
Abstract
Introduction
U
The de novo synthesis of BH4 is under strict control by the rate limiting enzyme guanosine triphosphate cyclohydrolase I (GTPCH1) (28). The catalytic activity of GTPCH1 is regulated by the inhibitory GTP cyclohydrolase I feedback regulatory protein (GFRP) (13). GFRP inhibits the catalytic activity of GTPCH1 by negative feedback (33) and thereby limits excessive BH4 biosynthesis under normal conditions. GFRP may be dysregulated under conditions of oxidative stress (11,16,17). For example, hydrogen peroxide-induced oxidative stress increases Gfrp mRNA expression in endothelial cells, leading to a decline in BH4 levels (17). These in vitro data suggest that cellular redox homeostasis plays a critical role in GFRP expression and hence may regulate BH4 bioavailability in vivo after exposure to ionizing radiation (IR).
Gfrp transgenic mice were generated to investigate the effect of in vivo Gfrp overexpression on 5,6,7,8-tetrahydrobiopterin (BH4) biosynthesis and on radiation response. This study showed that increased in vivo Gfrp expression in Gfrp+/Cre+ mice was accompanied by reduced BH4 levels, increased oxidative stress, differential mitochondrial functions, increased radiation-induced peroxynitrite formation, and decreased white blood cell counts compared with control mice. Moreover, irradiated control mice exhibited increased Gfrp mRNA expression. Because guanosine triphosphate cyclohydrolase I feedback regulatory protein tightly regulates biosynthesis of BH4, which plays a critical role in cardiovascular, neurological, and oxidative stress-induced diseases, this transgenic mouse model is a useful tool to understand the roles of BH4 in health and disease.
While the significance of GFRP in the regulation of GTPCH1 activity and BH4 levels has been investigated in vitro (18), there is a paucity of studies validating these concepts in vivo. To obtain further insight into the role of in vivo GFRP overexpression in the BH4 biosynthetic pathway and BH4 levels during the radiation response in vivo, a novel GFRP overexpressing Cre-Lox-regulated transgenic mouse model was generated. Our results demonstrate that in vivo overexpression of Gfrp induces changes in GTPCH1–GFRP interaction, decreases tissue BH4 levels, increases parameters of oxidative stress, and induces changes in mitochondrial bioenergetic functions. Moreover, exposure to radiation is associated with differential effects in redox homeostasis in transgenic mice and littermate controls. The novel Gfrp transgenic mouse model will likely become a valuable tool to study the involvement of BH4 in whole body radiation and is also suitable for studies of tissue-specific radiation responses and other conditions associated with oxidative stress.
Results
Generation and analysis of Gfrp transgenic mice
To determine the role of in vivo Gfrp overexpression on the BH4 biosynthetic pathway, a Gfrp transgenic mouse model was generated. Gfrp is a single exon gene and the 84 amino acid long GFRP's molecular weight is ∼10 kDa. Figure 1a illustrates the gene targeting strategy to generate Gfrp transgenic mice and the subsequent scheme of transgene expression used in this study. The targeting construct was delivered by pronuclear microinjection. Microinjections were performed in embryos obtained from C57BL/6 mice. The flox-stop vector containing Gfrp cDNA was used to generate the Gfrp transgenic “knock-in” founder lines in a C57BL/6 background. Upstream of the Gfrp cDNA and downstream of CAG promoter, a STOP cassette (neo cassette flanked by two loxP sites) was positioned to prevent transcription. Transgene expression was achieved upon Cre-mediated deletion of the STOP cassette allowing the promoter to transcribe the transgene constitutively in Gfrp overexpressing transgenic mice (Gfrp+/Cre+); while littermates that do not carry the Gfrp transgene were used as control (Gfrp−/Cre+).
