
Abstract
Introduction
Oxidative DNA Damage and Disease
It is clear that at elevated levels, the DNA-damaging effects of ROS have a clear impact on cellular function, genome stability, and human health. For example, oxidative stress has been demonstrated to play a significant role in the onset of cardiovascular and pulmonary diseases such as atherosclerosis (43), and has also been implicated in the pathogenesis of neurodegenerative diseases such as Alzheimer's, Parkinson's, and familial amyotrophic lateral sclerosis (13). Similarly, oxidative damage has been demonstrated to be associated with aging and age-dependent cognitive decline (26), as well as diseases associated with inflammatory conditions such as rheumatoid arthritis and hepatitis infection (46).
In addition to some of the disease states mentioned above that likely manifest as a consequence of DNA damage-induced cell death and organ dysfunction, ROS and the associated oxidative DNA damage can also induce mutations in our DNA. This genomic instability may act as causative agents in the initiation, promotion, and malignant conversion stages of carcinogenesis. Many studies have described elevated levels of oxidative DNA lesions in numerous tumor types, such as colorectal cancer, renal cell carcinoma, gynecological, and gastric cancers, thereby strongly implementing such damage in the etiology of cancer (7). However, a key question remains whether the presence of elevated levels of oxidative DNA damage in tumors is causal for cancer formation and progression or simply a consequence of the disease.
Several recent reports have provided evidence to support the causal theory that ROS and oxidative damage may promote cancer initiation and formation. Focusing on uterine myoma tissues, Foksinski et al. suggested that exposure to higher levels of estrogen can induce the oxidative DNA lesion, 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxo-G), with an increase in these lesions being a contributing factor to the malignant transformation of myoma cells (16). Furthermore, it has been suggested that such oxidative damage can also control the size of the tumor mass (16). Oxidative DNA damage has also been proposed as a causal mechanism in the formation of lymphomas and lung and ovarian tumors (48). In breast carcinomas, 8-oxo-G levels have been found to be elevated 8–17-fold compared with nonmalignant tissue (35). Karihtala et al. also showed that breast carcinoma cells themselves rather than, for example, inflammatory cells express high levels of 8-oxo-G. In addition, oxidative protein modification was significantly higher in invasive lesions than in preinvasive lesions, suggesting oxidative stress as a causal factor in the early steps of breast carcinogenesis (24). Corresponding results have also been reported from head and neck squamous cell carcinomas, where increased levels of oxidative damage were observed in the early stages of reactive, dysplastic, and carcinoma samples compared with samples of normal mucosa (2). Environmental exposure to oxidizing agents has also been reported to induce genetic mutations and genome instability. For example, human exposure to elevated levels of arsenic (an ROS-inducing agent) has been associated with an increased incidence of several cancers that include skin, lung, and liver (42). Additionally, benzo(α)pyrene reacts with the 2-amino-position of guanine to generate oxidative damage that can trigger an adduct and mutation profile in the TP53 tumor-suppressor gene that strongly correlates with the TP53 mutation profile in lung tumors from smokers, but not in nonsmokers (39).
Conversely, elevated levels of oxidative damage may occur as a result of the well-established characteristics of tumors such as increased metabolism or cell turnover, and thus be viewed as a product/consequence of cancer formation, rather than causal in tumor generation. For example, oxidative stress can be induced by thymidine phosphorylase, an enzyme that is overexpressed in the majority of breast carcinomas. Thymidine phosphorylase catabolizes thymidine to thymine and 2-deoxy-D-ribose-1-phosphate, the latter being a very powerful reducing sugar that rapidly glycates proteins to generate oxygen radicals within the cell. The frequent upregulation of thymidine phosphorylase in human breast tumors suggests that this enzyme may play an important role in generating oxidative DNA lesions in breast cancer (4).
Our current understanding of oxidative DNA damage therefore suggests that it may be both causal in the origin and/or progression of cancer, and also manifest simply as a consequence of carcinogenesis. What remains clear, however, is that oxidative DNA damage is a defining hallmark of many tumor types. Elucidating the molecular pathways that repair such damage, in addition to targeting these oxidative DNA lesions, may provide useful therapeutic avenues in the treatment of cancer.
