Antioxidants can protect the brain against oxidative damage by (i) removal of ROS/RNS, (ii) inhibition of ROS/RNS formation, (iii) binding metal ions needed for catalysis of ROS generation (93).
Superoxide radical
After TBI the microvascular superoxide radical (O2•−) production is increased (92). The scavengers of O2•− decrease the post-traumatic superoxide levels and protect against the loss of microvascular autoregulatory competency. Within the injured nervous system, a number of possible sources of O2•− may be operative during the first few minutes and hours after injury, including the arachidonic acid (AA) cascade enzymatic or autoxidation of biogenic amine neurotransmitters, “mitochondrial leak,” xanthine oxidase activity, and the oxidation of extravasated hemoglobin. Activated microglia and infiltrating neutrophils and macrophages provide additional sources of O2•− at later timepoints. O2•− is reactive, its direct reactivity toward biological substrates in aqueous environments is relatively weak. If formed, O2•− undergoes spontaneous dismutation to form H2O2 in a reaction that is accelerated by the enzyme superoxide dismutase (SOD):
In solution, O2•− exists in equilibrium with the hydroperoxyl radical (HO2•):
which is more lipid soluble and a far more powerful oxidizing or reducing agent. Since the pKa of the O2•−/HO2• is 4.8, as the pH of a solution falls the equilibrium between O2•− and HO2• shifts in favor of HO2•, which is much more reactive than O2•−, particularly toward lipids.
Iron and hydroxyl radical
Iron represents one of the most abundant components of CNS; its distribution varies in parallel with the sensitivity of various regions to oxidative damage (169). Under fisiological conditions, low molecular weight forms of redox-active iron are maintained at extremely low levels. In plasma, the iron transport protein transferrin tightly binds iron in the Fe+++ form. Intracellularly, Fe+++ is sequestered by the iron storage protein ferritin. While both ferritin and transferrin have very high affinity for iron at neutral pH and effectively maintain iron in a noncatalytic state (70) both proteins give up their iron at pH values of 6.0 or less, which is a level of acidosis that has been shown to have reached the injured brain. In traumatized brain, where pH in injured areas is lowered, conditions are favorable for the potential release of iron from storage proteins (70). A second source of catalytically active iron is hemoglobin. Hemorrhage resulting from mechanical trauma provides an obvious source of hemoglobin. While hemoglobin itself has been reported to stimulate oxygen radical reactions, it is likely that iron released from hemoglobin is responsible for hemoglobin-mediated oxidative damage (139). Iron is released from hemoglobin by either H2O2 or by lipid hydroperoxides (LOOH) and this release is further enhanced as the pH falls to 6.5 or below.
Free iron or iron chelates participate in free radical reactions at two levels. The autoxidation of Fe++ results in the formation of O2•
Second, Fe++ is oxidized in the presence of H2O2 to form •OH (Fenton reaction):
Lipid peroxidation
Lipid peroxidation (LP) is well established after stroke; it causes alterations in cell membrane fluidity, increased permeability of membranes, and a decrease in membrane ATPase activity, leading to cell injury. LP refers to the oxidative degradation of lipids. In this process, free radicals “steal” electrons from the lipids in cell membranes, resulting in cell damage. It most often affects polyunsaturated fatty acids (PUFAs), because they contain multiple double bonds in between which lie methylene -CH2- groups that possess especially reactive hydrogens. As with any radical reaction, the reaction consists of three major steps: initiation, propagation, and scission. Initiation represents the step in which a fatty acid radical is produced. The most notable initiators in living cells are ROS, such as OH• and HO2, which combines with a hydrogen atom to make water and a fatty acid radical. In this step, a radical species such as •OH reacts with and removes an allylic carbon and extracts a hydrogen and its single electron from AA (AA+R•→AA•+RH). The initiating radical is quenched by receipt of an electron from the polyunsaturated AA. This, however, converts the AA into a lipid or “alkyl” radical (AA•). This sets the stage for a series of “propagation” reactions, which begins when the alkyl radical takes on a mole of oxygen creating a lipid peroxyl radical (AA-OO•; AA•+O2→AA-OO•). The peroxyl radical then reacts with a neighboring AA within the membrane and steals its electron forming a lipid hydroperoxide (AA-OOH) and a second alkyl radical (AA•; AA-OO•+AA→AA-OOH+AA•). The fatty acid radical is not a stable molecule, so it readily reacts with molecular oxygen, thereby creating a peroxyl-fatty acid radical. This too is an unstable species that reacts with another free fatty acid, producing a different fatty acid radical and a lipid peroxide, or a cyclic peroxide if it had reacted with itself. This cycle continues, as the new fatty acid radical reacts in the same way. In particular, the alkyl radical takes on a mole of oxygen creating a lipid peroxyl radical (AA-OO•; AA•+O2→AA-OO•). The peroxyl radical then reacts with a neighboring AA within the membrane and steals its electron forming a AA-OOH and a second alkyl radical (AA•; AA-OO•+AA→AA-OOH+AA•).
Once LP begins the propagation phase, iron may participate in driving the process as lipid •hydroperoxides are decomposed by reactions with either ferrous iron (Fe++), or ferric iron (Fe+++). In the case of Fe++, the reaction results in formation of a lipid alkoxyl radical (AA-O•; AA-OOH+Fe++→AA-O•+OH−+Fe+++). If the reaction involves Fe+++, the AA-OOH is converted back into a lipid peroxyl radical (AA-OO•; AA-OOH+Fe+++→AA-OO•+Fe++). Both the reactions of AA-OOH with iron have acidic pH optima causing them to be augmented by tissue acidosis. Either alkoxyl (AA-O•) or peroxyl (AA-OO•) radicals arising from AA-OOH decomposition by iron can initiate so-called lipid hydroperoxide-dependent LP resulting in “chain branching” reactions:
Finally, the LP process leads to “scission” reactions in which the peroxidized AA breaks down to give rise to the neurotoxic aldehydes 4-hydroxynonenal (4-HNE) or 2-propenal (acrolein). The 4-HNE produces neurotoxicity by binding to basic amino acids such as lysine or histidine and sulfhydryl-containing cysteine residues in cellular proteins. This happens only when the concentration of radical species is high enough for there to be a high probability of collision of two radicals.
Peroxynitrite
The principal ROS involved in producing tissue injury in a variety of neurological disorders is the “reactive nitrogen species” peroxynitrite (PN; ONOO−). PN is an unstable structural isomer of nitrate, NO3−. Although peroxynitrous acid (ONOOH) is highly reactive, its conjugate base PN is stable in basic solution.