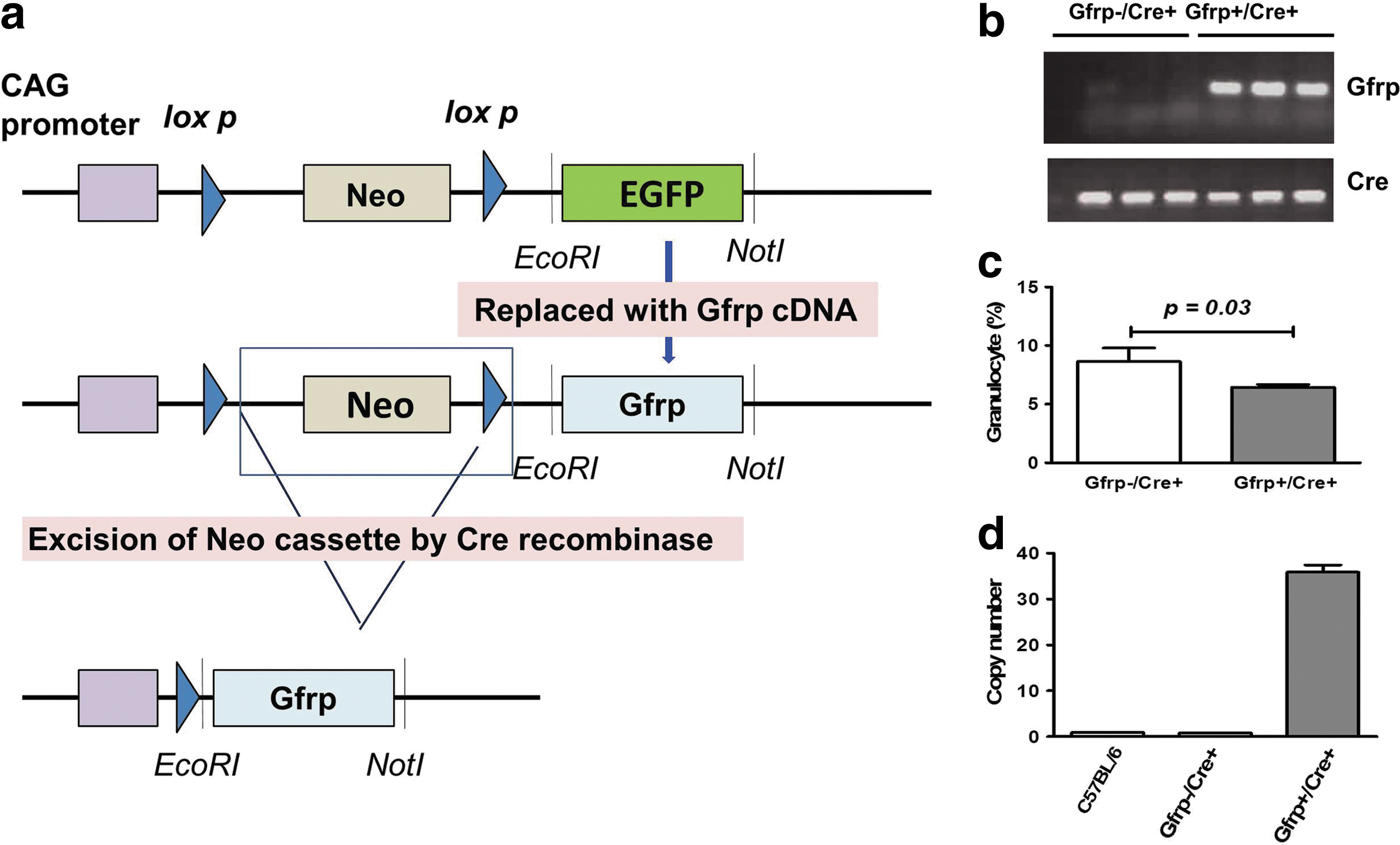
Polymerase chain reaction (PCR) analysis initially identified six different founder lines, which showed transmission of the Gfrp transgene to the germline. The germline transmission of the Gfrp transgene was assessed by genotyping (Fig. 1b) using transgene specific primers. Functionality of the transgene was tested by breeding all the transgenic founder male animals with EIIa Cre female mice expressing Cre recombinase to delete the STOP cassette. The level of Gfrp transgene expression in different tissues was measured by quantitative reverse transcription PCR (qRT-PCR) and western blot analysis. Two transgenic founder lines (706 and 712) were identified that exhibited particularly high Gfrp expression in all tissues examined and were used to develop the mouse colonies. The “706” transgenic founder line was used for this entire study. The Gfrp+/Cre+ mice and the control littermates were normal in terms of gross morphological features, development, and fertility (data not shown). No discernible difference in hematocrit values was observed between Gfrp+/Cre+ mice and Gfrp−/Cre+ littermates except for granulocyte percentage. Gfrp+/Cre+ mice had slightly lower granulocyte percentage (without a difference in total granulocyte count) compared with control littermates (Fig. 1c).
