
Abstract
Introduction
H
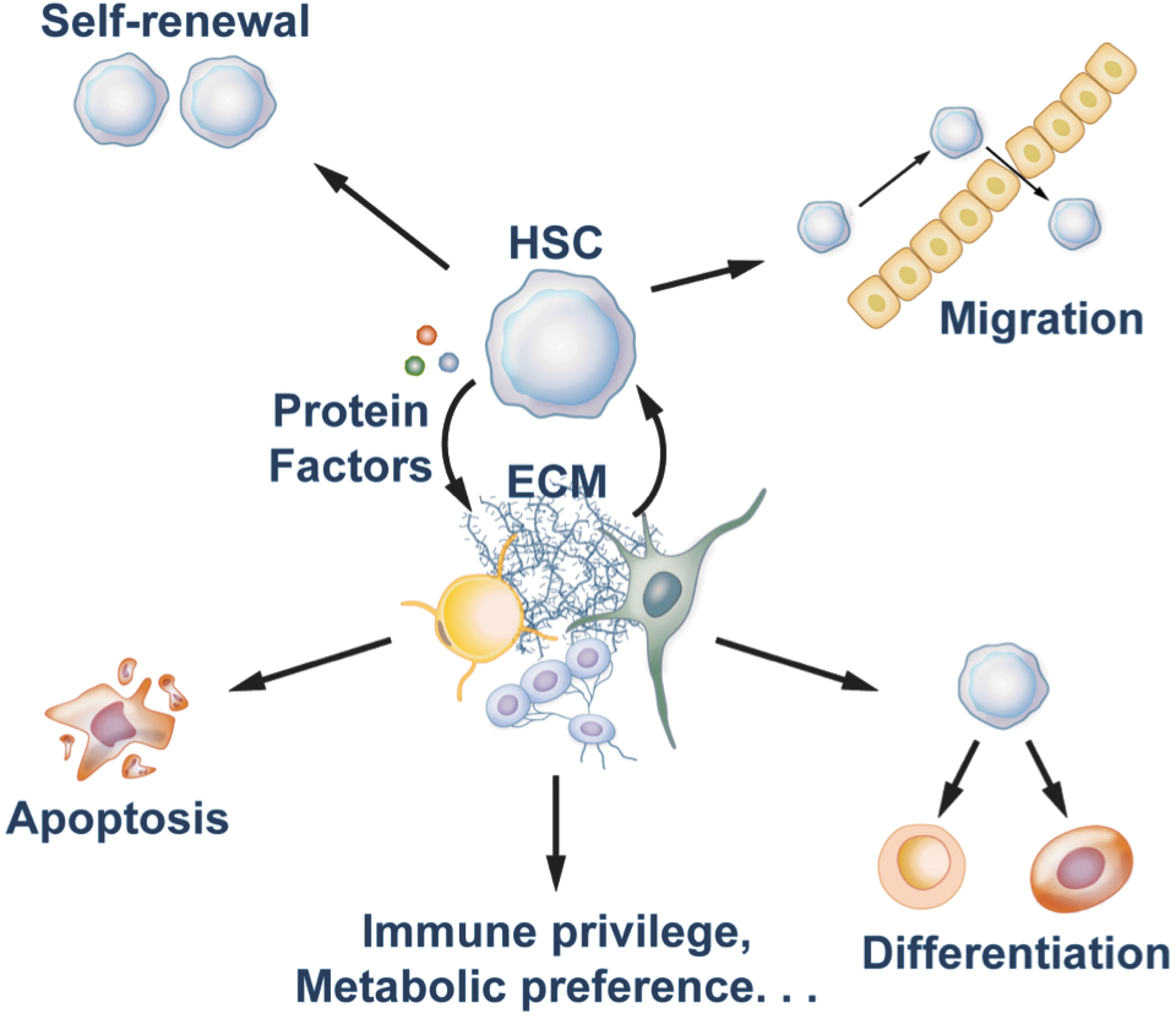
In vivo, stromal cells form a complex microenvironment for HSCs. This environment controls the multiple cell fates of HSCs, including quiescence, self-renewal, differentiation, apoptosis, and migration (21, 129). Depending on their residence in or out of the niche, new properties of HSCs such as their immune privilege (26, 44, 139) and metabolic preference (111, 116) have also been identified in recent studies (Fig. 1). Since extensive reviews are available on oxidative stress regulation of HSCs (28, 94), our review seeks to provide an update on the recent progress toward the understanding of the hypoxia-related metabolism of mouse HSCs.
