
Abstract
Introduction
Phosphorylation is a widely exploited mechanism of controlling cellular homeostasis. A transient and reversible modification, phosphorylation functions as a form of spatial and temporal regulation on macromolecules. It is thought that up to one-third of all cellular proteins are phosphorylated during their lifespan (21), demonstrating the ubiquity and importance of this modification in signal transduction. Two groups of enzymes, kinases and phosphatases, play a critical role in regulating phosphorylation and are thus implicated in a wide array of cellular processes, including survival and apoptosis.
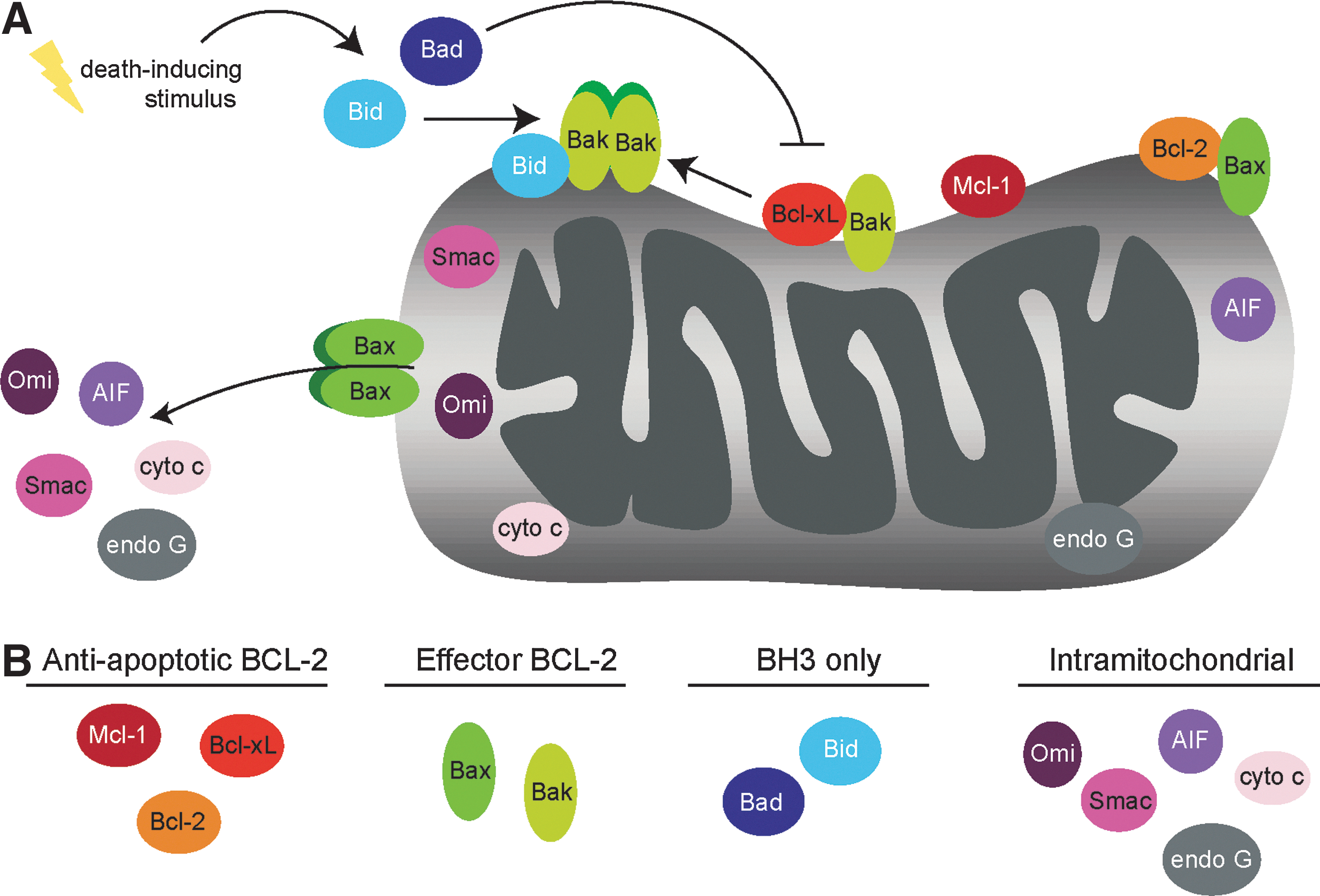
Mitochondrial apoptosis is a complex process in which the cell undergoes an intrinsic program leading to the death of the cell. Upon sensing extensive stress and/or irreparable damage, the cell utilizes a well-conserved signaling pathway to carry out this suicidal program. Apoptosis proceeds upon the compromise of the outer mitochondrial membrane (OMM), allowing the release of proteins that directly or indirectly activate caspases, proteases that facilitate the dismantling of the cell. Mitochondrial outer membrane permeabilization, often referred to as MOMP, is the point of no return in the intrinsic apoptotic process (17). Members of the Bcl-2 family of proteins control MOMP, and thus are the primary decision makers of apoptotic cell fate. Upon Bcl-2 family-orchestrated permeabilization of the OMM, several mitochondrial proteins resident in the intermitochondrial membrane space (IMS) are released, promoting various proapoptotic functions, the most important of which is caspase activation.