Next, the transgene copy number was estimated using a duplex TaqMan PCR-based method. A custom-made amplicon spanning part of the Gfrp transgene and part of the insertion vector was designed so that it amplified only the Gfrp transgene under the specific experimental setup without amplifying the endogenous Gfrp gene. TaqMan assays for the Gfrp transgene and the internal control were performed together for each animal in quadruplet to determine the number of transgene copies integrated in the genome. A significantly greater Gfrp transgene copy number was observed in Gfrp+/Cre+ mice, while Gfrp−/Cre+ exhibited similar transgene copy numbers as those of C57BL/6 mice. Approximately 35 copies of the Gfrp transgene were integrated in Gfrp+/Cre+ mice as estimated from the standard curve (Fig. 1d).
Increased GFRP expression in tissues of Gfrp+/Cre+ mice
Gfrp expression at the mRNA level was measured in multiple tissues of Gfrp+/Cre+ mice and the level of expression was compared to its Gfrp−/Cre+ littermates. As shown in Figure 2a, Gfrp mRNA expression was significantly higher in Gfrp+/Cre+ mice than in control littermates in every organ evaluated. However, variability in the level of Gfrp transgene expression was observed in different tissues of Gfrp+/Cre+ mice. Gfrp expression was highest in thymus followed by spleen, lung, intestine, kidney, and liver in Gfrp+/Cre+ mice. In the same tissues, no statistically significant change in Gtpch1 expression levels was found (Supplementary Fig. S1; Supplementary Data are available online at

We also measured GFRP protein expression in six different tissues from the Gfrp+/Cre+ mice and Gfrp−/Cre+ littermates. GFRP was detected in all tissues of Gfrp+/Cre+ mice that were investigated. As expected, the level of GFRP expression was significantly higher in Gfrp+/Cre+ mice compared with control littermates (Fig. 2b). In contrast, GFRP expression in tissues from the control group was very low or undetectable, except for liver tissue. Similarly, basal level of GFRP in control mice was relatively high in liver compared with other tissues. Immunohistochemical analysis further confirmed the presence of significantly higher levels of GFRP in different tissues of Gfrp+/Cre+ mice compared with the control group (Fig. 2c and Supplementary Figs. S2–S5). These data are consistent with electron microscopy studies performed by others (6).
Reduced BH4 levels and increased GTPCH1–GFRP interaction in Gfrp+/Cre+mice
Since GFRP is known to inhibit GTPCH1 catalytic activity by protein–protein interaction, we next investigated the effect of in vivo GFRP overexpression on the de novo BH4 biosynthetic pathway. BH4 level in lung sample of Gfrp+/Cre+ and control littermates was estimated by a liquid chromatography with tandem mass spectrometry (LC-MS/MS) method. The reason for selecting lung tissue in this study is that lung has maximum endothelial cells per unit area compared with other tissues, and that BH4 plays a particularly critical role in regulating endothelial function by modulating eNOS activity. Gfrp+/Cre+ mice exhibited significantly less BH4 level compared with control littermates (Fig. 3a). In addition, we observed significantly less BH2 level in transgenic mice, and, as a result, no significant difference in BH4/BH2 ratio was observed between two genotypes (Fig. 3a).

