
Abstract
Introduction
I
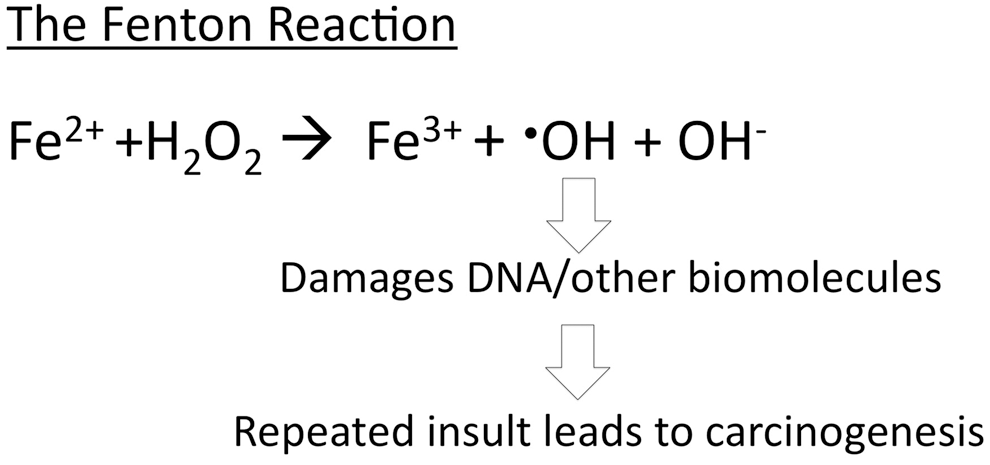
In normal cells, both iron and ROS are carefully managed by the cell to maintain homeostasis or to regulate their functions (1, 9, 15). However, in cancer cells, many of the regulatory processes that control iron and ROS are altered, which may enable the cells to over-proliferate and contribute to disease progression (15, 34, 44). Thus, better understanding the role of ROS and iron in cancer cell biology provides valuable information for the development of cancer therapeutics. In this review, both normal and cancer cell metabolism will be assessed in relation to the iron and ROS pathways or effects.
Normal Iron Metabolism and Cellular Iron Regulation
Over the years, more and more information has been discovered about the regulation of iron metabolism and how modification of some of these regulatory mechanisms may play a role in cancer progression. Under normal conditions, most of the iron for the body is obtained from the reticuloendothelial system, which involves recycling iron from old red blood cells engulfed by macrophages (1). Additionally, a smaller portion of iron comes from the diet (30). Many of the major players in dietary iron absorption are also relevant to the iron metabolism of individual cells that come from different tissues found throughout the body (1, 30).
Normal absorption of dietary iron begins in the duodenum, where it enters in the form of inorganic iron (II) (from vegetables/other sources) via the divalent metal transporter (DMT1) or in the form of heme iron (mainly meat/fish) by a potential heme carrier protein 1, all of which are located in the apical membrane of the enterocytes (1, 30). Iron can either be stored in these cells in the form of ferritin or exit the cell and enter into circulation by the iron exporter protein known as ferroportin (13, 30). Thus, DMT1 and ferroportin bring iron into and out of the cells of the duodenal enterocytes, respectively, and are not only centrally involved in regulating iron homeostasis of the body but also involved in the regulation of iron metabolism of many different kinds of cells. Gene expression of DMT1 was found in different tissues in the body, including the brain, kidney, and lung (46). Moreover, ferroportin is predominantly found in the macrophages, liver, and the placenta, but it has also been identified in breast and brain tissues (13, 36, 48).
Another regulator of dietary iron absorption is the hormone hepcidin, a protein secreted from the liver (33). This protein, also known as the master iron regulator, increases in high iron or inflammatory conditions and binds to ferroportin, which causes the iron exporter to internalize into the cell, degrade, and ultimately prevent iron absorption from the duodenum (31). However, when iron levels are low, hepcidin levels are low, and ferroprotin is available to allow absorption of iron to occur (31). Moreover, chronic low levels of hepcidin are associated with excessive iron absorption and iron overload (14). Although hepcidin is mostly expressed in the liver and associated with iron absorption, it is also detected at lower levels in other tissues, including those from the heart and the brain (24).