Hypoxic Bone Marrow Niche of HSCs
In 1978, the concept of the special microenvironment or niche of HSCs was introduced by Schofield (105). Since then, mounting evidence indicates that the niche plays a crucial role in quiescence, self-renewal, differentiation, apoptosis, migration, and immune privilege of HSCs (25, 26, 113). Currently, we know of the existence of several types of cells that form bone marrow HSC niches (71). The supportive cells in the niches produce growth factors and extracellular matrix components and provide other intercellular signals that can promote self-renewal rather than differentiation of HSCs. The endosteal HSC niche contains osteoblasts as the main supportive cell type for maintenance of hematopoiesis (12, 137). The initial suggestion that osteoblasts may be a cell type important for support of HSCs came from studies that showed bone-forming potential of stromal cell lines which can support HSC expansion in culture (115). Real-time imaging techniques demonstrated that HSCs are close to endosteum which supports HSC expansion in response to damage signals (69, 132). The vascular HSC niche is mainly composed of sinusoidal endothelial cells (54). In both bone marrow and spleen, many HSCs are associated with the sinusoidal endothelium (54). The role of endothelial cells in maintenance of HSCs was initially shown in co-culture experiments which indicated that endothelial cells from the yolk sac or the aorta-gonad-mesonephros maintain and expand HSC activity (13, 65, 87). Endothelial cells isolated from non-hematopoietic tissues do not have the ability to support HSC growth in culture. The role of the vascular HSC niche has not been fully elucidated, but it is hypothesized that HSCs in this niche constantly self-renew to maintain HSC numbers and, at the same time, contribute to normal hematopoiesis through asymmetrical divisions (128). Furthermore, the fact that extramedullary hematopoiesis can proceed in tissues such as spleen and liver that contain vascular endothelium but not in endosteum suggests that endothelial cells in different tissues can acquire an HSC-maintenance phenotype (13). Multiple groups have demonstrated that osteoblasts are in close apposition to sinusoidal endothelial cells, and, therefore, these two types of cells may establish a compound niche (35, 132). In addition, stromal cell-derived factor 1 (SDF-1) abundant reticular cells (114), CD146-expressing subendothelial stromal cells (103), Nestin+ mesenchymal stem cells (MSCs) (77), macrophages (16, 131), and the sympathetic nervous system (52) have also been demonstrated to represent components of HSC niches (71). Of these cell types, Nestin+ MSCs are in direct contact with HSCs or in clusters around them, and express HSC maintenance genes, the deletion of which significantly decreased numbers of bone marrow HSCs (77). Furthermore, regulatory T cells (Treg) co-localize with HSCs in the endosteal area of the bone marrow to protect HSCs from immune attack (26).
Bone marrow is considered a tissue with limited oxygen supply, and one of the hallmarks of the HSC niche is its low oxygen tension, hence the term “hypoxic niche” (23). Although the anatomic localization of the hypoxia niche for HSCs has not been determined, several levels of evidence suggest that HSCs prefer this hypoxic environment to more oxygen-rich locations. First, mathematical modeling of the oxygen pressure distribution within the bone marrow suggested that HSCs should be located in a hypoxic zone (17). In addition, in vitro hypoxic cultures with 1%–3% oxygen have been shown to promote the production of erythroid, megakaryocytic, and granulocytic-monocytic progenitors (10, 51, 59, 62) and to enhance HSC expansion and engraftment (19, 20, 41). Finally, and most importantly, in vivo studies confirmed that most quiescent and primitive HSCs reside in hypoxic bone marrow regions with diminished blood perfusion (61, 70, 91, 130). In these studies, hypoxia markers pimonidazole or Hoechst were used to show localization of HSCs to the low-oxygen endosteal bone marrow region. Additional studies also demonstrated that the maintenance and retention of primitive undifferentiated HSCs are accompanied by endosteal osteoblasts, endosteal macrophages, some endothelial cells, and Nestin+ MSCs, which produce chemokines, cytokines, growth factors, and adhesion molecules (12, 16, 77, 131, 137, 138). While the bone marrow osteoblasts and sinusoidal endothelial cells may form a compound niche (35, 132), the migration of HSCs out of the bone marrow, or HSC mobilization, was suggested to occur more through the vasculature [18]. It appears that this view is concordant with a recent study by Stefan Karlsson's group. In their experiments, Miharada et al. showed that LT-HSCs express surface receptor GPR78, and these GPR78+ HSCs reside in the bone marrow endosteal region and exhibit higher glycolytic flux and decreased mitochondrial potential. The block of GPR78 induces HSCs to move from the endosteal area to the central bone marrow (78). Nevertheless, it is still a question where the physical location of the hypoxia niche is, and it is possible that bone marrow sinusoidal endothelium is hypoxic. Needless to say, within the hypoxia niche, there may exist a narrow window of oxygen tension that provides the optimal environment for HSC maintenance and function, as suggested by the small range of the hypoxia-inducible factor-α (Hif-1α) levels which are necessary for the normal activity of HSCs reported by the Suda's group (116). Collectively, these studies indicate that the low oxygen microenvironment is not only tolerated by HSCs, but also essential for maintaining their stemness. The mechanism of hypoxic tolerance of HSCs, although not well understood, implies that they have unique metabolic characteristics.