To determine the reason for the lower BH4 levels in Gfrp+/Cre+ mice, we hypothesized that the interaction of GFRP with its protein partner GTPCH1 would be increased in Gfrp+/Cre+ mice compared with control littermates, thereby inhibiting GTPCH1 activity. To address this, we determined the interaction between these two proteins by co-immunoprecipitation (IP) assay. Significantly increased GTPCH1–GFRP interaction in Gfrp+/Cre+ mice compared with control littermates was observed (Fig. 3b and Supplementary Fig. S6).
Increased oxidative stress and elevated reserve respiratory capacity in Gfrp+/Cre+mice
Decreased BH4 bioavailability may lead to increased oxidative stress in tissues by NOS uncoupling in Gfrp+/Cre+ mice. To determine the status of cellular oxidative stress, total glutathione (GSH) and oxidized glutathione (GSSG) levels were measured in peripheral blood obtained from Gfrp+/Cre+ mice and Gfrp−/Cre+ littermates. Significantly lower total GSH levels and higher percentage GSSG, an endpoint indicative of oxidative stress, were found in peripheral blood samples of Gfrp+/Cre+ mice compared with Gfrp−/Cre+ littermates (Fig. 4a, b).
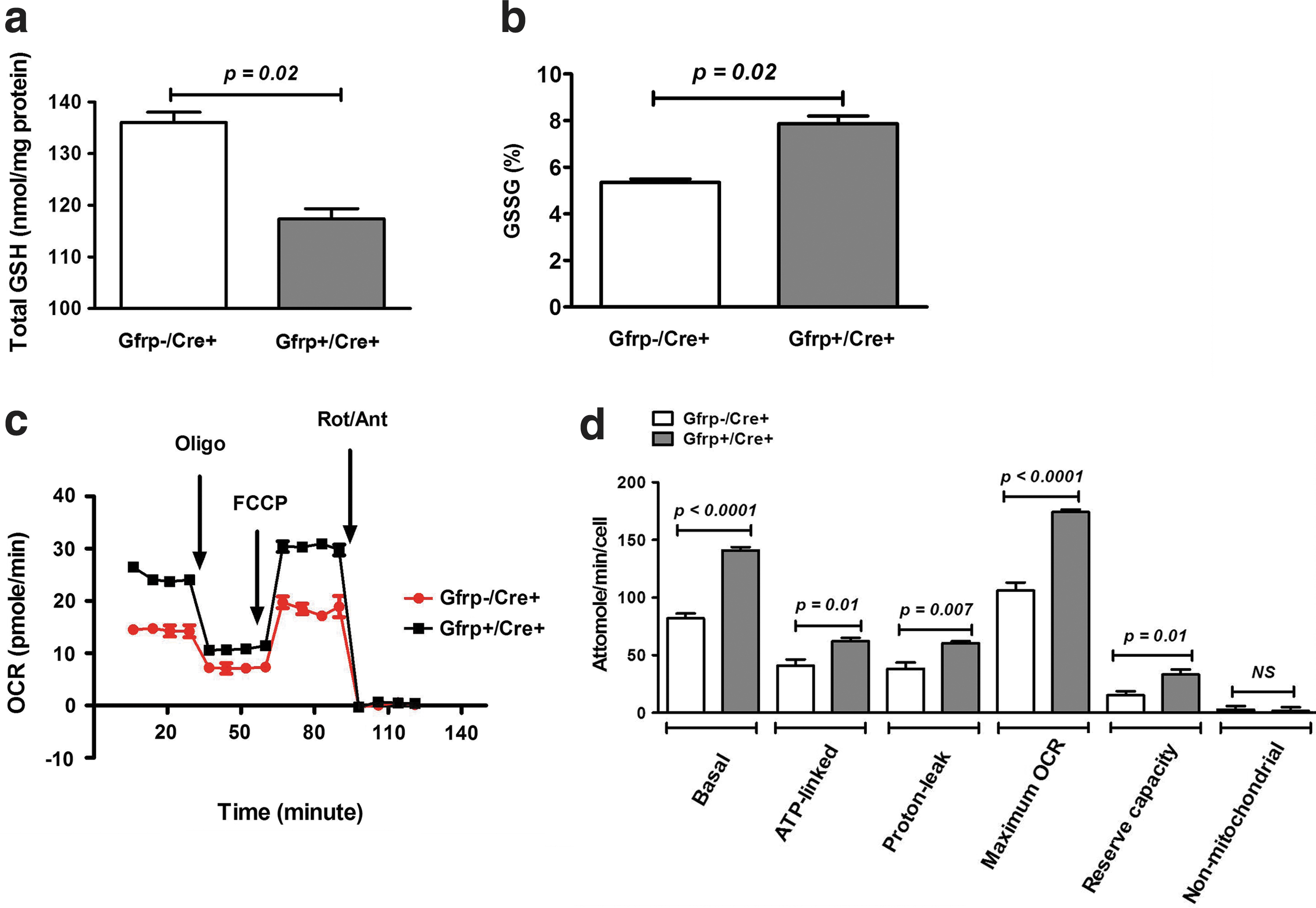
Because increased oxidative/nitrosative stress is strongly correlated with mitochondrial dysfunction, we next investigated the bioenergetic profile of mitochondria isolated from primary thymocytes of Gfrp+/Cre+ mice and their Gfrp−/Cre+ littermates. The function of individual components of the respiratory chain was examined by sequentially adding chemical inhibitors to primary thymocytes (Fig. 4c, d). Compared with Gfrp−/Cre+ controls, Gfrp+/Cre+ mice demonstrated increased basal oxygen consumption rate (OCR). Adenosine triphosphate (ATP)-linked OCR and proton-leaked OCR were also determined in thymocytes of Gfrp+/Cre+ and control Gfrp−/Cre+ mice by oligomycin addition to inhibit ATP synthase (Complex V). Although OCR decreased after oligomycin addition in both groups, increased ATP-linked OCR and proton-leaked OCR were observed in Gfrp+/Cre+ mice. To determine the maximal OCR, primary thymocytes were next treated with proton ionophore (uncoupler) carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP). As expected, OCR