Because the decision between life and death is critical to overall homeostasis, the proteins involved in modulating the apoptotic response are tightly regulated. Phosphorylation is vitally important in this regulation, affecting the induction of apoptosis by altering protein localization, stability, and/or enzymatic activity. In this review, we summarize the current literature regarding phosphorylation events on mitochondrial-localized proteins directly implicated in apoptosis, specifically focusing on 12 of these proteins identified in the MitoCarta (37) (Table 1). Strikingly, 11 of these proteins have documented phosphorylation events (Fig. 1), many of which have known effects on apoptotic function (Figs. 2 and 3). Additionally, we will briefly discuss mitochondrial-localized kinases and phosphatases linked to cell death and how these enzymes may regulate the apoptotic machinery (Fig. 4).


Twelve proteins directly involved in the execution of the apoptotic process were identified as mitochondrial through the query of the MitoCarta, an index of proteomically identified mitochondrial proteins. Gene symbol, aliases, accession numbers, known subcellullar localization, chromosomal location, protein size (in Da), and known post-translational modifications are listed. For characterization, all protein products are of human origin, and the longest splice isoform is reported. As this review focuses solely on phosphorylation, acetylation and ubiquitination events are not followed-up on within the article. Abbreviations: aK, acetylated lysine; uK, ubiquitinated lysine; pS, phosphorylated serine; pT, phosphorylated threonine; pY, phosphorylated tyrosine. For interpretation of the references to color in this and all other figures, the reader is referred to the electronic version of this dissertation.
PTM, post-translational modification; IMS, intermitochondrial membrane space; IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane.
Bcl-2 Family Proteins: Controlling Membrane Permeabilization
At the molecular level, antiapoptotic Bcl-2 family members function to inhibit apoptosis through two distinct MODES: MODE 1 involves the direct interaction of antiapoptotic Bcl-2 family members with the direct activator BH3-only proteins, resulting in BH3-only sequestration; MODE 2 involves antiapoptotic Bcl-2 proteins binding directly to activated proapoptotic effectors Bax and Bak, diminishing their ability to homo-oligomerize (31). Neutralization of the antiapoptotic proteins occurs through activation and/or stabilization of BH3-only proteins, which can displace sequestered proapoptotic proteins, allowing mitochondrial membrane permeabilization to proceed. The complex interplay between the proapoptotic and antiapoptotic family members is tightly controlled so as to ensure the survival or death of a cell within the appropriate cellular context. As such, phosphorylation of Bcl-2 family members affects their stability and interactions, which can shift the cell toward or away from apoptosis as necessary (Table 2).
Bcl-2 family members with both pro- and antiapoptotic functions are regulated by phosphorylation. Phosphorylation sites detailed in the article are listed in this table, with phosphoresidues corresponding to the human protein sequence. Proposed function, as well as regulatory molecules known to control phosphorylation (kinases and phosphatases), are detailed. Blank columns indicate that the function or regulatory molecules associated with the phosphosite are currently unknown.
Bcl-2
Numerous studies have linked phosphorylation of the antiapoptotic Bcl-2 family members with their ability to induce apoptosis, particularly in the context of chemotherapeutic-induced microtubule disruption (18). In most studies, the induction of apoptosis is accompanied by Bcl-2/Bcl-xL phosphorylation, suggesting that this modification blocks the prosurvival functions of these proteins.
The most well-characterized phosphorylation site on Bcl-2 is Ser70, which lies within an unstructured loop region (LR) between the BH4 and BH3 domains. Interestingly, the LR exists only within the antiapoptotic Bcl-2 family members, and its deletion has been shown to increase cell survival in cell culture models (2, 41). These data suggest that phosphorylation within this region may provide a switch by which cells can specifically inactivate the prosurvival functions of the antiapoptotic Bcl-2 family members.