Oxidative Stress-Induced DNA Modifications
DNA has a limited intrinsic chemical stability and is one of the most biologically important targets of ROS. In the majority of cases, DNA damage from ROS-generating agents is mediated by Fenton chemistry to give rise to the formation of chronic and persistent DNA damage such as oxidized bases, nucleotide base modifications, apurinic/apyrimidinic (AP) sites, DNA crosslinks, and single-/double-strand breaks (1). Approximately 100 different kinds of base and sugar damage have so far been identified after exposure to oxidative stress. We do not intend to detail the structure of every modified base or deoxynucleoside here, but will briefly discuss the major DNA modifications that are the primary targets of DNA repair pathways (Fig. 1). The most prevalent damage to purines is 8-oxo-G, while the most common damage to pyrimidines is the formation of thymine glycol (Tg). For the generation of 8-oxo-G, a highly reactive •OH initially reacts with guanine to form a C8-OH adduct radical. After this, the loss of an electron and a proton generates the oxidative lesion 8-oxo-G. Conversely, through the uptake of an electron and a proton, the C8-OH adduct radical may also be reduced to form 7-hydro-8-hydroxyguanine, which is subsequently converted to 2,6-diamino-4-hydroxy-5-formamido-pyrimidine (FaPy), a second major oxidation target of guanine (9). Several other oxidative DNA modifications also result as a consequence of ROS attack, such as the oxidized pyrimidines, cytosine glycol (Cyt glycol), and Tg. Initially, an •OH reacts with thymine and/or cytosine by addition to the C5- and C6-positions to yield C5-OH- and C6-OH-adduct radicals, respectively, and additionally by abstraction of a H

Oxidative DNA Damage Repair
To deal with oxidative damage to DNA from various endogenous and exogenous sources, mammalian cells have evolved many intricate mechanisms to first detect, and subsequently repair such damage. NER and BER are the two pathways responsible for repair of the majority of DNA lesions induced by ROS (Fig. 2). NER is a complex multistep pathway that repairs DNA when it is damaged by ROS in such a way as to create a major structural distortion. Essentially, NER involves six major steps: (i) recognition of the specific oxidative modified DNA bases such as Tg, 8-oxo-G and cyclodeoxyadenosine mediated by the repair factors xeroderma pigmentosum (XP) complementation group A (XPA), replication protein A (RPA), and XPC. These then in turn recruit the TFIIH transcription/repair factor that contains six polypeptides (including XPB, XPD, XPG, and XPF-ERCC1) essential to the repair process; (ii) DNA unwinding around the damaged site mediated by the helicases XPB and XPD; (iii) dual incisions bracketing the lesion. At this stage, the XPG and XPF-ERCC1 subunits are responsible for the 3′ and 5′ incisions, respectively; (iv) subsequent removal of the DNA damage; (v) repair synthesis to fill the resulting gap by replication factor C (RPC), proliferating cell nuclear antigen (PCNA), and DNA polymerases (POL) δ and ɛ using the other strand as a template; and (vi) finally, ligation of the newly synthesized strand by DNA ligase I (21, 41) (Fig. 2).

BER is a simpler version of NER, with this mode of repair operating when the oxidative damage is confined to a base. BER is essentially composed of four steps and can be divided into two subpathways: the short-patch and long-patch pathway. In the long-patch pathway, as many as up to 13 nucleotides may be inserted, whereas the short-patch pathway involves mostly the filling up of a single-nucleotide gap. Focusing on the short-patch pathway, in the first step, the oxidatively modified base such as 8-oxo-G or Tg is specifically recognized and cleaved from the sugar phosphate moiety by an appropriate DNA glycosylase, such as 8-oxoguanine glycosylase (OGG1) for the recognition of 8-oxo-G damage, or NEIL3 for Tg/Fapy damage. At the same time, the AP endonuclease (APE1) attaches itself to the 5′ side of the base to break the chain. After this, DNA POLβ fills up the one-nucleotide gap and also releases the 5′-deoxyribose phosphate (dRp). Consequently, the DNA ligase III/XRCC1 complex arrives at the site whereby DNA ligase III seals the nick and POLβ dissociates from the site. XRCC1 and DNA ligase III are released, leaving repaired DNA (41, 44) (Fig. 2).