HSCs in a Hypoxic Niche
Metabolism in a low-oxygen environment
In differentiated cells, mitochondrial oxidative phosphorylation is the main source of ATP in the presence of oxygen, producing 18 times as much ATP as cytoplasmic glycolysis (109). In the mitochondria, oxygen is used as the terminal electron acceptor for the respiratory chain. In the absence of oxygen, the proton gradient formed by the respiratory chain collapses and mitochondrial ATP production stops. Under these hypoxic or anoxic conditions, energy production is derived from cytoplasmic glycolysis through the fermentation of glucose, with the net production of 2 mol of ATP per mol of glucose. In the final step of anaerobic glycolysis, pyruvate is converted to lactate to replenish NAD+. In 1861, Luis Pasteur first reported that oxygen regulates the rate of glucose fermentation (93). Later, the stimulation of glycolysis in the absence of oxygen became known as the “Pasteur Effect.” The rate of glycolysis is controlled by modulating either the activity (30, 92, 100) or the expression (42, 125 –127) of regulatory glycolysis enzymes or both.
Although the aim of stimulation of glycolysis under hypoxic conditions is to increase energy supply, this is not as important as minimizing energy demand in most cells, where shutting down non-essential functions is crucial for energy preservation. The comparative physiologist Kjell Johansen referred to this process as “turning down the pilot light” (34). We demonstrated that HSCs are metabolically quiescent and may be constantly operating under “pilot light” conditions, content with the limited energy derived from glycolysis (111). This observation is consistent with the finding that most HSCs are quiescent in their niches in the homeostatic condition (3).
Metabolic phenotype and reactive oxygen species
The energy advantage of mitochondrial oxidative phosphorylation over glycolysis is, unfortunately, not without deleterious consequences, as the mitochondrion is considered the major source of free radical production (31, 79, 86, 120, 121). It is estimated that 2% of all electrons flowing through the mitochondrial respiratory chain result in the formation of oxygen-free radicals. The mechanism of production of oxygen radicals appears to be the leaking of electrons by the respiratory chain, which then interact with oxygen, thus partially reducing it to superoxide anion (O2 −•). Even though O2 −• itself is not a strong oxidant, it is the precursor of most other reactive oxygen species (ROS) (121), including the highly reactive hydroxy radical (OH•) (68) and peroxinitrite (NO•) (8). ROS overwhelm the natural antioxidant defense mechanisms over time and result in widespread cellular damage either directly or indirectly through the more stable lipid peroxidation products, rendering cells dysfunctional. This is believed to be an important mediator of aging and of numerous degenerative diseases, including HSC dysfunction and senescence (24).
In fact, within the HSC compartment, the repopulation capacity is localized to only those HSCs with low levels of free radicals (45). By contrast, elevated levels of ROS decrease the self-renewal and quiescence of HSCs, as evidenced by the phenotypes of the ataxia telangiectasia mutated-deficient and Forkhead Box O (FOXO)-deficient HSCs (39, 80, 119, 133) [also see review ref. (94)]. During the differentiation of HSCs into myeloid cells, ROS levels increase (89). Therefore, we hypothesize that the glycolytic metabolic phenotype of HSCs may not only protect them against hypoxic insults, but may also serve to minimize oxidant damage which results from mitochondrial oxidative phosphorylation.