Consistent with phosphorylation negating the prosurvival function of Bcl-2, transfection of prostate cancer cells with a phosphomimetic mutant of Bcl-2, S70E significantly enhances cell death in response to paclitaxel treatment relative to cells transfected with wild-type Bcl-2 (2). These data suggest that phosphorylation of Bcl-2 at Ser70 diminishes its proapoptotic activity in the presence of paclitaxel.
Interestingly, phosphorylation of Bcl-2 at Ser70 has also been demonstrated to enhance its survival function, particularly upon withdrawal of the growth factor interleukin 3 (IL3) in myeloid cells (23). In these studies, the phosphomimetic S70E enhances cell survival in response to IL3 withdrawal, while a nonphosphorylatable S70A mutant does not allow long-term survival of myeloid cells in response to either IL3 withdrawal or treatment with the chemotherapeutic etoposide (23).
A handful of experimental observations can perhaps address the paradox as to how phosphorylation at the same serine on Bcl-2 could have seemingly opposite functional effects. The first explanation is that Bcl-2 simply has tissue-specific functions, and that its phosphorylation in different cell types, such as prostate cancer cells and myeloid cells, results in opposite biological functions. A second potential reason for the different phenotypes could be the differential kinetics of Bcl-2 phosphorylation in these two experimental systems. Interestingly, paclitaxel-induced Bcl-2 phosphorylation is seen only after 2 h, while growth factor starvation induces Bcl-2 phosphorylation within minutes of IL3 withdrawal (2). These data suggest that different signaling pathways are responsible for the phosphorylation of Ser70, and that the regulation of this phosphorylation event may lead to differential survival capabilities within various cellular contexts. Finally, it has been noted that paclitaxel treatment induces not only phosphorylation of Ser70, but also proximal residues, Thr69 and Ser87 (2). Thus, phosphorylation on multiple residues, perhaps in a particular sequential order, may be necessary to negate the prosurvival functions of Bcl-2 in response to paclitaxel.
Multiple studies have sought to determine the functional outcomes of multisite phosphorylation on Bcl-2. In addition to Thr69, Ser70, and Ser87, Thr56 was also identified as a phosphosite which, upon mutation to a phosphomimetic in coordination with Ser87 (T56E and S87E) ablates cytochrome c release and growth factor deprivation-induced cell death (7). Additionally, a mutant form of Bcl-2 (S70A, S87A, T56A, and T74A) increases its effective inhibition of cell death, demonstrating that phosphorylation at one of these residues inhibits its apoptotic potential (42).
Mechanistically, phosphomimetic mutations can enhance the prosurvival function of Bcl-2 by affecting its potential to bind to proapoptotic proteins. A triple phosphomimetic Bcl-2 mutant (T69E/S70E/S87E) decreases p53 binding, and thus enhancing its antiapoptotic function relative to wild-type Bcl-2 (10). Additionally, phosphorylation of Bcl-2 may affect its stability: nonphosphorylatable T74A and S87A mutants showed significantly increased degradation relative to wild-type Bcl-2, suggesting that phosphorylation of these residues maintains its protein expression (3). Although both of these mutations were seen in the quadruple mutant (S70A, T74A, S87A, T56A), no altered stability effects were noted in this quadruple mutant, suggesting that this phenotype, like many others documented, may be context dependent.
While the exact functional outcomes of Bcl-2 phosphorylation seem to be tissue type-, time-, and/or insult-dependent, it is clear that the modification of this protein has stark effects on its function. Detailed analysis of the spatial and temporal regulation of these phosphorylation sites will be required to fully understand the global, and perhaps context-dependent, effects of phosphorylation on Bcl-2 function.
Bcl-xL
Like Bcl-2, the phosphorylation of Bcl-xL is most often linked to its inactivation, and is also seen frequently in the context of microtubule-disrupting agents (18). JNK has been proposed to phosphorylate Bcl-xL and lead to its inactivation in response to stress signaling. In particular, studies have demonstrated that JNK phosphorylates three specific residues: Thr47, Ser62, and Thr115, though studies often identify only one or two of these phosphosites in their analyses. JNK has been suggested to phosphorylate Ser62 of Bcl-xL in prostate cancer cells in response to taxol treatment. Additionally, JNK phosphorylates Bcl-xL on Thr47 and Thr115 in vitro and in vivo (24); simultaneous mutation of these residues to alanine results in increased apoptotic inhibition relative to wild-type enzyme, suggesting that these two phosphorylation events are inhibitory and promote apoptosis. Thus, as with Bcl-2, there is probably underlying context dependency regarding which residues of Bcl-xL are phosphorylated, and how these post-translational modifications affect the function of the protein. On a molecular level, phosphorylation of Bcl-xL has been suggested to inactivate the prosurvival function of the protein through the disruption of its proapoptotic-binding partners. One study demonstrates that a nonphosphorylatable mutant of Bcl-xL, S62A, renders cervical carcinoma cells unresponsive to chemotherapeutic treatment, consistent with this phosphorylation event being necessary for cells to undergo apoptosis (43). Further, it was shown that the interaction between the proapoptotic effector molecule Bax and the S62A mutant of Bcl-xL is much more robust than with wild-type Bcl-xL (43). This suggests that phosphorylation of S62 is required to interrupt the interaction between Bcl-xL and Bax, disengaging the sequestration of the proapoptotic Bax and allowing apoptosis to proceed. Thus, phosphorylation of Bcl-xL seems to have robust effects on its ability to enhance cellular survival through controlling the antiapoptotic/proapoptotic interaction of the Bcl-2 family members.