The DNA MMR Pathway
Maintaining the chemical integrity of DNA in the face of assault by oxidizing agents is a constant challenge for living organisms. In addition to BER and NER, the MMR pathway has also been shown to play an important role in preventing mutations associated with the oxidative DNA lesion, 8-oxo-G. Currently, however, less is known concerning the mechanisms by which MMR is able to execute such damage resolution. In the following sections, we focus on the MMR pathway, the emerging role of MMR in oxidative DNA damage repair, and its implications in cancer therapeutics.
Mismatch components: MutS and MutL homolog protein complexes
The eukaryotic MMR pathway is composed of two major components, both functioning as heterodimeric complexes, with names originating from their lower-organism orthologs, MutS and MutL. The most abundant mismatch-binding heterodimer, referred to as MutSα, is composed of MSH2 and MSH6. This binding factor initiates the repair of base–base mismatches and small insertion–deletion loops (IDLs) of one or two extrahelical nucleotides (25). MutSβ, a heterodimer of MSH2 and MSH3, repairs both small loops in addition to large-loop mismatches of ∼10 nucleotides. Current evidence indicates that MutSβ is unable to support the repair of single base insertion/deletion mispairs (17). Similarly, heterodimeric complexes of MutL-related proteins are able to recognize damaged DNA to excise and repair DNA mismatches. The primary MutL activity for mismatch correction is orchestrated by the MutLα complex (made up of MLH1 and PMS2 proteins), with the MutLβ heterodimer (composed of MLH1 and PMS1 proteins) being suggested to play only a minor role. Furthermore, MutLγ (MLH1 and MLH3) acts as a backup for MutLα in the repair of base–base mismatches and small IDLs (40).
Molecular mechanism of mismatch recognition
Genetic and protein–protein interaction experiments have led to the proposal of a model for eukaryotic MMR in which a cascade of proteins are responsible for the repair of postreplicative mismatches, in which an incorrect base has been incorporated into newly synthesized DNA. In addition, MMR also repairs mispaired bases that arise during homologous recombination or oxidative DNA damage (37). At the start of the process, mispaired bases of DNA are recognized by heterodimeric complexes of MutS-related proteins. The MutS heterodimer recognizes distortions in the DNA double-helix structure and subsequently binds to double-stranded DNA at the site of the mismatch. This specificity of binding is achieved by the enzymatic activity of MutS, provided by ATPase. The MutS ATPase does not directly enhance MutS mismatch association, but enables MutS to verify mismatch recognition by kinetic proofreading. In the presence of homoduplex DNA, MutS is able to hydrolyze ATP quickly; however, in the presence of a DNA mismatch, the burst of ATP hydrolysis is inhibited, thus allowing an MutS-DNA-ATP complex to form. It is only when MutS is associated with both ATP and a mismatch that the heterodimer is able to recruit the downstream repair protein MutL, which seems to act as the mediator for a series of subsequent protein interactions that facilitate MMR. In one model of MMR, the MutS/MutL complex leaves the site of the mismatch after the initial recognition event and subsequently slides up and down the flanking DNA sequence