It is prepared by the reaction of H2O2 with nitrite:
Formation of PN in vivo has been ascribed to the reaction of the free radical superoxide with free radical nitric oxide. PN is formed by the combination of NO synthases (NOS)-generated NO radical and O2•:
PN-mediated oxidative damage is caused by PN decomposition products that possess potent free radical characteristics. These are formed in different ways. One of this involves the protonation of PN to form ONOOH, which can undergo homolytic decomposition to form the highly reactive nitrogen dioxide radical (•NO2) and OH; (ONOOH→•NO2+•OH). More important physiologically, PN will react with carbon dioxide (CO2) to form nitrosoperoxocarbonate (ONOOCO2), which can decompose into NO2 and carbonate radical (•CO3); (ONOOCO2→•NO2+•CO3). Each of the PN-derived radicals (•OH, •NO2, and •CO3) can initiate LP cellular damage by abstraction of an electron from a hydrogen atom bound to an allylic carbon in PUFAs or cause protein carbonylation by reaction with susceptible amino acids. Additionally, •NO2 can nitrate the three positions of tyrosine residues in proteins; 3-NT is a specific footprint of PN-induced cellular damage. There are several lines of evidences that showed the implication of PN in post-TBI pathophysiology. First, three NOS isoforms (endothelial, neuronal, and inducible) are known to be upregulated during the first 24 h after TBI in rodents (64). Second, several laboratories have shown that the acute treatment of injured mice or rats with NOS inhibitors can exert a neuroprotective effect and/or improve neurological recovery (158). Third, biochemical footprints of PN-mediated damage have been documented in rodent TBI paradigms including an increase in 3-NT levels (115) and ADP ribosylation. Fourth, the notion that these markers of PN-mediated damage are pathophysiologically important is supported by the finding that the NOS inhibitor N-nitro-L- arginine-methylester (L-NAME) can lessen the accumulation of 3-NT in injured brains (115) at the same doses that improve neurological recovery (116).
Glutathione peroxidase
Glutathione peroxidase (GPx) and glutathione reductase (GR) are well-known intracellular antioxidant enzymes. GPx converts peroxides into nontoxic forms, often with the concomitant oxidation of reduced glutathione (GSH) into the oxidized form (GSSG), and GR recycles GSSG to GSH. GSH-S-transferase detoxifies deleterious substances and glucose-6-phosphate dehydrogenase (G-6PD) provides electron donors for the antioxidant defense system. Enzymes that remove both superoxide and H2O2 protect the cells from oxidative stress, but when the production of O2− and H2O2 crosses the normal threshold, the system becomes compromised. Catalase and GPx, acting in concert with SOD, constitute the major defense enzymes against superoxide radicals (130). As a consequence of oxidative stress, both the function and transport of mitochondria to synaptic regions are impaired, decreasing synaptic function (133), which results in neurodegeneration after brain injury (93).
Malondialdehyde
Reactive oxygen species-mediated damage is mainly characterized by the onset of LP and revealed by measuring tissue malondialdehyde (MDA). MDAis a highly reactive three carbon dialdehyde produced as a byproduct of PUFA peroxidation and AA metabolism. MDA mainly exists in the enol form:
MDA combines with several functional groups on molecules including proteins, lipoproteins, and DNA.
Starting from 1 min after trauma, the MDA level progressively increased up to the second hour, when its maximum concentration was recorded. Relatively high levels of this compound, originating from decomposition of peroxidized membrane phospholipids, persisted at 24 and 48 h after trauma. Oxidation of ascorbate paralleled MDA production, although the minimum ascorbate value was observed 6 h after trauma, showing a 60% decrease with respect to controls (79).
Glutamate
The pathophysiological cascade induced after TBI is initiated by an excitotoxic process triggered by excessive glutamate release. Glutamate is the major mediator of excitatory signals in the mammalian CNS and is involved in most aspects of normal brain function including cognition, memory, and learning. Glutamate does not only mediate a lot of information, but also information that regulates brain development and information that determines cellular survival, differentiation, and elimination and formation and elimination of synapses. Increased levels of extracellular glutamate following head injury causes overstimulation of metabotropic and ionotropic receptors that may result in secondary events leading to neuronal cell death. Activation of metabotropic receptors causes mobilization of calcium (Ca2+) from internal stores. Three types of ionotropic glutamatergic receptors are activated: N-methyl-D-aspartate (NMDA) receptor, 2-amino-3-(3-hydroxy-5- methylisoxazol-4-yl)propionic acid receptor (AMPAR), and kainate receptor. When activated, AMPAR leads to the entry of Na+ in cell contributing to membrane depolarization. Intracellular water increases inducing a cell swelling. When activated, NMDA receptor promotes the entry of Na+ and Ca2+ in cells contributing to membrane depolarization and to activation of Ca2+—dependent enzymes (168). Calcium overload can trigger many downstream neurotoxic cascades, including the uncoupling mitochondrial electron transfer from ATP synthesis, and the activation and overstimulation of enzymes, such as calpains, protein kinases, endonucleases, and NOS (60).
N-acetylaspartate
N-acetylaspartate (NAA) is the acetylated form of amino acid aspartate, and it is present in the nervous system. Indeed, NAA is one of the most highly concentrated chemicals found in the brain of humans and animals, and yet the functions served by this brain-specific metabolite remain elusive and controversial.
NAA became the most reliable molecular marker for the imaging of several pathologies in brain H-MRS studies. By measuring whole-brain NAA via HPLC (151) in three different levels of trauma, it was demonstrated that, at 48 h postinjury, NAA reduction was graded according to the severity of insult, showing spontaneous recovery with lower levels of trauma and irreversible decrease in the others (144). The findings were consistent with long-term behavioral observation in animals injured using the same model, showing slight differences from sham-injured animals, with the main differences being present at 1 day postinjury and consistent improvement occurring over time (11). These bench observations supported the clinical observations with proton MR spectroscopy with respect to a potential role of NAA in quantifying neuronal damage and predicting neuropsychological outcome after TBI (63).
Nitric oxide synthase
NOSs are a family of enzymes that catalyze the production of NO from L-arginine.
The canonical reaction catalyzed by NOS is:
NOS isoforms catalyze many other leak and side reactions such as superoxide production at the expense of NADPH. The unusual stoichiometry reflects the three electrons supplied per NO by NADPH; NO is a free radical with an unpaired electron.