Mcl-1
Mcl-1 is an antiapoptotic Bcl-2 family member that differs from Bcl-2 and Bcl-xL at its N-terminus, which is larger and is enriched in residues important in its regulation and turnover (Fig. 2). Mcl-1 is phosphorylated during mitotic arrest at Thr92 by the CDK1/cyclin B complex, promoting its ubiquitination and degradation via the E3 ubiquitin ligase activity of the anaphase promotion complex (APC/C). IL3 withdrawal or PI3K inhibition causes GSK-3-mediated phosphorylation of Mcl-1 at Ser159 in lymphocytes, resulting in its destabilization and the induction of apoptosis (32). GSK-3 has also been shown to dually phosphorylate Ser159 and Thr163, promoting the interaction of Mcl-1 with the E3 ubiquitin ligase SCF-FBW7. Loss of FBW7 is sufficient to drive tumorigenesis in an Mcl-1-dependent fashion, and downregulating Mcl-1 is sufficient to restore chemotherapeutic response to both microtubule destabilization agents (49) as well as the Bcl-2/Bcl-xL antagonist ABT-737 (22). As GSK-3 phosphorylation of Mcl-1 is required for this interaction, dysregulation of these phosphorylation sites could also have direct therapeutic consequences.
Interestingly, phosphorylation of Mcl-1 at Ser64 has been proposed to enhance its antiapoptotic activity; reconstitution of cells with the phosphomimetic S64E renders cells resistant to TRAIL-induced apoptosis (26). Mcl-1 S64E does not affect protein stability, but rather increases its affinity to BH3-only proteins, allowing their sequestration and effectively increasing its antiapoptotic activity. Thus, phosphorylation of Mcl-1 can influence its protein stability or interactions, modulating its apoptotic capacity accordingly.
Bax
Bax is a proapoptotic effector molecule that resides in the cytosol in healthy cells, but is recruited to the OMM upon induction of apoptosis. At the OMM, Bax undergoes conformational changes that allow its membrane insertion and oligomerization, which are points of regulation by phosphorylation. Many of the same kinases that inhibit the antiapoptotic activity of Bcl-2 and Bcl-xL (JNK, p38, and GSK-3) also function to increase the proapoptotic activity of Bax. In hepatocytes, JNK- and p38-dependent phosphorylation of Bax facilitates its mitochondrial localization and apoptotic function (25). Ser163 lies within a canonical GSK-3β phosphorylation motif, and mutation or deletion of this motif inhibits the mitochondrial localization of Bax in apoptotic neurons (30). In yeast, a phosphomimetic substitution at Ser60, which lies within a predicted PKA phosphorylation motif, increases mitochondrial localization of Bax, but, importantly, does not induce cytochrome c release (1). This suggests that phosphorylation of this N-terminal residue primes Bax for activation, but is not sufficient to impart full apoptotic activity.
Bax phosphorylation has also been linked to a decrease in apoptotic activity: AKT-mediated phosphorylation of Ser184 decreases mitochondrial localization and conformational changes associated with its activation (16). These results were confirmed in a second study showing that nonphosphorylatable Bax mutant S184A had increased apoptotic functions, while a phosphomimetic Bax (S184E) could not insert into the mitochondrial membrane (47). Additionally, mutation of S184 to a charged residue (His, Asp, Glu, or Lys) increases cytosolic distribution, whereas substitution with a valine or a deletion of S184 increases mitochondrial localization of Bax (34). Interestingly, Ser184 falls into a hydrophobic stretch of amino acids allowing the insertion of Bax into the OMM (Fig. 2). As a phosphoryl group is charged, this modification could inhibit insertion into the hydrophobic lipid membrane, blocking this step in Bax activation. Collectively, these data demonstrate that kinase cascades downstream of proapoptotic signals (JNK, p38, or GSK-3β), and antiapoptotic signals (AKT) can modulate the apoptotic potential of Bax through the phosphorylation of distinct and conserved residues.