After iron is absorbed into the bloodstream, it is transported by the protein transferrin (Tf), which allows for iron to be circulated throughout the body safely without causing toxicity (1, 30). Tf binds to the transferin receptor 1 (TfR1) in the membrane of cells of various tissues to provide iron to these environments (1, 10, 30). Through receptor-mediated endocytosis, iron enters the endosome, where acidification allows for its release from Tf (1, 16, 30). The six-membrane epithelial antigen of the prostate 3 (STEAP3) then reduces iron (III) to iron (II) before it is released to the cytosol via DMT1 (12, 32). Once in the cytosol, it is believed that an iron transport molecule shuttles iron to different parts of the cell, including the mitochondria for heme biosynthesis and ferritin for storage (16). These ligand transporters (e.g., ATP and citrate) are believed to transport biologically active iron (II), and contribute to what is known as the labile iron pool (LIP) (16, 44).
Altered Iron Metabolism of Tumor Cells and Therapeutic Targets
Many studies have investigated the role of altered iron metabolism in tumor cells, which are summarized in several review articles (38, 44). Regulatory iron proteins, such as Tf receptors, are highly elevated in many cancer cells compared to normal tissues and may indicate the increased iron demand for tumor cell survival (10). This is illustrated in a study that showed high levels of TfR1 expression in breast cancer tissues correlated with poor prognosis of patients and was associated with resistance to tamoxifen, a common drug used to treat breast cancer (19). Studies have also indicated that members of the STEAP family, which facilitate iron uptake into the cytosol from the endosome, were highly expressed in prostate tumors (32).
Other studies on breast cancer cells from patients indicated that ferroportin protein levels were decreased, and hepcidin levels increased compared to normal breast epithelial cells (36). Patients who had low ferroportin gene expression in the tumor cells were associated with a significant reduction in metastasis-free and disease-specific survival (36). These results imply that the tumor cells retain more iron than normal cells and that elevation of cellular iron contributes to increased tumor survival (Fig. 2). Additionally, the surrounding microenvironment and other extracellular functions may also contribute to increased iron uptake. Ferritin, known for its role in iron storage and detoxification, is reportedly elevated in some tumor tissues and may promote angiogenesis (8). Interestingly, ferritin also exists in extracellular compartments and can even bind to specific cell surface receptors such as TfR1 (27). Thus, ferritin might also play a role in iron delivery to the cells. Other extracellular mechanisms that may increase iron content in cancer cells by feeding or delivering iron to the cell, may involve certain types of macrophages and proteins, such a lipocalins that bind to ligands that recruit iron from extracellular regions (44).

All of these studies indicate that an increase in iron availability to tumor cells provides a favorable environment for the growth of these cells and suggests an increase in the LIP (Fig. 2). This also suggests the potential therapeutic value of depleting the LIP, as well as inhibiting regulatory proteins that increase iron uptake. One way to exploit the mechanisms that cater to the increased iron demand of cancer cells would be to target or alter the effects of TfR1, a protein more prevalent on tumor cells (10). Anticancer drugs (e.g., doxorubicin) or toxins (e.g., ricin) that either piggy-back as conjugates to Tf or masquerade as a similar chemical that binds to TfR1 have been explored as potential therapies to selectively and more effectively target tumor cells (10).
ROS Regulation and Effects in Normal Cells
ROS are a group of reactive chemical species that result from the incomplete reduction of oxygen and include superoxide anion (O2 −), hydrogen peroxide (H2O2), and the hydroxyl radical (HO•) (9). The principle source of ROS comes from the mitochondria, where it is a natural byproduct of normal metabolism of oxygen (2). Although endogenous antioxidants such as superoxide dismutase, glutathione (GSH), and catalase may protect the cell from ROS damage, excess ROS can overwhelm the cellular defense mechanisms and function as lethal oxidants that can damage DNA as well as oxidize polyunsaturated fatty acids, amino acids, and specific enzymes (2).
On the other hand, ROS may also function as a signaling molecule that may trigger many beneficial effects for the cell and organism (9). ROS can induce host defense mechanisms, developmental processes, and cell survival pathways. Platelets release ROS to recruit additional platelets to the sites of injury (25), and certain amounts of ROS are required for sperm to develop and function properly (9). Moreover, at low levels, ROS contribute to cellular signaling that is important for proliferation and survival of the cell (Fig. 3).