TBI induces the activation of NOS leading to the production of NO in brain (1). NO production has been also demonstrated in cerebrospinal fluid (CSF) and in brain tissue after human TBI (152) and the end products of NO, nitrate/nitrite, levels are correlated with TBI severity (76). NO can combine to cellular thiols compounds leading to the formation of S-nitrosothiols (62). Bayir et al. (10) demonstrate that increase in S-nitrosothiols is correlated with intracranial pressure decrease, suggesting a neuroprotective role of S-nitrosothiols following TBI. NO can be synthetized by three isoforms of NOS: NOS1 (neuronal) and NOS3 (endothelial) that are both constitutive and Ca2+-dependent enzymes. The third one is NOS2 (inducible) that produces large quantity of NO in inflammatory situations. The first study demonstrating the role of NOS in TBI has been published by Mesenge et al.,(116). Indeed, post-treatment with L-NAME, a nonselective NOS inhibitor, decreases the post-traumatic neurological deficit with a therapeutic window of opportunity of 60 min. The same results have been observed with 7-nitroindazole, a selective NOS1 inhibitor (116). In addition, pretreatment with this inhibitor decreases the brain lesion volume following TBI (157). Treatment with L-arginine, reduces the brain lesion volume and this is associated with NOS3 activation and improved CBF (47). NOS3−/− mice have a decreased CBF compared with wild-type animals (77), and NOS3 is necessary to induce beneficial vascular effect of L-arginine after TBI (78). Considering these data, NO produced by NOS3 plays a beneficial role after TBI. If the roles of NOS1 and NOS3 are quite well understood, the one of NOS2 is still controversial in TBI. Some studies suggest that early after TBI, effects of NOS2 may be detrimental as aminoguanidine (101) a selective NOS2 inhibitor, has been shown to reduce neuronal cell death following TBI (149). Sinz et al. (145) have shown that NOS2−/− mice present more important cognitive deficits than wild-type mice at 17–21 days after TBI. In addition, NOS2 knockout (KO) mice have been demonstrated to have greater loss of brain levels of ascorbate, an endogen antioxidant, compared with wild-type animals at 72 h after TBI (7).
Oxidative and nitrosative stress
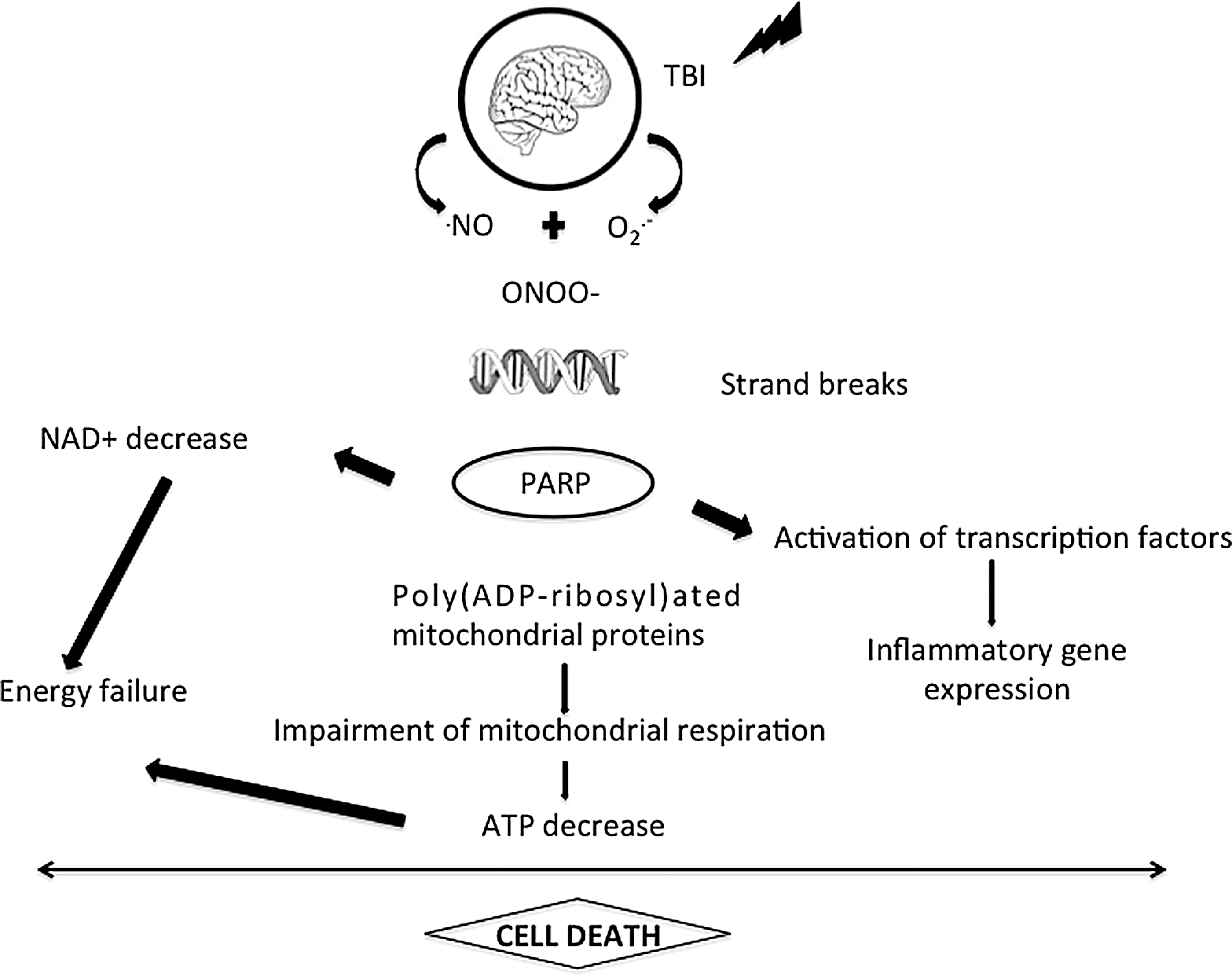
The oxidative stress is defined as an imbalance between free radical production and endogen antioxidant systems. In humans, TBI increases plasma and CSF levels of MDA, as early as 2–3 h that persists at least until 7 days after injury (9). In addition, SOD activity decreases at 24 h and during 7 days after severe TBI (43). After experimental TBI, hydroxyl radicals OH and superoxide anions O2− increase early after injury (59). Antioxidant strategies aiming at decreasing oxidative stress showed to reduce post-traumatic neurological deficits, cerebral edema, and brain lesion volume. This has been observed after different treatments, including polyethylene glycol (PEG)-conjugated SOD (PEG-SOD), an antioxidant enzyme (72); melatonine mimicking GPx (115); OPC-14117 (7-hydroxy-1- [4- (3- methoxyphenyl) - 1- piperazinyl] acetylamino -2, 2,4, 6-tetramethylindan), a scavenger of superoxide anions (3); phenyl-tert-butylnitrone, a free radical scavenger (97); mesylate tirilazad, an inhibitor of LP (69); or in SOD1 (117) or SOD3 (132) transgenic mice. PN is a strong oxidant that can directly react with tyrosine of proteins producing nitrotyrosine (131). Treatment with L-NAME decreases nitrotyrosine levels after TBI, demonstrating the involvement of NO in PN formation (114). The deleterious role of PN is supported by reports demonstrating the beneficial effects of PN scavengers such as penicillamine, penicillamine methylester, and tempol (69). Although not a free radical in nature, PN is much more reactive than NO and O2− (131). Further, deficiency of NOS1 but not NOS2 and NOS3 attenuates SOD2 nitration after experimental TBI (8). Nitration and inactivation of SOD2 could lead to self-amplification of oxidative stress in the brain progressively enhancing PN production and secondary damage. LP has been demonstrated with 4HNE staining in experimental models of diffuse and focal TBI (46). PN can alter DNA by introducing oxidative damage on guanine producing 8-oxoguanine. The formation of PN -mediated DNA breakage represents one of the most deleterious aspects of PN anions as they represent the trigger for the activation of the nuclear DNA repair enzyme, poly (ADP-ribose) polymerase (PARP).