via ADP-ATP hydrolysis. Initially, MutS binds to mismatched DNA in an ADP-bound state. After mismatch binding, MutS first undergoes a conformational change that allows an ADP-ATP exchange, which then promotes a second conformational change allowing MutS to be released from the mismatch and form a sliding clamp (18). The sliding-clamp movement of the MutS/MutL complex up and down the DNA double helix eventually allows MutS/MutL to encounter a single-strand break in the DNA sequence that is tagged with other MMR factors, such as PCNA and RFC (22, 45). As the function of postreplicative MMR is to repair errors that are present in newly synthesized DNA, the MMR machinery is required first to distinguish between parental DNA and daughter DNA, and subsequently to remove and replace the aberrant daughter DNA sequence. Although the discriminatory signal on leading-strand DNA in mammalian cells is currently unclear, it is thought that intermittent gaps in DNA in between Okazaki fragments on lagging-strand DNA may represent a signal that distinguishes parental from daughter strand in MMR (34). Regardless, it appears that the presence of the sliding clamp with PCNA and RFC, in addition to the presence of a single-strand gap in the DNA, allows the identification of the daughter strand and a 5′ ->3′ DNA exonuclease (EXO1) to enter the DNA structure (45). EXO1, guided by the MutS/MutL sliding clamp, then starts removing daughter-strand DNA toward and then beyond the site of the mismatch. Once the mismatch is removed, the activity of EXO1 is suppressed by MutL, thus terminating DNA excision. Upon completion of this process, a DNA polymerase synthesizes DNA in the place of the excised sequence with a DNA ligase and then joining any gaps in the DNA sequence (22, 32) (Fig. 3).
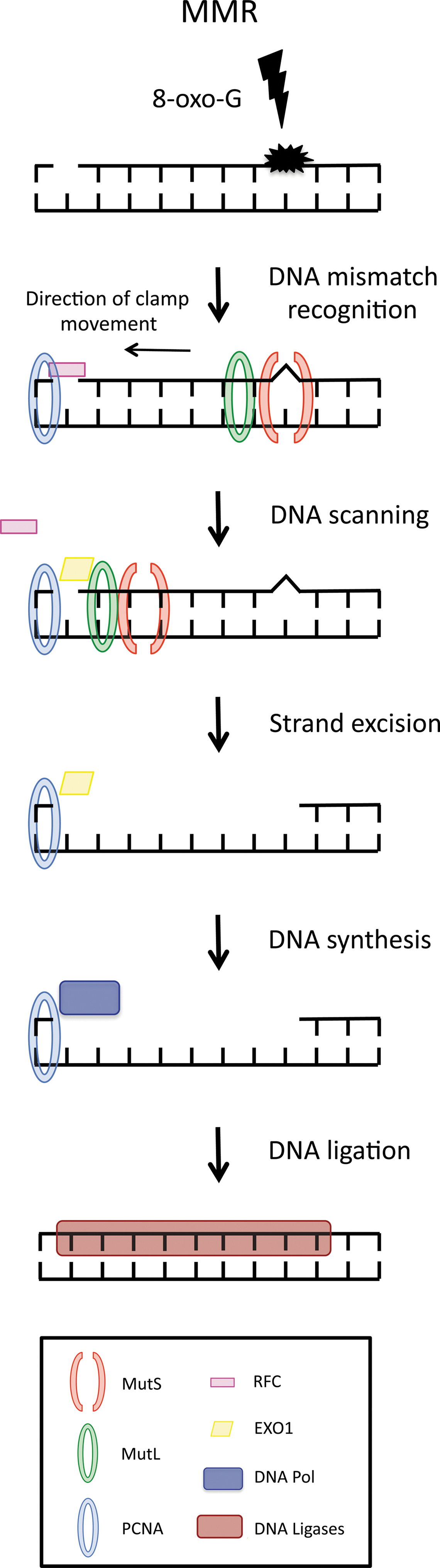
MMR of Oxidative DNA Damage
Bacteria, yeast, and human cells contain overlapping and redundant oxidative damage recognition systems. Although predominantly studied for its role in the recognition and repair of DNA mismatches and IDLs, emerging evidence has also implicated the MMR pathway in recognizing and resolving other abnormalities within the DNA heteroduplex, such as base modifications due to oxidative damage. Studies in Escherichia coli have demonstrated mismatch correction to be a key pathway in the repair of oxidative damaged bases (47). The authors demonstrated that MMR has the ability to prevent oxidative mutagenesis by removing 8-oxo-G directly, by removing adenine misincorporated opposite 8-oxo-G, or by a combination of both (47). Furthermore in Saccharomyces cerevisiae, it has been shown that transcription-coupled repair of thymine glycols depends on the MMR system, therefore implicating MMR in the recognition of at least some forms of oxidative damage (27). Additionally, Earley and Crouse demonstrated a clear reduction in mutation rates in MMR-defective mutants grown under anaerobic conditions, thereby suggesting that the high reversion rates observed in MMR-deficient strains are caused by misincorporations opposite oxidative damaged bases, suggesting that MMR is required for the repair of these mutations (12). Later work by Ni et al. confirmed these observations and demonstrated that in yeast, MSH2-MSH6-dependent MMR is the major mechanism by which misincorporation of A opposite 8-oxo-G is corrected (36).