Bak
Bak, a second effector apoptotic protein, is constitutively localized within the OMM and is thus primed for activation. Bak undergoes significant dephosphorylation in response to apoptotic activation; in vitro assays with phosphatases of differential specificity suggest that Bak is phosphorylated at serine, threonine, and tyrosine residues in healthy cells (14). Of these residues, the dephosphorylation of Tyr108 is necessary, but not sufficient for the activation of Bak. This dephosphorylation promotes proapoptotic conformational changes in the N-terminus of Bak, allowing it to oligomerize and destabilize the OMM. An RNAi screen of tyrosine phosphatases identified PTPN5 as the enzyme that specifically dephosphorylates Tyr108, and importantly, it was shown that downregulation of this PTPN5 directly affects the proapoptotic conformational change of Bak. Despite the importance of Bak phosphorylation in cell survival, the kinase responsible for phosphorylating Tyr108 has yet to be identified, and represents an important next step in understanding the regulation of this phosphorylation event in apoptosis.
Bad
BH3-only proteins play an important role in controlling the onset of the apoptotic response. Bad functions to bind and derepress antiapoptotic Bcl-2 proteins and numerous studies have demonstrated that Bad phosphorylation can affect these interactions. AKT was the first kinase associated with Bad phosphorylation, modifying Ser112 and Ser136 of murine Bad (Ser75 and Ser99 of human Bad; Fig. 2) (5). Phosphorylation of these sites creates a consensus motif for an interaction with 14-3-3 proteins, which sequester Bad from antiapoptotic Bcl-2 family members and thus promote survival. A second study demonstrated that mitochondrial-anchored PKA can also phosphorylate Bad at murine Ser112 (human Ser75), inhibiting its apoptotic activity in a fashion similar to AKT-mediated inactivation. (19) The phosphatase calcineurin dephosphorylates these two residues, causing dissociation of the Bad/14-3-3 complex and resulting in Bad translocation to the mitochondria, where it induces apoptosis (46).
Phosphorylation on other Bad residues, such as murine Ser155 (human Ser118), also seems to inhibit its apoptotic effects (45). PKA can phosphorylate Ser155 which, along with its homologous human residue Ser118, reside within the BH3-only domain of Bad (45). This phosphorylation disrupts the ability of Bad to bind to Bcl-xL, disallowing the displacement of the effector molecules Bax/Bak, and thus interrupting Bad-initiated apoptosis. These data demonstrate that prosurvival signaling pathways can directly block apoptosis through the phosphorylation and inactivation of BH3-only proteins such as Bad.
Bid
Bid is a unique BH3-only protein, in that it is activated by caspase-8 in response to death receptor activation. The caspase-mediated cleavage of Bid frees its C-terminus, containing a newly exposed BH3 domain, which is then able to directly interact with and activate Bax/Bak at the OMM. Thus, caspase-8-mediated cleavage of Bid is a key regulatory step in the activation of this protein. Multiple studies have documented phosphorylation of Bid, which is associated with diminished activation potential by caspase-8. One study found that a specific peptide of murine Bid, mapping to amino acids 41 to 65, contains three potential phosphorylation sites (Thr58, Ser61, and Ser64, mapping to human Thr59, Gln62, and Ser65, respectively) (12). Interestingly, these phosphorylation sites are proximal to the cleavage site for caspase-8 (Asp59), which ultimately controls Bid activation. Follow-up experiments suggested that Ser64 and Ser61 are sequentially phosphorylated by casein kinases I and II, respectively, preventing caspase-8 cleavage and thus prohibiting the activation of Bid. Interestingly, these sites are constitutively phosphorylated in vivo, suggesting that Bid must be dephosphorylated before it can be activated by caspase-8. The phosphatase responsible for this dephosphorylation has not yet been identified. Importantly, these results were reproduced with human Bid, with both Thr59 and Ser65 phosphorylation linked to decreased caspase cleavage in vitro (8). Collectively, these data demonstrate that phosphorylation of Bid results in a robust inhibition in its cleavage by caspase-8, and suggest that dysregulation of these phosphorylation events can alter Bid-mediated activation of apoptosis.
Intermembrane Space Proteins: Facilitating Caspase Activation upon Release
Upon permeabilization of the OMM, proteins residing in the IMS are released to perform proapoptotic functions within the cytosol and nucleus. Cytochrome c promotes apoptosome formation, resulting in caspase activation; Smac/DIABLO and Omi/Htra2 function to free caspases from inhibitor of apoptosis proteins (IAPs), facilitating their activation. AIF and Endonuclease G translocate to the nucleus upon release from the mitochondria, where they initiate DNA-associated apoptotic changes. Interestingly, these proteins are dynamically regulated by phosphorylation events, which can occur either within the mitochondria (before MOMP occurs) or upon exposure to their new cellular compartment (after MOMP). The details of these post-translational modifications and how they affect apoptotic induction are discussed in this section.