Poly (ADP-ribose) polymerase
PARP is a constitutive nuclear enzyme present in eukaryotes. It belongs to an expanding family of 17 members implicated in such physiological processes as DNA repair, maintenance of genomic integrity, gene transcription, cell division, and apoptosis (142). PARP-1 is the major isoform present in the nucleus. When activated PARP initiates an energy-consuming cycle by transferring ADP-ribose units from NAD to a set of nuclear acceptor proteins, including histones, several chromatin-binding proteins, and PARP itself (73). To study the role of PARP in pathological conditions, numerous inhibitors of PARP have been synthetized (52) and three different KO mice for PARP-1 have been generated (106). Using these pharmacological tools, it has been shown that PARP mediates necrotic cell death in response to excessive DNA damage under pathological conditions. This process results in rapid depletion of intracellular NAD, in a loss of ATP as it is used to synthesize new NAD, and finally to cell death. Oxidative stress induced in vivo in brain has been demonstrated to promote PARP activation, which contributes to neuronal cell death (Fig. 5). PARP inhibitors have been shown to prevent in vitro neuronal cell injury from glutamate (50), NMDA, direct NO, and NO donors (105). PARP is activated as early as 30 min after TBI and its activation persists for 72 h after TBI (140). Tyrosine nitration and PARP activation are both found to be persistently increased compared with normal brain, with relative peaks seen at 8 and 72 h (140). Poly(ADP-ribose) glycohydrolase rapidly degrades polymers of ADP-ribose (52); this suggests that ADP-ribosylation, that is, PARP activation, is a prolonged phenomenon. Cleavage of PARP by caspase-3, occurring only 7 days after TBI (95), is one of the mechanisms to inactivate PARP, thus preserving cell energy stores required during apoptosis. Following TBI, NO leads to PN formation, via its combination with superoxide anion, which in turn induces DNA strand breaks rendering PARP active. PARP is activated by brain trauma as poly(ADP- ribose)-modified proteins are increased at 24 h and until 48 h in CSF of pediatric TBI patients (61). Two PARP inhibitors, 5-iodo-6-amino-1,2-benzopyrone (INH2BP) (140) and INO-1001, prevent NAD depletion and improve post-TBI deficits, demonstrating PARP- mediated energy failure as a contributor to the pathological sequelae of TBI. In vitro PARP inhibition protects hippocampal slices against percussion-induced loss of CA1 pyramidal cell-evoked response (160). Whalen and colleagues (164) showed that motor and cognitive deficits of mice submitted to TBI are less severe when the PARP-1 gene is inactivated. The prototypical PARP inhibitor, 3-aminobenzamide, and other benzamide derivatives induce neuroprotective effects on the neurological deficit and the brain lesion after closed-head injury in mice (116) and after TBI induced by fluid percussion (12). It is important to emphasize that the protective effect of PARP inhibition on neurological function is a lasting one, which remains significant even 7 (12) or 21 days (140) after TBI. Finally, an NOS1 inhibitor, 3-bromo-7-nitroindazole, significantly decreases the production of poly(ADP-ribose) in damaged cerebral cortex after cryogenic lesion, demonstrating that TBI-induced PARP activation depends, at least in part, on prior activation of NOS1 (52). Deleterious mechanisms of PARP activation toxicity in brain are multiple. First, PARP activation mediates cell death. When DNA is damaged, PARP is activated resulting in high consumption of NAD and ATP and finally necrosis. Second, PARP regulates inflammation as it acts also as a co-activator of the transcription factor nuclear factor-kappa B resulting in the synthesis of pro-inflammatory mediators. In addition, PARP is able to directly poly-ADP- ribosylate other transcription factors including STAT and activator protein-1 and -2 (87). By this way, inhibition of PARP has been shown to mediate many anti- inflammatory effects in various inflammatory diseases (52) and acute brain injuries including stroke (66). PAR synthesis induced by PARP promotes translocation of apoptosis-inducing factor from mitochondria to the nucleus, subsequent DNA fragmentation and caspase-independent programmed cell death (2).
Brain Responses After TBI
The brain is a plastic system that derives its functional organization from interaction with environmental factors. ROS are generated during cellular respiration, and their levels are increased as a result of abnormal cell metabolism (117). Increase in ROS production has been identified as an important mechanism by which neuronal plasticity is compromised during injury (148). A crucial principle involved with hormesis is that the re-establishment of disrupted homeostasis can result in an adaptive condition that is more beneficial than the prior stage.
Hormesis and Neurodegenerative Diseases
Hormesis is a dose response phenomenon that is characterized by a low dose stimulation and a high dose inhibition (27 –29). This phenomenon may occur when a low dose of a physical or chemical agent that is toxic at higher concentrations induces an opposite effect, usually but not always a beneficial effect, at lower concentrations. This type of dose response is induced by a large number of endogenous agonists that affect a very broad range of receptor systems (Table 1).
Modified from (27).
Of particular importance is that hormetic dose responses display specific quantitative features concerning the amplitude and width of the stimulatory response. These specific set of quantitative features make the hormetic dose response a unique type of biphasic dose response. These low dose stimulatory responses are generally modest being at maximum only 30%–60% greater than the control value (Fig. 6).