HSC metabolism
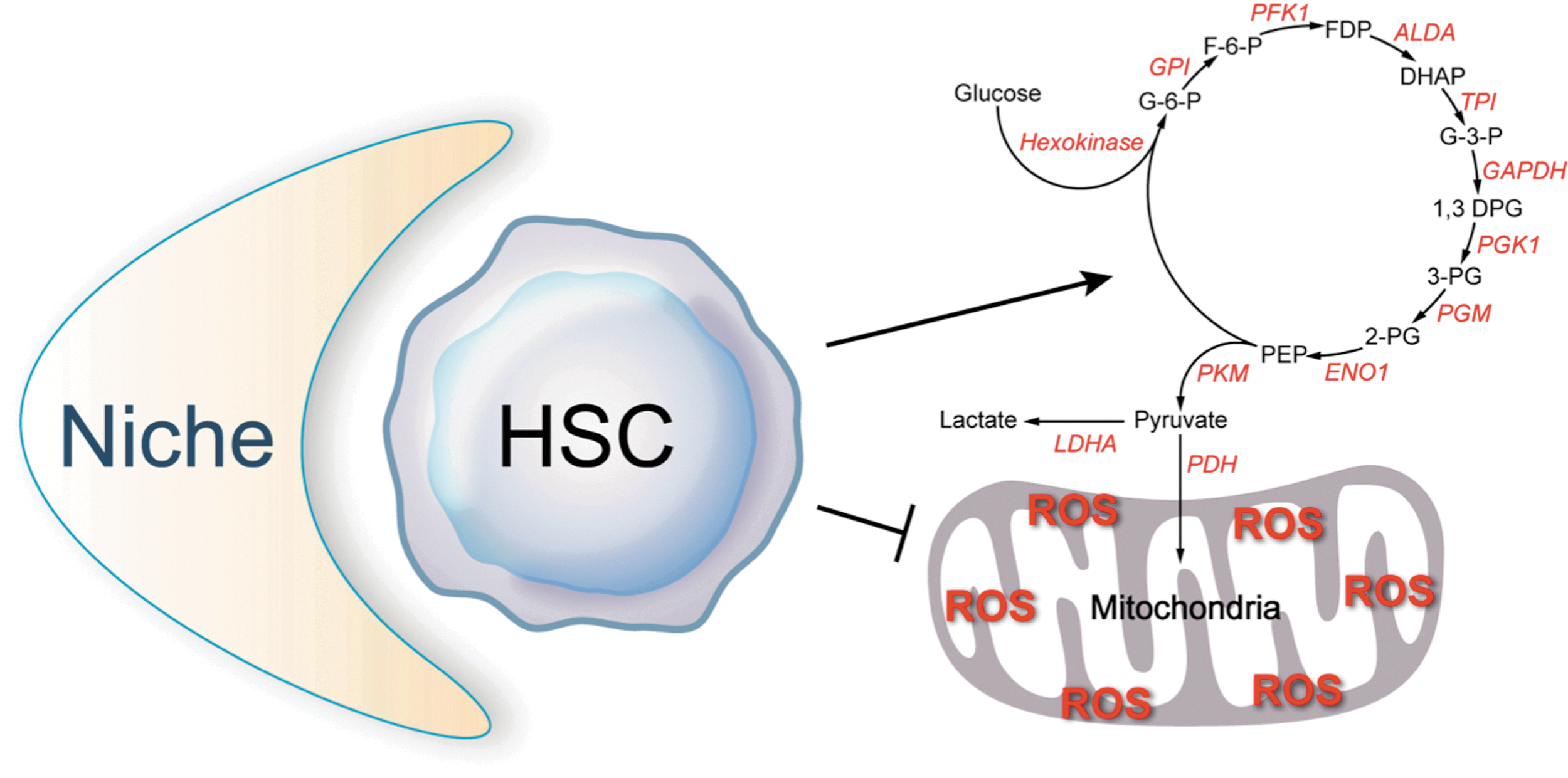
In 1990, Spangrude and Johnson showed that hematopoietic cells with higher clonogenic capacity display low staining with a mitochondrial dye rhodamine-123 (112). Until recently, however, the metabolic properties of HSCs have been largely unknown. Unwin et al., using an elegant proteomics approach, demonstrated that out of 145 dysregulated proteins identified when LSK+ and LSK− cells were compared, a large percentage were involved in metabolic adaptation favoring anaerobic glycolysis in LSK+ cells and mitochondrial oxidative phosphorylation in LSK− cells (122). We recently showed (by direct measurement of the incorporation of 13C from glucose into lactate) that LT-HSCs rely on anaerobic glycolysis (Fig. 2) and have lower rates of oxygen consumption and lower ATP levels than other cells in the bone marrow (111). Moreover, we demonstrated that separation of bone marrow cells solely based on their metabolic profile markedly enriches for HSCs, which confirms that LT-HSCs are localized to a low mitochondrial profile gate. Consistently, a recent proteomic study indicates that multipotent HSCs and progenitors obviously have a higher abundance of proteins in the glycolytic pathway than more differentiated precursors (57).