ROS regulate cellular processes by targeting kinases and phosphatases (4); redox-sensitive transcription factors (28); and cell cycle regulators (5). ROS stimulate signaling cascades, such as mitogen-activated protein kinases that are involved in regulating cell growth and cell death (4). ROS also control proliferation of the cell and prevent apoptosis by the transcription factor nuclear factor kappa light-chain enhancer of activated B-cells (NF-κB). Essentially, H2O2 activates NF-κB and increases antiapoptotic or survival proteins, such as Bcl-2 and inhibitor of apoptosis proteins family members (28, 40). ROS may also play a role on the effects of NF-κB regulation of cellular proliferation or differentiation through involvement of cyclin D1 (5, 40). Moreover, ROS are involved in other survival pathways that activate the hypoxia-inducible factor α (HIF-1α), a transcription factor that is induced by hypoxia (low oxygen) (43). H2O2 inhibits prolyl 4-hydroxylase activity, which leads to HIF-1α stability and thereby increased glycolysis and survival signaling (43).
Although certain ROS levels may stimulate survival and proliferation, relatively intermediate levels may induce apoptosis or differentiation, and even higher levels may induce necrosis (Fig. 3). Low levels of H2O2 are reported to activate the tumor suppressor p53 to produce an antioxidant response, but at high levels may cause p53-dependent apoptosis (9, 43). Additionally, H2O2 activates transcription factors of the class-O forkhead box (FOXO) family and can induce either apoptosis or tolerance to higher levels of oxidative stress (26). ROS can also induce perturbation of the mitochondrial permeability transition pores and disrupt electron transfer that can result in apoptosis or cell necrosis (43). Usually, in more severe stress conditions, when there are very high amounts of ROS and ATP depletion, cells become necrotic and die in a less-regulated manner than apoptosis (43).
Although high levels of ROS may be detrimental to the cell, this may at the same time be beneficial to the host organism that is effectively eliminating an aberrant or possibly malignant cell. Hence, ROS appear to have multiple functions that are involved in various pathways of maintenance for the cell or host organism, and only recently are these mechanisms beginning to be elucidated.
Altered ROS in Tumor Cells
Studies have indicated that cancer cells have higher basal ROS levels than normal cells (34, 37, 49) (Fig. 3). These effects may be due to oncogenic stimulation, increased metabolic activity, mitochondrial malfunctions, and other dysregulated activities in the cells (34). It appears that high levels of ROS confer a growth advantage to tumor cells by facilitating mitogenic signaling through the activation of several stress–kinase pathways (4). For instance, ROS can mediate amplification of the survival AKT/mTOR signaling pathway, which, in absence of Foxo3 (a member of the FOXO family), is associated with myeloproliferative syndrome in mice (50). In addition, tumor cells may adapt to high ROS levels as a result of the presence of the oncogene c-Myc, which increases ROS tolerance by activating the transcription of genes involved in GSH biosynthesis in response to H2O2 (3). Altogether, these data corroborate the notion that increased levels of ROS, without an adequate and normal physiological response, are associated with abnormal cell metabolism and tumorigenesis.
It is also apparent that oncogenic transformation leads to an increase in ROS that is high enough to render the cells sensitive to additional ROS production (49). Several chemicals that increase ROS have been shown to selectively target transformed cells, but not normal cells. In particular, natural compounds such as phenylethyl isothiocyanate (PEITC) found in many cruciferous vegetables (Fig. 4) and piperlongumine (Fig. 4) found in the pepper plant (Piper longum L.) have indicated ROS-induced apoptosis in transformed cells lines, but not in the normal or parental nontransformed cells (37, 49). PEITC was found to disable the antioxidant activity of the cell by transporting glutathione out of the cell and also by reducing glutathione peroxidase 1 (45). Furthermore, the addition of the antioxidant N-acetyl-
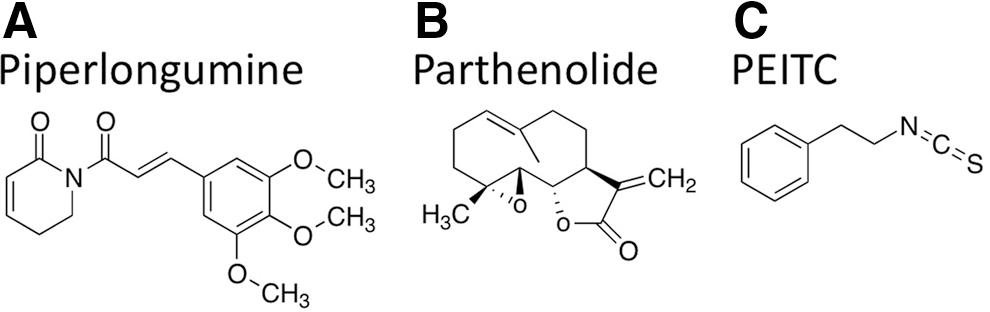
Piperlongumine also selectively induced apoptosis in a cancer genotype by targeting the ROS–stress response pathway (37). More specifically, piperlongumine was found to bind to glutathione S-transferase pi-1 and carbonyl reductase 1, both of which are involved in antioxidant activity. Such effects indicate that this compound modulates ROS homeostasis (37). Furthermore, piperlongumine selectively increased ROS in tumor cells in vitro and dramatically reduced the tumor size in numerous in vivo models, suggesting that this drug effectively works by targeting a vulnerable feature of the cancer phenotype (37). Moreover, another natural drug known as parthenolide (Fig. 4) found in the plant feverfew (Tanacetum parthenium L.) increased ROS and targeted cancer cells and cancer stem cells (CSCs) (18).