Cytochrome c
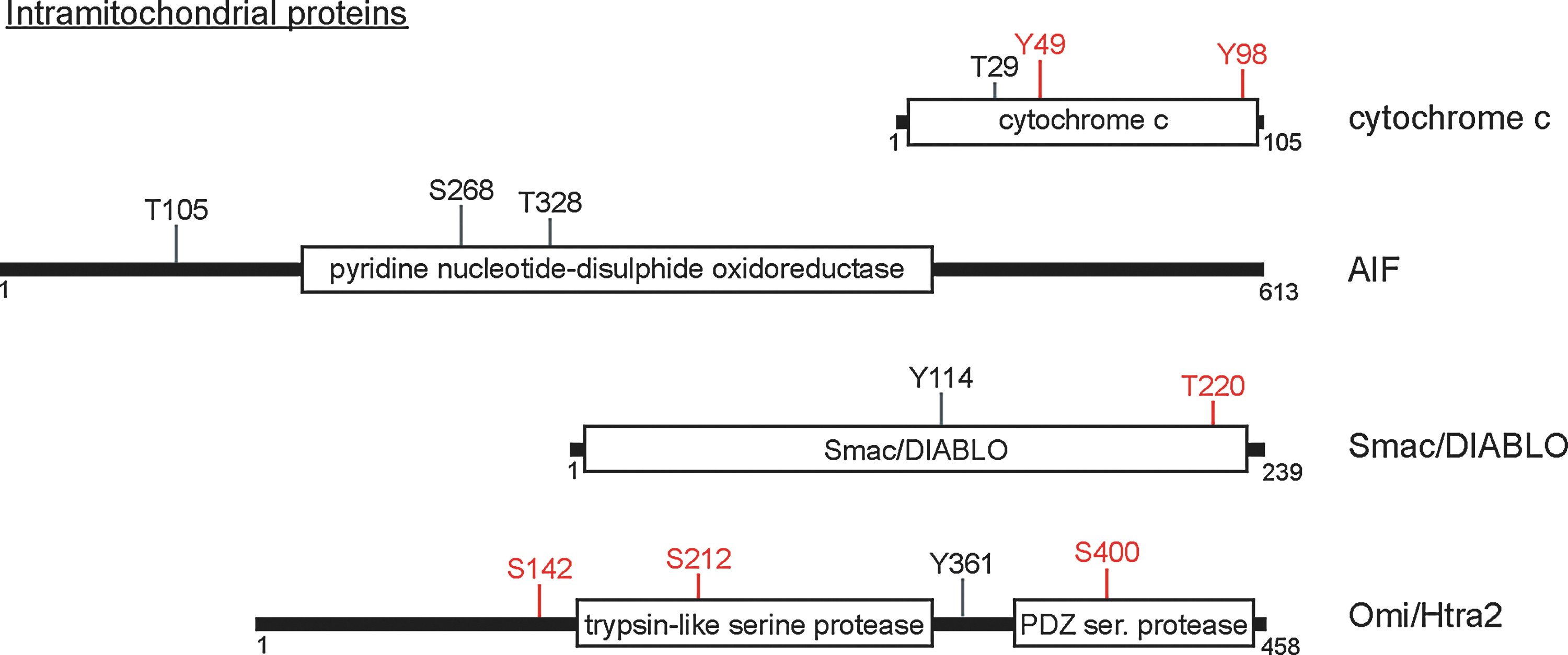
Native cytochrome c purified from bovine tissue is tyrosine phosphorylated, though perhaps in a tissue-specific context: cytochrome c isolated from the bovine heart is phosphorylated specifically at Tyr97, while Tyr48 phosphorylation is identified when cytochrome c is purified from bovine liver tissue (28, 51). Phosphorylation of either residue may play a role in apoptotic induction. The key residue of cytochrome c that participates in apoptosome formation, Lys7, is spatially proximal to the Tyr97 side chain. It has been postulated that phosphorylation of Tyr97 could sterically hinder apoptosome formation, though experiments proving this have yet to be performed. The phosphorylation of Tyr48 does directly affect apoptosome formation in vitro, as the Tyr48 phosphomimetic, Y48E, significantly decreased caspase-9 activation relative to wild-type cytochrome c (15). These data demonstrate that cytochrome c is a bona fide tyrosine kinase substrate, and suggest that phosphorylation at specific tyrosine residues can influence its ability to induce apoptosis.