Hif-1α As a Master Regulator of Hypoxia in HSCs
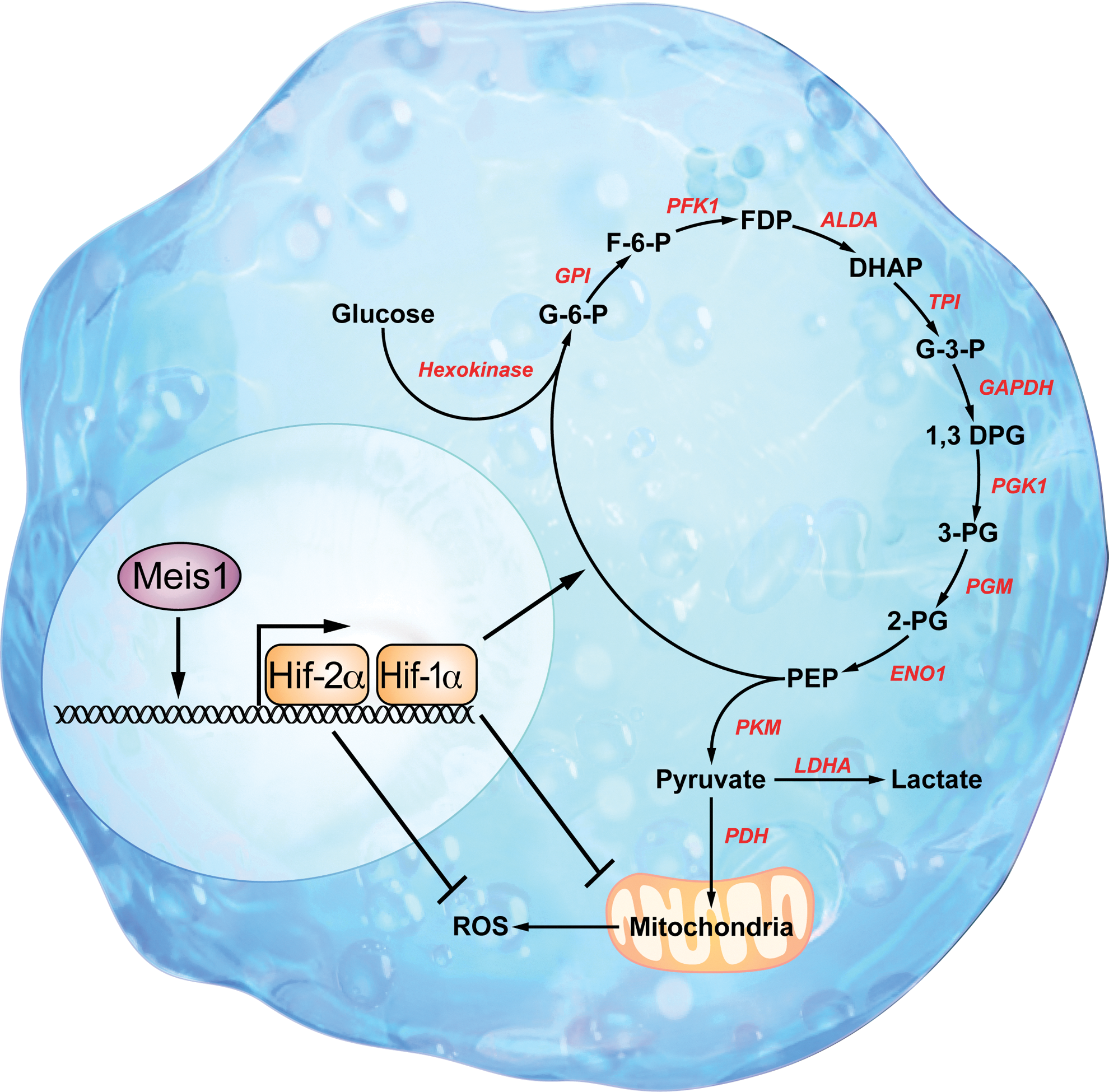
We demonstrated that Hif-1α, the oxygen sensor, is highly expressed in LT-HSCs, especially those that prefer to using glycolysis as their main energy source (111). We also found that the homeobox transcription factor myeloid ecotropic viral integration site 1 (Meis1) regulates HSC metabolism through transcriptional activation of Hif-1α (111). These findings revealed an important transcriptional network that regulates HSC metabolism.