These studies indicate the value of exploring therapeutic approaches that increase ROS for cancer therapy. In fact, protecting against ROS with antioxidant therapy may even promote cancer. Dietary depletion of the antioxidants (e.g., vitamins E and/or A) have been shown to inhibit the growth of several cancer cell lines, as well as tumor development and metastasis in mouse models (39). Although increased ROS may be detrimental for tumor cell survival, questions still remain about the least toxic level necessary for ROS induction, as well as what kinds of complementary therapies that might enhance the antitumor effects. Different degrees of intensity of ROS can contribute to different effects, and thus specific levels and mechanisms of ROS that are necessary to target tumor cells need to be better clarified.
ROS and CSCs
Stem cells are undifferentiated cells that can differentiate into an array of specialized mature cells or self-renew to maintain the stem cell population. ROS play an important role in stem and progenitor cell function by regulating self-renewal and differentiation (11, 42). In the case of CSCs, it has been shown that some human and murine breast CSCs contain lower ROS levels than normal cells, and as a result are protected from radiation therapy that generally targets mature cancer cells (11). These effects may be due to enhanced ROS defense systems or mechanisms of CSCs to maintain lower ROS levels (11). Thus, a potential therapeutic strategy may involve modulation of ROS levels in CSCs to induce cell death or differentiation. Potential for such a therapy is exemplified in a study that showed that the natural compound parthenolide selectively targeted acute myelogenous leukemia CSCs, but not normal hematopoietic stem cells, by increasing ROS levels as a part of its mechanism of action (18).
Iron Chelators and ROS
Iron chelators are best known for their ability to sequester iron away from the cell or organism and to function as an antioxidant by preventing the Fenton reaction (17, 22). Cancer cells have high iron requirements and therefore are sensitive to iron depletion (38). This information is what originally prompted researchers to investigate the antitumor effects of iron chelators. Interestingly, researchers found that several iron chelators selectively induced apoptosis in cancer cells in vitro and reduced the size of tumors in vivo (22, 38). However, the mechanisms are still not fully elucidated, and the optimal iron chelator structures are currently under investigation.
One of the mechanisms reported to contribute to the cytotoxic effects of iron chelators is the ability of these compounds to deplete iron and thereby inhibit the iron-containing enzyme ribonucleotide reductase, a rate-limiting step for DNA synthesis (7). More recently, it has been found that iron chelators may also cause cytotoxic effects in tumor cells by increasing ROS levels through redox-active iron complexes (7, 22). Depending on the structure of iron chelators, these compounds can intercept iron-sensitive pools for DNA synthesis and/or increase oxidative stress in the cells by a toxic iron complex (Fig. 5). Chelators that form iron complexes that are redox reactive appear to be best at facilitating the generation of ROS. For example, the iron chelator Triapine® and di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT) are redox active, whereas desferoxamine (DFO) is redox inactive (Fig. 6) (22). Hexadentate iron chelators (ligands with six atoms that bind to iron) such as DFO prevent the iron from being exposed to oxygen and H2O2, whereas tridentate chelators (e.g., Dp44mT) render iron exposed and are able to generate ROS (21). Thus, the structural differences of the chelators appear to contribute to the different modes of action of these chemicals that induce apoptosis or cell death (Figs. 5 and 6).