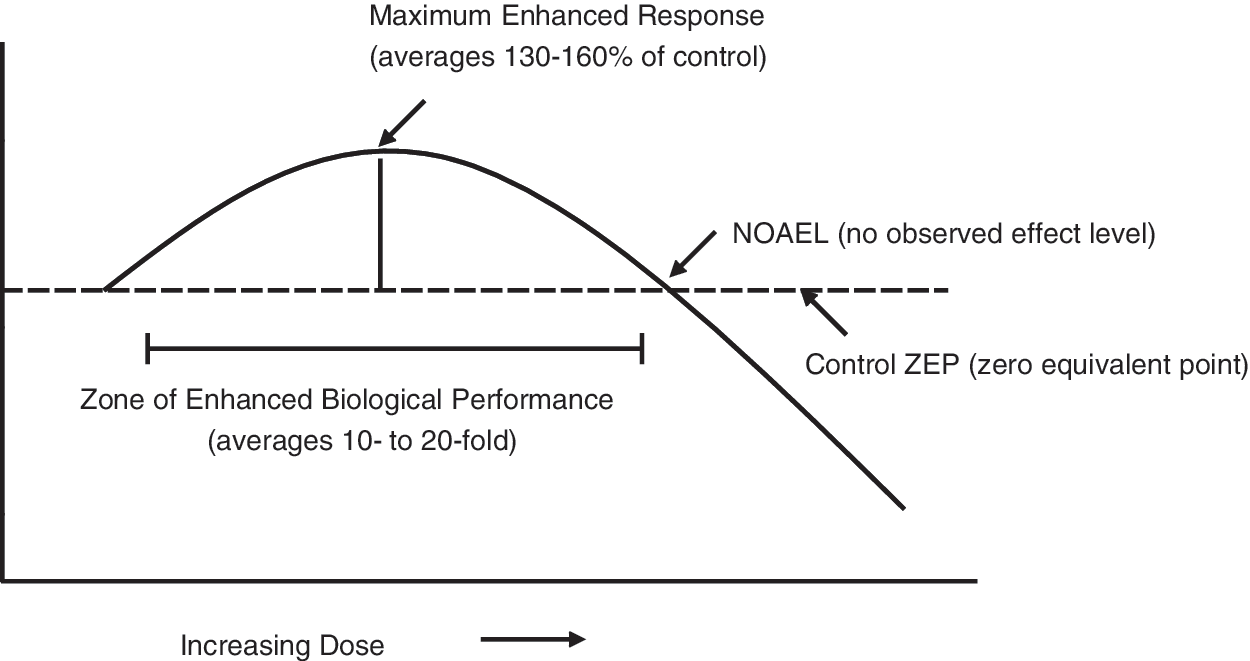
The wide spread occurrence of hormetic dose responses and their extensive generalizability are often overlooked within the biological and biomedical sciences due to the high degree of disciplinary specialization (24, 108). Hormetic dose responses are reported for key endpoints in neurosciences such as neuronal survival (26) and neurite outgrowth (23), prevention of damage in models for Alzheimer's disease (AD) (134) and Parkinson disease (PD) (111), in addition to stroke and traumatic brain injuries and (22) pain (25).
Since the hormetic dose response is similar regardless of receptor or signaling pathway it is likely that hormesis may provide a quantitative index of neuroplasticity (30).
Cellular Stress Response and the Vitagen Network
Protein thiols play a key role in redox sensing, and regulation of cellular redox state is a crucial mediator of multiple metabolic, signaling, and transcriptional processes (36). Under optimal conditions long-term health is maintained by protein homeostasis, a highly complex network of molecular interactions that balances protein biosynthesis, folding, translocation, assembly/disassembly, and clearance (37, 38). The ability of a cell to counteract stressful conditions is also known as cellular stress response or heat shock response (37, 104). Production of heat shock proteins (Hsp), including protein chaperones, is essential for the folding and repair of damaged proteins, serving thus to promote cell survival conditions that would otherwise result in apoptosis (5). There is significant interest in the discovery and development of small molecules that modulate heat shock responses and parallel stress response pathways for therapeutic purposes (33, 35, 38, 121, 154). The major regulator of the heat shock response genes is the heat shock transcription factor 1 (HSF1), which is kept in a latent state by an inhibitory complex of stress-proteins, and plays a key regulatory role in response to environmental stress, development, and many pathophysiological conditions, including cancer, ischemia-reperfusion injury, diabetes, and aging (163).
Mammalian cells contain three HSF family members, HSF1, HSF2, and HSF4 (19, 120). Protein stress results in conversion of HSF1 from inactive monomer to DNA binding trimer and remodeling of the inhibitory molecular chaperone complex (39). The major activator of HSF1 is proteotoxic insults, like heat shock. Misfolded proteins displace HSF1 and form the inhibitory chaperone complex; HSF1 trimerizes, becomes phosphorylated, and is translocated to the nucleus where it is able to bind to the heat shock element of Hsp genes (119, 163).
Cellular stress response requires the activation of pro-survival pathways and production of molecules endowed with antioxidant and anti-apoptotic activities, which is under control of protective genes called vitagens (32, 34 –36). The molecular chaperones help hundreds of signaling molecules to keep their activation-competent state, and they regulate various signaling processes ranging from signaling at the plasma membrane to transcription. Recent studies have revealed that chaperones act as genetic buffers stabilizing the phenotypes of various cells and organisms (20). Among the cellular pathways involved in the so-called “programmed cell life” conferring protection against oxidative stress, a key role is played by the products of vitagenes (33 –36, 38, 109). These include members of the Hsp family, such as heme oxygenase-1, Hsp72, sirtuins, and thioredoxin/thioredoxin reductase (33, 35). Heme oxygenase-1, also referred to as Hsp32, degrades heme, which is toxic if produced in excess, into free iron, carbon monoxide, and biliverdin (33, 38). Sirtuins are a group of proteins linked to aging, metabolism, and stress tolerance in several organisms (110). Mammalian sirtuins are histone deacetylases, requiring NAD+ as a cofactor to deacetylate substrates ranging from histones to transcriptional regulators. Through this activity, sirtuins are shown to regulate biological processes, such as apoptosis, cell differentiation, energy transduction, and glucose homeostasis.
DNA Damage and Repair in the Brain During Aging: Neurodegenerative Diseases
Elderly individuals are more vulnerable to TBI; in fact, they have clinically worse outcomes after TBI, including increased morbidity and mortality and reduced functional recovery (Fig. 7). Age-associated increases in levels of ROS, especially during the last quarter of life, result in excessive oxidative damage to macromolecules, including DNA. Guanine is prone to oxidation due to its low reduction potential among DNA bases. It is modified by the hydroxyl radical at or near diffusion-controlled rates (56, 57, 86, 135, 143, 156, 159). More than 20 oxidation products of guanine base have been identified and among them the most abundant and well studied is 8-oxo-7,8 dihydroguanine (8-oxoG) (15, 125). 8-oxoG levels in DNA increase with ischemia/reperfusion, acute exercise, neurodegenerative diseases, and aging (84, 135). When 8-oxoG is not repaired it is mutagenic, as it has been shown to pair with adenine (A) instead of cytosine (C) inducing G:C→T:A transversions (125). 8-oxoG is excised from DNA by formamidopyrimidine-DNA glycosylase (Fpg) in Escherichia coli and by its functional homolog 8-oxoguanine DNA glycosylase (OGG1) in mammals during base excision repair (BER) (74, 122, 126).