Hif-1α: a master regulator of metabolism
Hif-1 is a master regulator of cellular metabolism (Fig. 3). With hundreds of downstream target genes, it regulates various aspects of metabolism from the oxidant stress response to the regulation of glycolysis and mitochondrial respiration. Hif-1 function is mainly controlled by the Hif-1α subunit that forms a complex with the constitutively active Hif-1β subunit. Hif-1α undergoes rapid degradation during normoxia and is stabilized during hypoxia in non-stem cells, whereas normoxic stabilization of Hif-1α occurs in HSCs (56, 95). Hif-1α undergoes hydroxylation by prolyl hydroxylases during normoxia (40, 43), which results in ubiquitination (50, 75) and proteasomal degradation (104). Under hypoxic conditions, however, both the protein level and the DNA-binding activity of Hif-1α increase dramatically (47).

Stabilization of the Hif-1α protein can occur by numerous mechanisms such as inhibition of hydroxylation by prolyl hydroxylase domain protein 2 (PHD2), inhibition of prolyl hydroxylase domain protein 3 (PHD3) by the ubiquitin ligase seven in absentia homolog 2, or deubiquitination of Hif-1α by the VHL deubiquitinating enzyme 2 (66, 84, 99, 108).
In addition to well-known upstream regulators of Hif-1α [see review ref. (6)], the energy sensor 5′ AMP-activated protein kinase (AMPK) and redox sensor sirtuin 1 (SIRT1) also regulate the stability of Hif-1α. For example, AMPK activation stabilizes Hif-1α indirectly by decreasing the intracellular ATP level (46). SIRT1 appears to inactivate Hif-1α activity through deacetylating Lys674 of Hif-1α, and this is reversed during hypoxia when SIRT1 is down-regulated owing to decreased NAD+ levels (67). Furthermore, the Aryl hydrocarbon receptor (AhR) also down-regulates Hif-1α signaling by forming an AhR/aryl hydrocarbon receptor nuclear translocator (ARNT) complex that decreases ARNT binding to Hif-1α and results in lower vascular endothelial growth factor (VEGF) levels (36). An antagonist of AhR was shown to support ex vivo expansion of human HSCs (9).
Hif-1α is also regulated by transcriptional activation in HSCs (32, 63). We have recently shown that Meis1 regulates Hif-1α transcription in HSCs and is required for optimal Hif-1α transcription (111). Up-regulation of Hif-1α in HSCs by protein stabilization or by transcriptional activation enables HSCs to survive under hypoxia insults and to minimize oxidative damage that results from oxidative phosphorylation. Consequently, glycolysis is used as the main energy source for HSCs in the bone marrow niche.
Hif-1α regulates the expression of hundreds of downstream genes with roles in angiogenesis, metabolism, proliferation and survival, autophagy, proteolysis, and pH regulation [see reviews refs. (53, 64, 76)]. Some of the Hif-1α target genes are also known to be important players in HSC function and metabolism; these include VEGF, ADM, SDF1, SCF, Ang2, Angptls, IGF-2, IGFBPs, p16, p19, p21, FOXOs, and others (53, 64, 76, 116, 136). In particular, Hif-1α stimulates transcription of glucose transporters, a number of glycolytic enzymes, and glycolytic inducing factors such as Cripto (42, 78, 106). Of note, many of these Hif-1α target genes (such as SDF1, SCF, Angptl, p21, FOXOs, and Cripto) are critical regulators of the quiescence of HSCs in the bone marrow niche (15, 78, 80, 85, 117, 119, 133, 138).
Hif-1α also crosstalks with Notch and Wingless/Integrated (Wnt) pathways that play important roles in regulation of HSC activity. Hif-1α interacts with the intracellular domain of the Notch receptor, which stabilizes NICD and potentiates the transactivation of Notch target genes (22). The interaction between Hif-1α and β-catenin reduces the transcription of some of the Wnt target genes and enhances the transcription of certain Hif-1α target genes (49). These studies suggest a complex relationship among metabolism, mitochondria, hypoxia, and stem cell function.