increased in both groups after FCCP treatment as mitochondrial inner membrane became permeable to protons; however, the maximal OCR and reserve capacity was higher in Gfrp+/Cre+ mice. Finally, to determine non-mitochondrial OCR, a mixture of rotenone and antimycin A was injected to inhibit electron flux through complexes I and III causing drastic suppression of OCR. No difference in non-mitochondrial OCR was observed in either group.
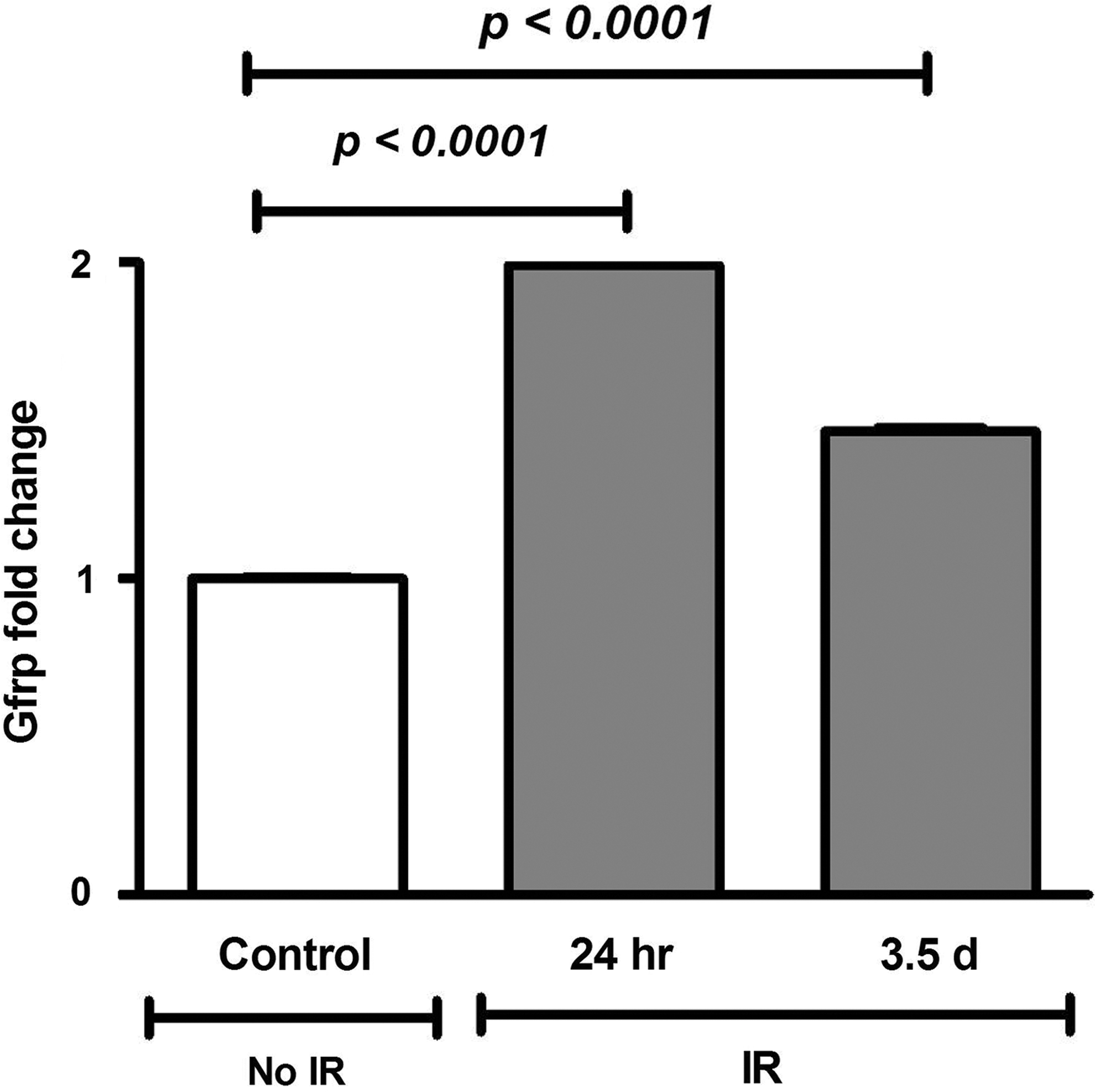
Increased Gfrp mRNA expression in irradiated C57BL/6 mice
To determine the effect of IR on in vivo Gfrp expression, C57BL/6 male mice were exposed to 8.5 Gy of total body irradiation (TBI). Gfrp expression at the mRNA level in lung samples was measured by qRT-PCR at 24 h and 3.5 days after irradiation. As shown in Figure 5, Gfrp expression in lung samples increased after radiation. Gfrp expression increased ∼2-fold and 1.5-fold at 24 h and 3.5 days after radiation exposure, respectively. These data indicate that radiation-induced oxidative stress modulates in vivo Gfrp expression, which may play a role in de novo BH4 biosynthesis.