Impaired function and accumulation of DNA damage in neurons have been suggested to be major factors related to brain aging and neurodegenerative diseases (14, 141). Using a rodent model, there are a number of investigations that show that aging results in elevations in DNA damage (42, 129, 150).
Mecocci and co-workers have reported that aging results in significant increases in nuclear and mitochondrial 8-oxoG levels in human cerebral cortex and cerebellum (113).
Mitochondrial dysfunction is increased in the aged TBI brain, with enhanced oxidative damage of synaptic mitochondria contributing to significant impairments in bioenergetic function. After TBI, aged animals show increased evidence of oxidative stress and reduced antioxidant capacity when compared with younger animals. It was demonstrated that neurons are sensitive to accumulating 8-oxoG (162) and it appears that the capacity to repair 8-oxoG is dependent on brain regions (41), suggesting a link between the incidence of those neurodegenerative diseases where initiation occurs in a brain region-specific manner. Age-associated decreases in the DNA repair activity of synaptosomes is associated with reduction in the levels of DNA repair enzymes and reduced mitochondrial axonal transport (68). Since upregulation of DNA repair suppresses DNA damage (41), methods that can elevate the activity of OGG1 could potentially decrease the pathology of certain diseases. Mitochondrial DNA is more exposed to ROS and the age-associated increase in 8-oxoG level is a magnitude higher in mtDNA compared with nDNA (123). The failure of mtDNA repair is associated with chronic recurrent seizures in rat brain (99) suggesting that the consequences of seizures could impair mitochondrial function.
Brain is rich in metals and the concentration of metals increases with aging (107). Li et al. (98) noted the relationship with metal levels and BER. AD is associated with the accumulation of metals and decreased levels of DNA repair (49, 128). Indeed, it has been recently shown that 8-oxoG-OGG1 complex can activate the Ras family GTPases to catalyze replacement of GDP with GTP, to serve the guanine nuclear exchange factor. In addition, 8-oxoG-OGG1 complex-induced activation of Ras further activates mitogen-activated kinases MEK1,2/ERK1, which can lead to ROS production (16).
It is known, that the activity of certain DNA repair proteins, including OGG1 can be modified by acetylation. The effects of aging and neurodegenerative diseases on the protein acetyl transferases and/or protein deacetylases could be important. Acetylation of OGG1 is mediated by p300/CBP (13, 136), while the deacetylating agents are not known. Preliminary data suggest that SIRT1 silencing increases the acetylation of OGG1 (Sarga et al., unpublished observation) suggesting that SIRT1 I could be actively involved in DNA repair. Aging increases the content of OGG1 in the hippocampus of rats, but the acetylation of OGG1 is drastically decreased with aging, which could be the reason for the age-associated increase in 8-oxoG level in old rats (90).
Redox Regulating Sirtuins in Aging and Neurodegenerative Diseases
Enzymes from the sirtuin family are ubiquitously distributed in mammals with seven homologues (SIRT1–7). Powerful protein deacetylase activity of SIRT1, SIRT2, SIRT3, and SIRT5 has been reported toward histones, whereas SIRT4, SIRT6, and SIRT7 have no detectable enzymatic activity on a histone peptide substrate (80, 127). Mammalian sirtuins are involved in metabolism (80, 85, 137), which is linked to the mitochondrial generation of ROS (96, 155). NADPH oxidase (nicotinamide adenine dinucleotide phosphate-oxidase) is implicated in the ongoing pathology of several age- associated neurodegenerative diseases such as AD and PD and it has been shown to play a major role in oxidative stress and neuroinflammation after ischemic brain injury and TBI. Moreover, metabolic challenges, such as ischemia, or oxidization could result in decreased NAD and nicotinamide phosphoribosyltransferase (NAMPT) contents, and reduced activity of sirtuins (18). It has been shown that glutamate-induced excitotoxicity in neurons and ischemia in mice brain result in marked decreases in NAD+ levels, which is a precausative step before cell death (100). Indeed in TBI model, intranasal administration of NAD+ (20 mg/kg) right after TBI, rescued neurons in dentate gyrus (DG) of the hippocampus. According to this finding, NAMPT inhibition increased the damage size in brain following cerebral ischemia in rats, while NAMPT overexpression significantly reduced the infarct size. The protective effects of NAMPT overexpression were associated with the activity of SIRT1 and its interaction with serine/threonine kinase 11, which is an upstream kinase of AMPK. These results demonstrate that ROS associate reduction of levels NAD leading to decreased activity of sirtuins and often results in death of neurons.
However, sirtuins readily deacetylate p53, forkhead homeobox type O (FOXO)-s, and peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) among others.
Activation of p53 by CBP/p300 could lead to enhanced production of ROS by mitochondrial dysfunction, or target genes that would produce ROS, such as PUMA, NOXA, PIG3, and stress-activated protein kinases (67, 94). p53 can indirectly activate Nrf2 via p21, and Nrf2 binds to the antioxidant response element and controls response to oxidative challenge (81). Hence, SIRT1, by its interaction with p53, could exert both pro- and antioxidant effects. In a transgenic mouse model of AD and tauopathies, has been shown, that resveratrol significantly ameliorated neuron viability in the hippocampus, prevented loss of learning, and decreased the acetylation of p53 and PGC-1alpha as a result of activation of SIRT1.
PGC-1α regulates the transcription of genes, encoding proteins in the mitochondrial electron transport chain, and key antioxidant enzymes such as Mn-SOD, GPX, catalase, uncoupling protein 2 (UCP2) and UCP3 (147) and being a target of SIRT1. An early study on sirtuins and DNA repair and telomere silencing was done on saccharomyces cerevisiae, and revealed that Ku protein, which is associated with DNA double-strand break and telomeric length maintenance, could interact with sirtuins (17, 118). SIRT6 is important for other DNA repair mechanisms, since on SIRT6 ablated cells, overexpression of a single strand break gap-filling DNA Polβ rescued cells, pointing out that SIRT6 plays a crucial roleApurinic/apyrimidinic endonuclease-1 (APE1) is also regulated by SIRT1(167). It is important to note that samples obtained from human brain regions of healthy subjects and others with different degrees of AD suggest that SIRT1 expression and protein levels are decreased in parietal cortex of AD patients, while cortical expressions were not affected. Global cognition scores obtained before death would suggest that the decrease in SIRT1 levels is related to the accumulation of amyloid-beta and tau in the cerebral cortex of persons with AD (82).