Hif-1α and HSCs
As described earlier, Hif-1α regulates the expression of a number of genes that are critical for HSC activity and quiescence and interacts with other proteins involved in HSC signaling. Hif-1α also plays an important role in hematopoiesis during development. Hif-1α knockout is embryonically lethal due to multiple neural tube, cardiovascular, and hematopoietic defects (42, 102). The yolk sac [the first source of definitive hematopoietic progenitors (90)] from the Hif-1α-null mutants is smaller and has significantly lower numbers of cells than wild-type embryos (42, 134). A recent cDNA microarray analysis comparing gene expression in human embryonic stem cells, HSCs, and MSCs found that Hif-1α was the only transcription factor that these cell have in common (55). Interestingly, Hif-1α protein is stabilized in mobilized human hematopoietic progenitor cells after they leave their hypoxic microenvironment (95). We have recently demonstrated that Hif-1α is markedly enriched in LT-HSCs (111), and a report from Suda's group (using Mx-Cre) (116) showed that adult Hif-1α deletion results in the loss of quiescence and a decrease in the repopulation activity of HSCs. Suda's group also demonstrated that the Hif-1α expression should be within a certain range for optimal HSC maintenance and activity (116). Furthermore, a mutated vegfa promoter that prevents HIF binding leads to impaired repopulation activity of HSCs (101), underscoring the importance of HIF in HSC function. However, despite the fact the Hif-1α is a master metabolic regulator, until recently the effect of Hif-1α deletion of HSC metabolism has not been determined. Recently, we demonstrated that conditional deletion of Hif-1α in adult HSCs results in a metabolic shift toward oxidative metabolism with increased rates of oxygen consumption and decreased rates of glycolysis. Interestingly, we found that Hif-1α deletion also results in profound up-regulation of Hif-2α in HSCs, likely secondary to increased oxidative stress as a result of shift to oxidative metabolism (58).
Transcriptional regulation of Hif-1α in HSCs
We have recently demonstrated that Meis1 is required for transcriptional activation of Hif-1α in HSCs (111) (Figs. 4–5). Meis1 belongs to the Hox family of homeobox genes, an evolutionarily conserved set of genes that encode DNA-binding transcription factors (60). Meis1 is expressed in fetal HSC compartments and in primary sites of definitive hematopoiesis (5). In addition, Meis1 is expressed in the most primitive hematopoietic subpopulations and is down-regulated on differentiation (4, 37, 97). Targeted Meis1 knockout is embryonically lethal by embryonic day 14.5 with multiple hematopoietic and vascular defects (5, 33, 37). The transcriptional regulation of Hif-1α by Meis1 suggests that the anaerobic metabolism and increased Hif-1α expression of HSCs is somewhat an intrinsic feature of stem cells. Until recently, the precise role of Meis1 in adult HSCs was poorly understood. In a recent report, we demonstrated that conditional deletion of Meis1 results in down-regulation of both Hif-1α and Hif-2α in HSCs, with the resultant increase in ROS, loss of HSC quiescence, increased HSC apoptosis, and profound HSC dysfunction. Intriguingly, and in a testament to the central role of ROS in the Meis1 phenotype, we demonstrate that systemic administration of the ROS scavenger N-acetylcystein restored HSC function (58). These findings further our understanding of mechanisms of adaptation of HSCs to their environment and could have a great impact on HSC enrichment and expansion techniques, in addition to providing novel tools for understanding HSC dysfunction and aging.

Possible Role of Mitochondrial Metabolism in HSCs
The up-regulation of Hif-1α in HSCs may enable these important cells to survive under hypoxic conditions and minimize oxidative damage from oxidative phosphorylation. Accumulation of mitochondria in mutant HSCs such as those deficient for autophagy protein Atg7 is associated with higher mitochondrial superoxidase, increased DNA damage, apoptosis, and proliferation (82).
Although HSCs appear to use glycolysis as their main energy source, there is certainly measurable mitochondrial respiration even in the most purified HSC population. Therefore, the role of mitochondria in HSC metabolism is not fully understood. Although it was shown that epidermal progenitors are independent of mitochondrial respiratory chain (7), it is likely that a basal level of mitochondrial respiration is essential for HSC energy homeostasis. Alternatively, oxidative phosphorylation might be used by HSCs in cell fates other than quiescence. For example, mitochondrial biogenesis has been linked to HSC differentiation (96), and a recent study found that mitochondria appear to be abundant, but inactive, in HSCs, possibly in preparation for the energy production on HSC expansion or differentiation (73).
Targeted deletion of liver kinase B1 (LKB1), an important promoter of mitochondria biogenesis, results in a severely compromised HSC phenotype. Interestingly, the phenotype of LKB1-knockout HSCs is similar to that of the Hif-1α-deficient HSCs. In both, there is loss of HSC quiescence and increased HSC/multipotent progenitor cell proliferation. This phenotype is associated with defects in mitochondrial number and function (27, 29, 83). Similarly, the knockout of a guanidine nucleotide binding protein (GTPase of the immunity-associated protein 5) also results in decrease of mitochondrial potential and loss of quiescence of HSCs (14). It should also be noted that dysfunctional mitochondrial activities (such as caused by deletion of Stat3) can be associated with overproduction of ROS, which coincides with pronounced HSC defects (74). Intriguing questions are whether these HSCs with defective mitochondria have any defects in glycolysis and whether any relationship between the knockout genes and Hif-1α exists. Overall, it is important to further elucidate the role that mitochondrial oxidative phosphorylation plays in HSC fates.