Decreased BH4/BH2 ratio in irradiated Gfrp+/Cre+mice
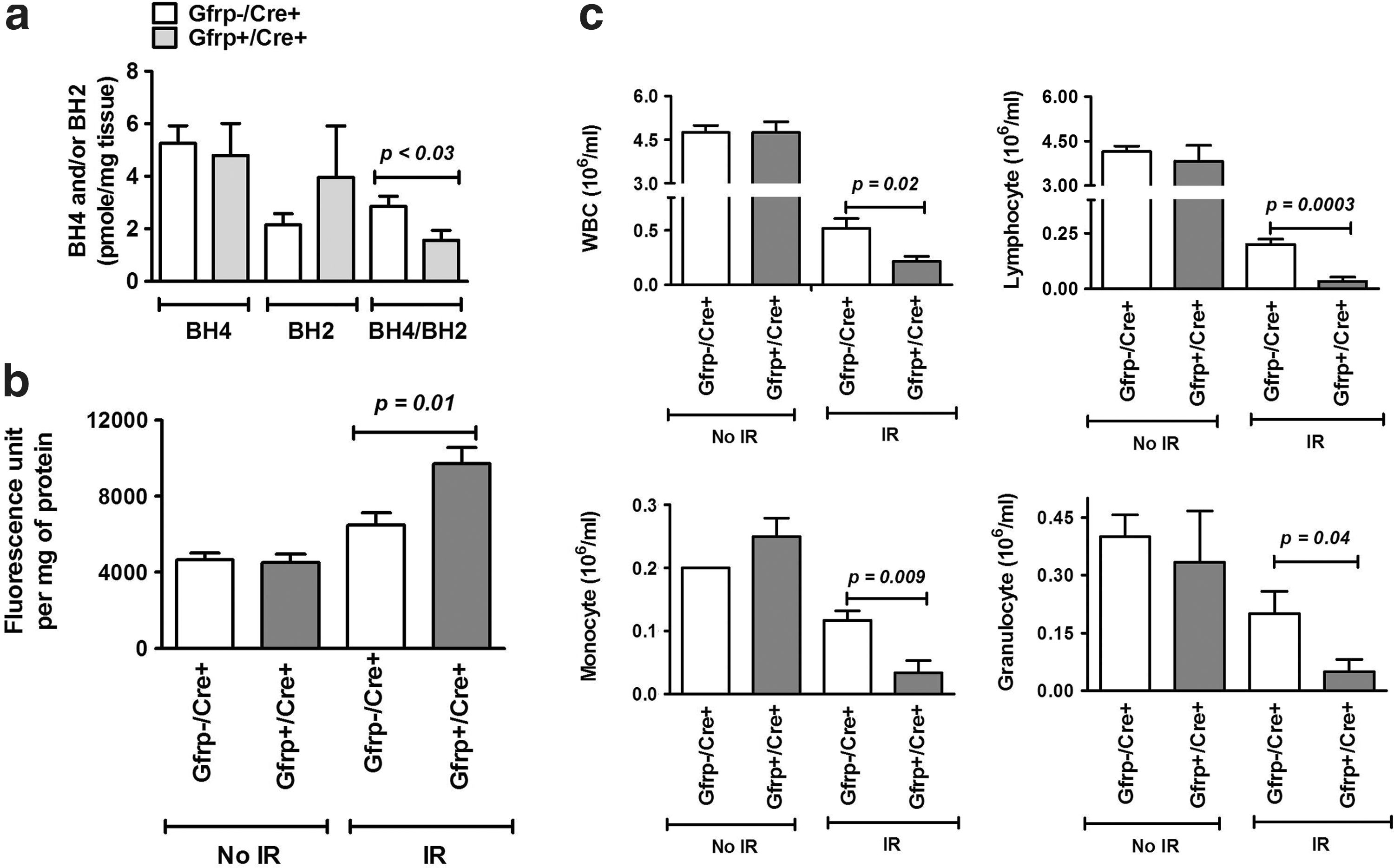
Next, we investigated the effect of IR on the BH4 level, BH2 level, and BH4/BH2 ratio in Gfrp+/Cre+ and Gfrp−/Cre+ mice. Although we did not observe significant difference in BH4 and BH2 level between Gfrp+/Cre+ and Gfrp−/Cre+ mice after irradiation, however, significantly low BH4/BH2 ratio was observed in irradiated Gfrp+/Cre+ mice compared with irradiated Gfrp−/Cre+ (Fig. 6a). We also observed significant decrease in BH4 level and BH4/BH2 ratio, while increase in BH2 level in both Gfrp+/Cre+ and Gfrp−/Cre+ mice after radiation exposure compared with unirradiated group. These data are consistent with the notion that oxidative stress including IR has a critical role in BH4-BH2 homeostasis and irradiation causes further decrease in BH4/BH2 ratio in Gfrp+/Cre+ mice compared with the Gfrp−/Cre+ group.