Several lines of evidence have also indicated that MMR is involved in the repair of oxidative stress-induced DNA damage in mammalian cells. Work in mouse embryonic stem (ES) cells highlighted that MMR-deficient ES cells respond abnormally to oxidative DNA damage induced by ionizing radiation. In particular, Msh2−/− ES cells displayed an increased survival after protracted radiation exposure compared to wild-type Msh2 cells (8). Similarly, focusing on mouse fibroblasts, Young et al. demonstrated that the MSH2-MSH6 complex promoted cellular apoptosis after UV-induced oxidative DNA damage and protected cells from UV-induced malignant transformation (49). Furthermore, Colussi et al. reported that both steady-state and H2O2-induced levels of DNA 8-oxo-G were increased in Msh2−/− mouse embryonic fibroblasts (6). The same group further demonstrated that mismatch recognition by MutSα can prevent frameshift mutations induced by 8-oxo-dGTP incorporation (28). This is also supported by the preferred binding of MutSα to mismatched duplexes containing 8-oxo-G, but not to 8-oxo-G:C and the preferential activation of MutSα by 8-oxo-G (33). The demonstration of a direct interaction between hMYH and hMSH6 in the hMSH2/hMSH6 heterodimer suggests however that MMR proteins may also be involved in the removal of A misincorporated opposite 8-oxo-G (20). It has also been suggested that PCNA, which may interact with both hMYH and MMR proteins, might act as a coordinator of 8-oxo-G:A repair (19, 20).
Recent data from our laboratory have further corroborated a role for MMR proteins in the repair of oxidative DNA damage. Small-interfering RNA (siRNA) screens performed to identify synthetic lethal interactions with the loss of MMR genes have demonstrated that MSH2 deficiency is synthetically lethal with inhibition of DNA POLβ in a series of cellular models, such that silencing POLβ is toxic to MSH2-deficient cells, while correspondingly in DNA POLγ-silenced cells, MLH1 deficiency is synthetically lethal (Table 1). Interestingly, the MSH2/POLβ synthetic lethal relationship correlated with an accumulation of 8-oxo-G DNA lesions in the nucleus, while the MLH1/POLγ synthetic lethal interaction resulted in an accumulation of mitochondrial 8-oxo-G DNA lesions (29). Indeed, POLγ encodes the catalytic subunit of the mitochondrial DNA polymerase, and mutations in this gene are associated with several mitochondrial diseases. Furthermore, we have also shown that silencing PTEN-induced putative kinase 1 (PINK1) is synthetically lethal in MMR-deficient cell lines (31). Inhibition of PINK1, a mitochondrial serine/threonine protein kinase, in an MMR-deficient background resulted in an elevation of ROS and subsequently the accumulation of both mitochondrial and nuclear oxidative DNA lesions (Table 1). Taken together, these studies suggest that in cells with proficient MMR, inhibition of POLβ, POLγ, or PINK1 initially leads to the formation of 8-oxo-G lesions that are eventually repaired by/or in collaboration with the MMR pathway. However, in the absence of MMR, inhibition of the same proteins leads to the accumulation of 8-oxo-G lesions, which are not repaired efficiently and ultimately reach a threshold that is incompatible with cell viability. Interestingly, these studies also demonstrate for the first time a potential role for MLH1 in the repair of 8-oxo-G lesions in mitochondrial DNA.