AIF
AIF is a proapoptotic protein released from the IMS upon MOMP, after which it translocates to the nucleus to promote DNA fragmentation and other nuclear-associated apoptotic changes. Phosphorylation of AIF has been documented in several large-scale proteomics studies (6, 11, 36). Interestingly, all of these studies looked at phosphorylation events correlated with cell cycle progression and identified multiple AIF phosphosites (Fig. 3). While the functional significance of these residues is unknown, it is worth noting that most are conserved at least through mammals, suggesting that these residues could be important to protein function. Follow-up studies will be required to determine whether these phosphorylation events are dynamically regulated, in which cellular compartment they occur, and which enzymes are responsible for their regulation to determine what effects, if any, AIF phosphorylation plays in its apoptotic function.
Smac/DIABLO
Smac/DIABLO is a resident of the IMS that is released upon the permeabilization of the OMM. Only two phosphorylation sites have been documented on Smac; these two residues, Tyr114 and Thr220, were identified in large-scale proteomic studies (20). The functional significance of these phosphorylation events, as well as their dynamic regulation in cells, has yet to be defined. Phosphorylation of Smac, however, has been suggested to impact its apoptotic effects; a recent study demonstrated that JNK3 directly phosphorylates Smac, diminishing its capacity to bind and neutralize the prosurvival caspase inhibitor XIAP in in-vitro caspase activation assays. This suggests that Smac may be phosphorylated by cytosolic prosurvival kinases to diminish its apoptotic capacity. More work will be required to determine the consequences of this phosphorylation in a cellular context, and to determine whether it is exploited as a cellular mechanism to dampen the apoptotic response.
Omi/Htra2
The serine proteinase Htra2/Omi resides in the IMS and is released into the cytosol after MOMP, where it interacts with IAPs, allowing the activation of caspases. Mutations in Htra2 are associated with familial Parkinson's disease (PD), a disease in which dopaminergic neurons undergo inappropriate cell death. The phosphorylation of Htra2 at three specific residues has been linked to its apoptotic functions in cells. Htra2 is phosphorylated at S142 upon activation of p38, and interestingly, this phosphorylation is dependent on PINK1, a mitochondrial kinase also associated with the etiology of PD (39). S142 phosphorylation increases the proteolytic activity of Htra2 in vitro, which is suggested to enhance its proapoptotic function (39). Cdk5 phosphorylates Htra2 on Ser400 in a p38-dependent manner, which protects cells against cellular stress through currently undefined mechanisms (13). Htra2 can also be phosphorylated after release from the mitochondria; AKT phosphorylates Htra2 at Ser212, diminishing its protease activity and subsequently decreasing its proapoptotic function (50).
Interestingly, two recurrent mutations associated with PD fall proximal to putative phosphorylation sites, suggesting that dysregulation of Htra2 phosphorylation could alter disease susceptibility and/or progression. A PD-associated mutant of Htra2, P143A, is hyperphosphorylated relative to wild-type Htra2 (29). This proline residue is next to Ser142, whose phosphorylation regulates the apoptotic function, suggesting that dysregulation of this residue could contribute to the pathology of PD.
Mitochondrial Kinases and Phosphatases—Intrinsic Regulators of Mitochondrial Apoptosis?
For phosphorylation events to be dynamically regulated, enzymes must control the placement and removal of phosphate onto specific substrates to trigger an intentional cellular response. For apoptosis-involved substrates localized to the OMM, these kinases and phosphatases can have one of two localization patterns: they may be resident in the OMM, placing the enzyme–substrate pair in constitutive proximity, or they may be recruited to the OMM upon proper stimulation to activate or inactivate the substrate as necessary. Both of these patterns have been exploited in the control of cell death; PGAM5 was recently identified as a resident OMM phosphatase whose dephosphorylation of DRP-1 is critical in the onset of programmed necrosis (48). The latter scenario is also employed; as previously outlined, numerous kinases activated by growth factor signaling have been implicated in the phosphorylation of proapoptotic proteins, causing their inactivation (16, 50).
Regulators of apoptosis that lie within the mitochondria are also phosphorylated, but the phosphorylation of these proteins can occur within the mitochondria itself—via a requisite mitochondrial-localized kinase—or in the cytosol, after MOMP. Both of these schemes have been documented, even on the same protein: Htra2 is thought to be phosphorylated within the mitochondria by PINK1 (39), but can also be inactivated by the cytosolic prosurvival kinase AKT upon apoptotic release (50). Importantly, these phosphorylation events can have differential consequences; phosphorylation of Htra2 by PINK1 increases its proapoptotic effects, while AKT phosphorylation dampens its activity. Thus, the phosphorylation of apoptotic proteins is further regulated by localization constraints and access to different sets of kinases and phosphatases. A handful of mitochondrial-specific kinases and phosphatases have been implicated in cell death, and are briefly discussed in this section (Fig. 4).