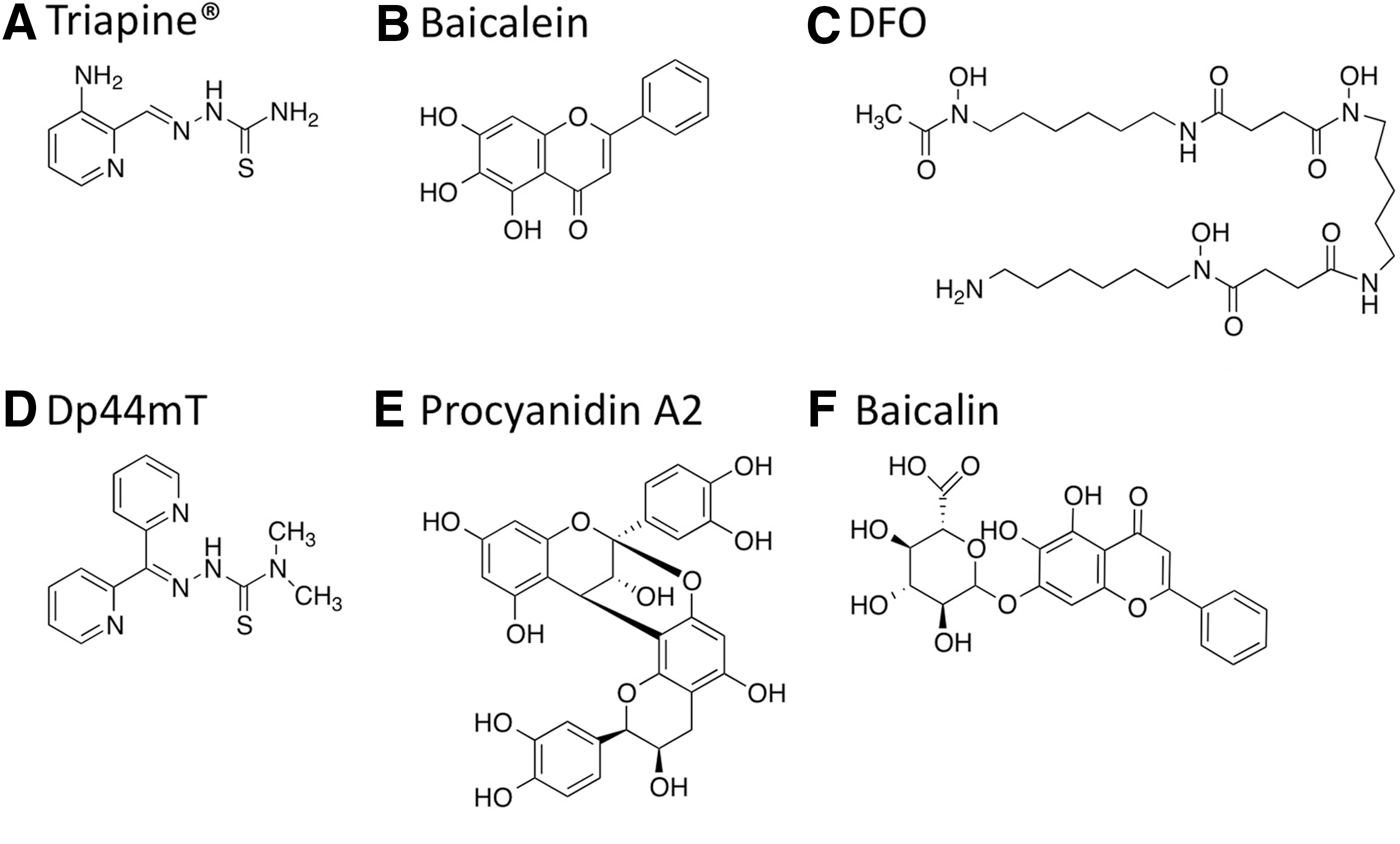
Many of the natural polyphenolic compounds (e.g., catechin derivatives) found in natural foods and botanicals may be known for their antioxidant effects, but many of these compounds are also redox reactive and form iron complexes that produce pro-oxidant effects (20). In particular, some of the polyphenolic compounds known as flavonoids possess ortho-hydroxyphenol iron-binding motifs that enable the compounds to function as iron chelators (17). Flavonoids with these type of functional groups include A-type proanthocyanidins (A-PACs) from cranberry species such as Vaccinium macrocarpon (e.g., procyanidin A2 in Fig. 6) and are the major component of cranberry fractions with antitumor activity that involved mechanisms that increased ROS for both ovarian and neuroblastoma cancer cells (23, 41). Thus, A-PACs may produce ROS-induced antitumor activity similar to other synthetic redox-active iron chelators. Other flavonoids such as baicalein and its glycoside baicalin (Fig. 6) are bioactive components of the Chinese medicinal plant Scutellaria baicalensis that are known to function as iron chelators (35). Interestingly, baicalin has been reported to be a potential therapeutic agent for iron-overload disease as well as possess the ability to induce apoptosis by ROS in HL-60 leukemia cells (29). Such effects indicate that baicalin may have several different mechanisms for targeting tumors cells that involve iron chelation.