8-oxo-G, 7,8-dihydro-8-oxo-2′-deoxyguanosine; PINK1, PTEN-induced putative kinase 1; POL, DNA polymerase.
Oxidative DNA Damage as a Therapeutic Strategy for MMR-Deficient Disease
When the genes that mediate MMR, such as MLH1, MSH2, MSH6, and PMS2, are mutated or epigenetically silenced, the predisposition to cancer is vastly increased. MMR deficiency is currently thought to be present in 15%–17% of all primary colorectal cancers and ∼30% of endometrial cancers, due to inherited or somatic MMR gene alterations, epigenetic suppression of MMR gene expression, or a combination of these factors (3, 38). The heritable disorder associated with MMR deficiency in adults is known as hereditary nonpolyposis colon cancer (HNPCC). HNPCC is an autosomal-dominant condition, associated with a predisposition to multiple cancers. Patients with HNPCC have an 80% lifetime risk of developing colorectal cancer, and women with HNPCC have a 40%–60% lifetime risk for endometrial cancer. Therefore, the identification of synthetic lethal relationships that result in selective killing of MMR-deficient cells may prove clinically beneficial (23). Such an approach has been exploited successfully in the clinic using inhibitors of the DNA repair protein poly(ADP-ribose) polymerase (PARP) in the treatment of patients with germline mutations in the tumor suppressor genes BRCA1 and BRCA2 (5, 15).
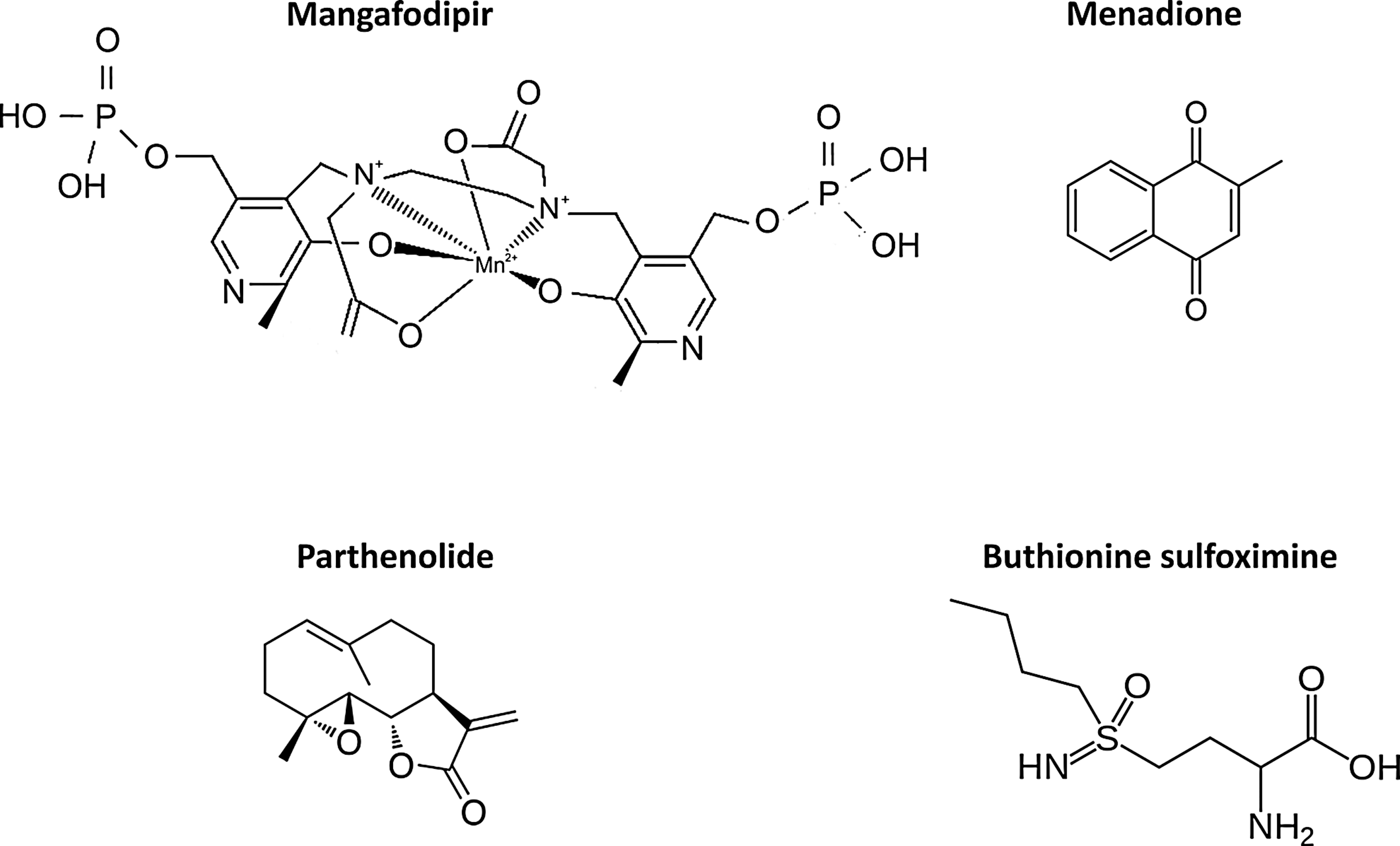
Therefore, based on this approach and using the knowledge gained from identification of synthetic lethal interactions with MMR deficiency, we postulated that treating MMR-deficient tumor cells with an oxidative DNA damage-inducing drug should cause selective lethality. To this end, we have identified the oxidative damage-inducing chemotherapy drug methotrexate as a selective agent in MSH2-deficient cell lines both in vivo and in vitro through the accumulation of nuclear 8-oxo-G lesions (30) (Fig. 4). These findings have been directly translated to the clinic, in a randomized phase II trial in metastatic colorectal cancer (MESH, NCT00952016), incorporating 