PINK1
PINK1 is a mitochondrial-localized serine/threonine kinase, and, like Htra2, is genetically linked to the etiology of PD. Most studies suggest that PINK1 is neuroprotective, with loss-of-function mutations linked to increased apoptosis in dopaminergic neurons (44). RNAi-mediated downregulation of PINK1 causes apoptosis in neuronal cell lines, whereas overexpression of the kinase confers increased survival (9, 44). Importantly, the prosurvival activity of PINK1 depends on its kinase activity, suggesting that the regulation of its phosphosubstrates is critical to its role in cell death (38). As previously discussed, PINK1 is thought to phosphorylate and alter the activity of Htra2; other substrates for PINK1 have been proposed, but the contribution of these substrates to the survival activity of PINK1 has yet to be fully characterized. Interestingly, PINK1 has been proposed to phosphorylate another PD-associated gene, Parkin, to regulate mitophagy, a process in which mitochondria are turned over via autophagy during times of cellular stress (Fig. 4) (33). As functional mitophagy is required for cellular viability, the survival effects of PINK1 may not directly dependent upon phosphorylation of the apoptotic machinery, but rather may reflect a cellular requirement for intact mitophagic flux.
AK2
Adenylate kinase 2 (AK2) is localized specifically to the mitochondria, where it is proposed to initiate the conversation of adenosine diphosphate to adenosine triphosphate within the IMS in healthy cells (Fig. 4). Its functional effects on non-nucleotide substrates are unknown. RNAi-mediated knockdown of AK2 dampens chemotherapeutic-induced intrinsic apoptosis (27). AK2 translocates to the cytoplasm upon apoptotic induction in a manner that is blocked by Bcl-2 overexpression, suggesting that it is released upon MOMP (Fig. 4). Interestingly, AK2 promotes caspase-3 activation in an FADD/caspase-10-dependent manner, which is distinct from the canonical mitochondrial pathway of apoptosis. It remains to be seen whether the kinase activity of AK2 plays a key role in intrinsic apoptosis, and, if so, what its relevant substrates may be.
DUSP18
DUSP18, a mitochondrial-specific phosphatase, is widely expressed in tissues but is relatively uncharacterized. Interestingly, DUSP18 is released upon apoptotic induction in a similar fashion to the currently characterized IMS proteins (Fig. 4) (40). This redistribution of DUSP18 may be important in its function, as it could dephosphorylate a distinct set of substrates upon postapoptotic release into the cytosol. The identification of both mitochondrial and cytosolic substrates will be critical in understanding the role of DUSP18 in apoptosis.
MK-STYX
MK-STYX is a catalytically inactive phosphatase that, when knocked down in HeLa cells, promotes robust chemoresistance (35). Mechanistically, this occurs through a block in cytochrome c release, suggesting that MK-STYX knockdown cells have a defect in MOMP (35). Interestingly, MK-STYX does not localize proximal to the molecular machinery currently known to affect MOMP, and thus is proposed to function through a novel pathway to control apoptosis (Fig. 4). The molecular mechanisms of this gene, including its effects, if any, on mitochondrial phosphorylation, have yet to be elucidated.
Conclusions
Apoptosis is a complex cellular process orchestrated by numerous proteins whose strict regulation is critical for proper initiation and completion of cell death. Phosphorylation events have been documented on proteins controlling nearly all steps of apoptosis: Bcl-2 family members are intricately modified to inhibit or enhance apoptosis, while intramitochondrial proteins are phosphorylated to modulate their effects on caspase activation. These post-translational modifications are complex in nature; many proteins can be phosphorylated to either enhance or inhibit their function, and thus are fine-tuned to appropriately respond to life and death stimuli. Interestingly, numerous mitochondrial kinases and phosphatases have also been linked to cell death, but, except in a few cases, little is known about their substrates and how these substrates may contribute to cell death. Intensive proteomic efforts have revealed the presence of novel phosphoresidues on these proteins, yet little is known about their regulation or function. The characterization of these phosphorylation events, as well as the discovery of additional phosphoresidues, will be paramount in understanding the complex regulatory network underlying the switch between life and death.
Footnotes
Acknowledgment
This work was supported by the Award Number R01CA138651 from the National Cancer Institute to J.P. MacKeigan.