Many of the natural and synthetic chelators may selectively target tumors cells by their pro-oxidant activities and/or iron-depletion effects because of the high iron and high ROS levels in tumors cells (Fig. 7). The high levels of LIP in tumor cells may suggest that there is more iron present to interact with redox-active chelators to induce high levels of ROS. Additionally, tumor cells are more sensitive than normal cells to iron depletion because of their high iron requirements. Tumor cells may also be more sensitive to increased ROS levels than normal cells because of their higher basal ROS levels. Moreover, iron chelators may also cause differentiation of tumor blasts by altering both iron metabolism and ROS levels. This has been illustrated in a study using iron-deprivation therapy (i.e., iron chelators or Tf receptor-blocking antibodies), which resulted in differentiation of leukemic cells into monocytes by mechanisms that involved modulation of ROS levels (6).

Conclusions
Both iron and ROS are important to the homeostasis of normal cells, and their dysregulation can contribute to tumorigenesis. Many forms of iron regulation occur in cells of different tissues from various organs in the body. Aberrant regulation of iron metabolism may be involved in carcinogenesis and may also be required to maintain the transformed state of tumor cells. The especially high iron demand of tumor cells indicates a vulnerable feature of these of cells in comparison to their nontumorigenic cell equivalents.
On the other hand, moderate changes in ROS levels are a part of differentiation and self-renewal programs both in normal and CSCs. However, robust increases of ROS levels are detrimental to tumor cells, as malignant cells exist at higher basal ROS levels than their normal counterparts. In addition, CSCs have resilient ROS defense systems and by targeting any of these defenses that are unique to CSCs, while concomitantly elevating ROS levels, may selectively ablate CSCs and not target normal stem cells. Thus, high iron demands or vulnerabilities to high ROS levels may distinguish cancer cells, and to some extent CSC, from normal cells in many different types of tissues. Therefore, targeting either of these characteristics of the cancer phenotype may have therapeutic value.
Interestingly, iron chelators have the potential to target at least one of these vulnerable features of cancer. Combinations of different iron chelators and drugs that target ROS or iron metabolism may also prove to be effective antitumor medicines. However, the exact levels of tolerance for ROS and iron depletion need to be further explored in a tissue-specific context. Moreover, resilient CSCs, which often escape the effects of chemotherapy and contribute to cancer relapse, are also prospective targets of iron chelators or ROS-inducing drugs that should be further investigated. Thus, in tumor metabolism, changing the relationship of ROS and iron from friends to foes might prove to be an interesting area of research that may lead to possible new therapeutic options for cancer patients in the future.
Footnotes
Acknowledgments
This work was supported by the Children's Cancer and Blood Foundation and U.S. National Institutes of Health grants 5K01DK063992 and NHLBI-5R01HL102449 (to S. Rivella). M.L.G. was funded by the U.S. National Institutes of Health (NIH) through the NIH Director's New Innovator Award Program, 1 DP2 OD007399-01, National Cancer Institute (R21 CA158728-01A1), Leukemia and Lymphoma Foundation (LLS 6330-11), and M.L.G. is a V foundation Scholar. L.M.B. was funded by the NIH/National Cancer Institute R25 career development and training grant for the program in Nutrition and Cancer Prevention at Weill Cornell Medical College 5R25 CA105012-04 and was funded by the National Center for Complementary Medicine and Alternative Medicine of NIH under the award number F32AT007112.
Author Disclosure Statement
S. Rivella is a consultant for Novartis, Isis and Biomarin Pharmaceuticals. In addition, he is a coinventor for the patents US8058061 B2 C12N 20111115 and US7541179 B2 C12N 20090602. The consulting work and intellectual property of S. Rivella did not affect in any way the design, conduct, or reporting of this research. No competing financial interests exist.