Neuroprotective Agents and Therapeutic Approach
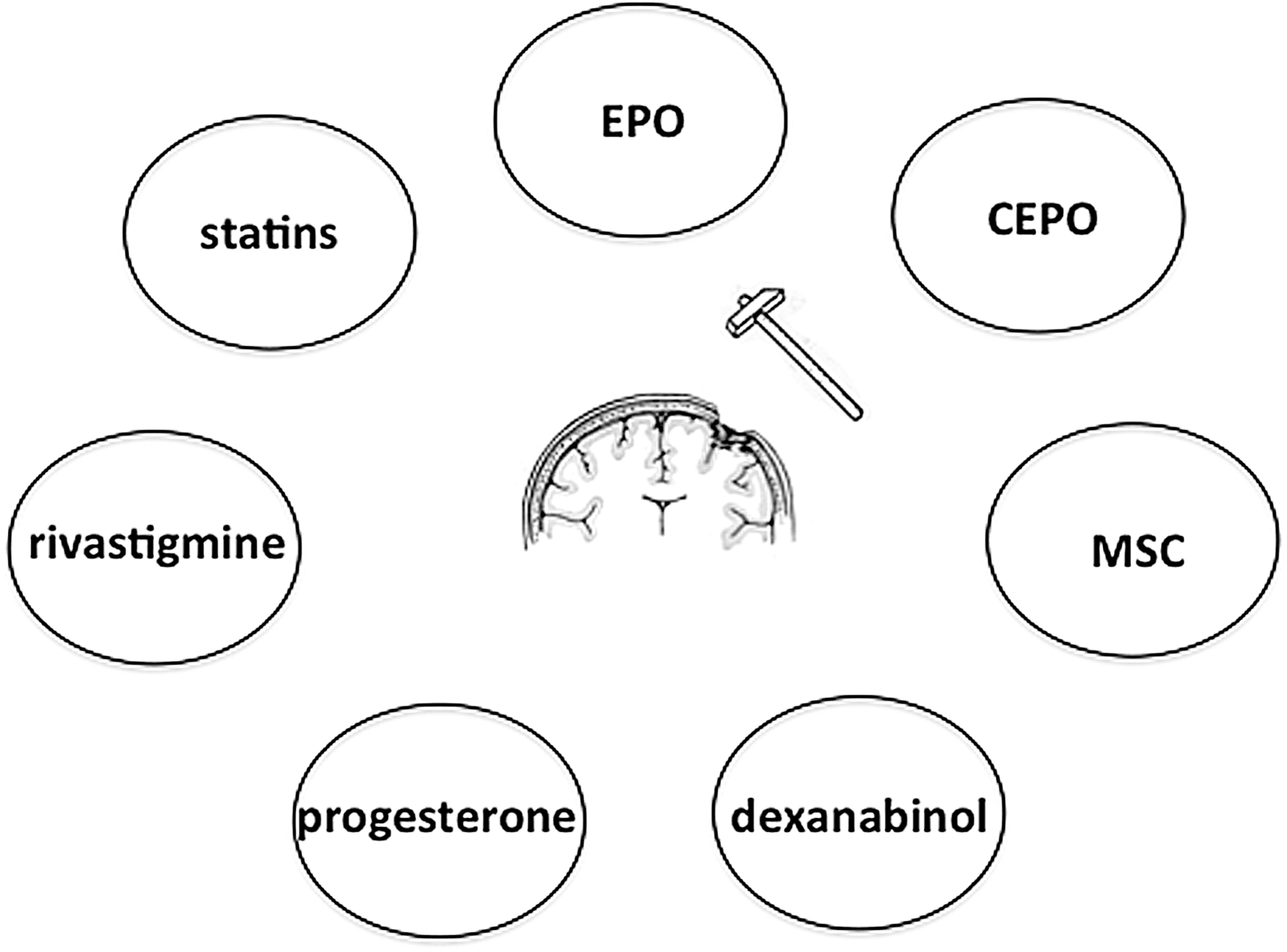
Several research lines have been implicated in the role that different pharmacological agents may have in neuroprotection and neurofacilitation. From beanch to clinical practice and based on pathophysiological extrapolations, multiple substances have been tested as possible neuroprotective agents in TBI. Preclinical studies have tested therapeutic efficacy of drugs in animal TBI models by targeting secondary injury mechanisms including calcium channel blockers, corticosteroids, excitatory amino acid inhibitors, NMDA receptor antagonist, free radical scavengers, magnesium sulfate, and growth factors (124). Different laboratories have identified several therapeutic classes showing promise for the treatment of TBI (161). Among these we included: erythropoietin (EPO), carbamylated form of EPO (CEPO), statins, bone marrow stromal cells (MSC), methylphenidate, progesterone, dexanabinol, and rivastigmine (161). (Fig. 8.) It was demonstrated that TBI induces neurogenesis in the subgranular zone of the DG in rat and mouse and treatments that enhance neurogenesis also stimulate cognitive function after TBI (102). Previous studies show that treatment of TBI with EPO, S100B, MSC, or other manipulations such as environmental enrichment (65) enhance neurogenesis along with functional improvement. In addition to neurogenesis, the brain remodeling after TBI includes angiogenesis, axonal sprouting, and synaptogenesis (166).
PEG-SOD
Administration of SOD prevented post-traumatic microvascular dysfunction (91). This leads to clinical trials in which the more metabolically stable PEG-conjugated SOD was examined in moderate and severe TBI patients when administered within the first 8 h after injury. Although an initial small phase II study showed a positive trend, subsequent multicenter phase III studies failed to show a significant benefit in terms of increased survival or improved neurological outcomes (54). Although many explanations for these negative results may be postulated, one reason may be that a large protein like SOD is unlikely to have much brain penetrability and therefore its radical scavenging effects may be limited to the microvasculature. A second reason may be that attempting to scavenge the short-lived inorganic radical O2•− may be associated with a very short therapeutic window. Indeed, the time course of measurable post-traumatic •OH formation in the injured rodent brain has been shown to largely run its course by the end of the first hour after TBI (79). Transgenic mice that overexpress Cu/Zn SOD are significantly protected against post-TBI pathophysiology and neurodegeneration (112).