Perspective and Questions on Hypoxia, Metabolic Property, and Cell Fates of HSCs
New studies should address many related questions and elucidate the link between HSC metabolism and their stem cell function.
1. Given that previous studies have not addressed the anatomic location of the “hypoxia niche” of HSCs, it will be important to clarify the relationship between the hypoxic niche and other HSC niches (endosteal niche, vascular niche, etc.).
2. Hypoxia may induce quiescent metabolic features in HSCs in the homeostatic bone marrow niche, and HSCs in the bone marrow hypoxic niche use glycolysis instead of mitochondrial oxidative phosphorylation. However, it is not clear whether quiescence is required for activation of the hypoxia-related metabolic features of HSCs or vice versa. We speculate that these two seemingly independent features may be closely related. It is possible that both cell cycle quiescence and metabolic quiescence of HSCs are regulated by the HSC niche. Alternatively, glycolytic metabolism may be required for maintenance of the quiescent state, and, therefore, HSCs preferentially reside in hypoxic niches to help stabilize Hif-1α and maintain glycolytic metabolism. It is not clear whether glycolysis rates of HSCs in the bone marrow differ from those of HSCs in other locations. What happens to HSCs during mobilization when oxygenation is increased? Do they express the same level of Hif-1α as in the bone marrow niche? Do they prefer to use oxidative phosphorylation instead of glycolysis as energy? Do HSCs change metabolic preference before leaving this niche or change after leaving? Do HSCs always use glycolysis in environments such as the extramedullary hematopoietic sites and during ex vivo culture?
3. It appears that hypoxic niche supports HSC quiescence and that increased mitochondrial biogenesis and ROS levels are linked to HSC differentiation. It would be interesting to study the relationship between metabolic preference and self-renewal/quiescence/differentiation using mutant mice with known skewed stem cell self-renewal/differentiation, such as those deficient for Angptl receptor signaling and many others (140). What other cell fates does hypoxia regulate? Does hypoxia suppress HSC differentiation and mobilization?
4. Is constitutive Hif-1α expression a feature for HSCs independent of niche? Or is it possible that a niche-independent high expression of Hif-1α is an indicator of transformation? We do not have a clear answer to either question. While there exist evidences which support the hypothesis that HSCs may utilize different energy sources in different environments, the transcriptional regulation of Hif-1α by Meis1 suggests that the glycolytic metabolism may be an intrinsic property of HSCs. In addition, the high level of Hif-1α expression maintained in acute myeloid leukemia stem cells is no longer dependent on a hypoxic environment (124).
5. Since a hypoxic environment and Hif-1α stability are critical for HSC quiescence, the disruption of this homeostasis could potentially lead to diseases. Indeed, some diseases are related to ROS increase, and it is well known that Hif-1α promotes neoplastic growth (1, 107). It is important to understand how the alteration of HSC metabolism contributes to the disease development.
7. The “Warburg hypothesis” suggests that cancer cells mainly generate energy by glycolysis but not by oxidation within the mitochondria. Nevertheless, when Otto Heinrich Warburg made this hypothesis in 1924, people were not aware that the cancer mass is, in fact, heterogeneous. What are the metabolic properties of cancer stem cells and differentiated cancer cells? Is there any difference? Do different types of cancer have different metabolic properties?
Footnotes
Acknowledgments
The authors thank Jose Cabrera in Eric Olson's laboratory for producing the artwork for the figures. C.C.Z. is the Michael L. Rosenberg Scholar in Biomedical Research at UT Southwestern Medical Center. C.C.Z. is supported by NIH grant K01 CA 120099, American Society of Hematology Junior Faculty Award, American Heart Association 09BGIA2230372, DOD PR093256, CPRIT RP100402, March of Dimes Foundation 5-FY09-146, Robert A. Welch Foundation I-1701, and the Gabrielle's Angel Foundation. H.A.S. is supported by grants from the American Heart Association (Grant in Aid) (Sadek), the Gilead Research Scholars Program in Cardiovascular Disease (Sadek), Foundation for Heart Failure Research, NY, and the NIH (1R01HL115275-01) (Sadek).