8-oxo-G levels as a biomarker of response. These data highlight the potential for focusing upon oxidative damage as a therapeutic target, not only in MMR-deficient cancers such as HNPCC, but also in a host of other cancers where the tumor cells manifest oxidative damage (7). Currently, there are a plethora of pharmacological agents that induce the overproduction of ROS to generate oxidative damage; examples include menadione, parthenolide, mangafodipir, and buthionine sulfoximine (Fig. 5). It may be that such compounds can refine clinical treatment by providing a novel therapeutic strategy in targeting MMR-deficient tumors.

Innovation
A number of DNA repair pathways exist within the cell to eliminate the accumulation of oxidative DNA lesions and resulting genomic instability. Our identification of a synthetic lethal interaction between increased oxidative DNA damage accumulation and mismatch repair deficiency suggests a novel approach to exploit the oxidative DNA repair machinery in cells as a novel anticancer therapeutic strategy.
Concluding Remarks
Oxidative stress can induce structural damage in cellular DNA. This DNA damage may be both cytotoxic and/or miscoding and is believed to be at the origin of cell lethality, tissue degeneration, aging, and cancer. To counteract the deleterious effects of such lesions, cells have evolved a number of DNA repair mechanisms. So far, the role of MMR in mediating DNA repair to oxidative damage has been explored less extensively than other pathways, despite emerging evidence that demonstrates a key role in resolving 8-oxo-G lesions. Novel synthetic lethal approaches have aided further elucidation of this pathway, revealing key components and functional relationships. Such findings suggest that oxidative DNA-damaging agents may be utilized to selectively target MMR-deficient cancers and potentially other tumor types deficient for oxidative DNA repair molecules.
Footnotes
Acknowledgments
We thank Barts and the London Charity, Cancer Research UK, and the Medical Research Council for supporting our research.