Statins
Statins are drugs used to lower cholesterol levels by inhibiting the enzyme HMG-CoA reductase, which plays a central role in the production of cholesterol in the liver (51) that make them potentially attractive neuroprotective agents (165). Statins increase endothelium-derived nitric oxide production, reduce vascular inflammation, limit hemorrhagic stroke at the microvascalture level, and protect cortical neurons from NMDA-induced excitotoxic death (55). They decrease interleukin (IL)-1β, tumor necrosis factor α (44), IL-6, and intracellular adhesion molecule 1 (44) expression levels. Several preclinical studies demonstrate that statins target multiple secondary injury pathways and improve functional outcome after TBI (44). The therapeutic window for this class of drugs is relatively large, with treatment 24 h after TBI resulting in long-term functional improvements and reduced neuronal cell loss.
Superoxide dismutase
SOD are enzymes that catalyze the dismutation of superoxide into oxygen and H2O2. Under physiologic conditions, reactive hydroxyl (OH) radicals are continuously produced and removed from the brain by natural antioxidant systems that consist of a numerous antioxidant compounds and enzymes. Three forms of SOD are present in humans: SOD1 (cytoplasm), SOD2 (mitochondria), and SOD3 (extracellular).
SOD converts O2, to H2O2. This molecule is converted to water and molecular oxygen by GPx. Copper=zinc SOD (SOD1) is a dimeric cytosolic enzyme that is coded by the human SOD1 gene and that needs copper and zinc cofactors. SOD3 is mainly localized in extracellular space, cerebral fluid, and cerebral vessels and its cofactors are copper and zinc. SOD1 is associated with a dose-related decrease in brain edema, tissue infarction, and BBB distruption in models of cold and contusion brain injury following transient cortical ischemia (6).
Erythropoietin
EPO, is widely recognized for its role in stimulating the maturation, differentiation, and survival of hematopoietic progenitor cells (164). It has been found to offer some protection to the brain when brain cells are deprived of their normal oxygen supply causing cells to die or be impaired. While EPO and its receptor (EPOR) are only weakly expressed in normal adult brain, expression of EPO and the EPORs is increased in neurons, neuronal progenitor cells, glia, and cerebrovascular endothelial cells in response to many different types of cell injury (164). Intraperitoneal administration of human recombinant erythropoietin (rhEPO) crosses the blood-brain barrier to protect against brain injury (71). When EPO binds to its receptors, it causes dimerization of receptor, autophosphorylation of Janus-tyrosine-kinase-2 (JAK-2), and receptor activation. JAK-2 activation leads to phosphorylation of several downstream signaling pathways such as phosphatidylinositol 3-kinase (PI3K) (88). PI3K then activates v-akt murine thymoma viral oncogene homolog (Akt) (89). These pathways are crucial for the therapeutic efficacy of EPO, since specific inhibitors of the PI3K pathway largely abolish the EPO-increased neuronal survival in a model of hypoxia (89). EPO activates PI3K/Akt and extracellular signal-regulated kinase (ERK1/2) and promotes neural progenitor cell migration in cultured mouse brain endothelial cells (171). rhEPO administration for 14 days increases the number of BrdU-labeled cells in both the contralateral and ipsilateral DG after TBI and promotes restoration of spatial memory after TBI. A significant increase in brain-derived neurotrophic factor expression and improvement in spatial learning are seen in animals treated with rhEPO or CEPO after TBI. CEPO devoid of hematopoietic bioactivity has also been shown to improve functional recovery after stroke and TBI (153).
Carnosine
Carnosine is a natural antioxidant dipeptide composed of the amino acids histidine and alanine found in brain, heart, skin, muscles, kidneys, gut, and other tissues. Hipkiss (75) proposed that carnosine is a naturally occurring suppressor of oxidative damage in olfactory neurons. De Marchis et al. (53) found carnosine in mature olfactory receptor neurons, a subset of glial cells, and neural progenitor cells in rat brain. Carnitine is also present in olfactory neurons and visual system (53). The involvement of nitrosative and oxidative stress in the development of neurodegenerative disorders is no longer a matter of question (31). The efficacy of carnosine in reducing both PARP-1 and PARP-2 activation after the oxidative stress induction has been also proved (146).
Nitric oxide
NO activates soluble guanylyl cyclase, leading to the formation of cyclic guanosine monophosphate (cGMP). As a second messenger, cGMP is involved in several cellular processes, including regulation of cellular proliferation. Increases in cGMP levels enhance proliferation of endothelial cells and motor neurons (45). cGMP levels in brain may be increased by cGMP production via increases in NO or inhibition of cGMP hydrolysis using phosphodiesterase 5 inhibitors (45). In the CNS, NO is an important messenger involved in the modulation of sensory motor functions, control of CBF, neuroprotection, and neurotoxicity after cerebral synaptic formation and remodeling, brain development, synaptic plasticity, neuroendocrine secretion, sensory processing, and CBF (40). While the proliferative effect of NO on endogenous progenitor cells in adult brain could be mediated through an increase in the tissue levels of cGMP (170), the antiproliferative effect of NO depends on the inhibition of cyclin-dependent kinases and transcription factors by p53 and the Rb protein, respectively. NO donors increase cGMP levels via activation of soluble guanylyl cyclase (170). TBI alters the migration pathway of SVZ progenitor cells from the rostral migratory stream to the striatum and corpus callosum. Treatment with NO donor, DETA/NONOate, enhances these responses. DETA/NONOate increases progenitor cell migration, induces differentiation of the progenitor cells, and enhances the survival of the newly generated cells in the striatum, corpus callosum, and boundary zone of the lesion. DETA/NONOate treatment improves the neurological outcome in rats subjected to TBI (103).
Conclusion
Trauma is responsible for sudden biochemical changes occurring at the time of impact, and the severity of brain insult can be graded by measuring these biochemical modifications, specifically, ROS-mediated damage, energy metabolism depression, alteration of gene expression, and ultimately variation of NAA concentration, a surrogate marker of neuron dysfunction. TBI combines mechanical stress to brain tissue with an imbalance between CBF and metabolism, excitotoxicity, edema formation, and inflammatory and apoptotic processes. Understanding the multidimensional cascade of injury offers therapeutic options including the management of mechanical (hyper-) ventilation, kinetic therapy to improve oxygenation and to reduce intracranial pressure, and pharmacological intervention to reduce excitotoxicity and intracranial pressure. The unpredictability of the individual's pathophysiology requires monitoring of the injured brain to tailor the treatment according to the specific status of the patients. It will be important to better facilitate bidirectional translational research between preclinical and clinical investigators, which should serve to improve both approaches to animal modeling and the design of clinical trials. Future advances in clinical data sharing should improve TBI classification in ways that may lead to delineation of specific patient subgroups that may benefit from better targeted neuroprotective strategies.