Increased peroxynitrite formation and decreased white blood cell counts in irradiated Gfrp+/Cre+mice
Next, we determined the effect of IR on nitrosative stress by measuring peroxynitrite formation in Gfrp+/Cre+ and Gfrp−/Cre+ mice. Peroxynitrite formation, a marker of oxidative/nitrosative stress, was measured in abdominal aorta at 24 h and 3.5 day after TBI. Vascular peroxynitrite formation significantly increased after irradiation compared with un-irradiated groups and significantly higher peroxynitrite formation was observed in Gfrp+/Cre+ mice compared with Gfrp−/Cre+ littermates at day 3.5 after exposure to 8.5 (Fig. 6b). On the other hand, no statistically significant difference in peroxynitrite formation between two groups was observed at 24 h after irradiation (data not shown). These results indicate that Gfrp+/Cre+ mice are more prone to radiation-induced oxidative/nitrosative stress, probably due to GFRP overexpression mediated BH4 unavailability.
Blood cell counts were also assessed in Gfrp+/Cre+ mice and its Gfrp−/Cre+ littermates at 24 h and 3.5 days after exposure to 8.5 Gy of TBI. No significant decrease in red blood cell counts and hemoglobin levels were observed at 24 h, while both the parameters were significantly decreased at 3.5 days; however, no statistically significant differences between Gfrp+/Cre+ mice and Gfrp−/Cre+ control group were observed (data not shown). White blood cell (WBC) count was also significantly decreased (p<0.001) at 24 h in irradiated groups compared with unirradiated groups. Notably, significant decrease in WBC counts were observed in Gfrp+/Cre+ mice after irradiation compared with irradiated Gfrp−/Cre+ littermates as shown in Figure 6c. The counts of all WBC subtypes were also significantly lower in irradiated Gfrp+/Cre+ mice compared with irradiated controls (Fig. 6c). WBC were undetectable at 3.5 days after irradiation, both in Gfrp+/Cre+ mice and Gfrp−/Cre+ controls. Consistent with these results, limited TBI experiments, while not reaching statistical significance, showed 30 day survival in Gfrp+/Cre+ mice to be 0%, compared with 25% in Gfrp−/Cre+ littermates (Supplementary Fig. S7).
Discussion
This study reports the generation and initial characterization of a novel Cre-Lox-driven Gfrp overexpressing knock-in transgenic mouse model. A key role for GFRP in the maintenance of BH4 and redox homeostasis in vivo was revealed. Moreover, we showed that in vivo Gfrp overexpression is an important factor in modulating aspects of radiation-induced oxidative damage, thus lending further support to the notion that BH4 is critically involved in radiation toxicity in normal tissues.
Although GTPCH1 is ubiquitously expressed in most eukaryotic tissues, GFRP expression is more tissue specific, with the level of expression and interaction with GTPCH1 varying from organ to organ (9,20). Consistent with previous studies from other laboratories (20), our study showed higher basal level of Gfrp expression in liver.
The role of GFRP in the regulation of BH4 biosynthesis in vivo is not known and the in vitro data are somewhat controversial. Numerous studies have contributed strong evidence that GFRP inhibits GTPCH1 activity in an allosteric manner resulting in reduced BH4 biosynthesis (22,31,32). Moreover, several in vitro studies have revealed that GFRP overexpression significantly decreases BH4 biosynthesis, while lowering GFRP expression with lipopolysaccharide increases BH4 level in endothelial cells (17,22). While these data are consistent with the notion that GFRP negatively modulates BH4 biosynthesis, other investigators report no role for GFRP in the regulation of BH4 biosynthesis (27). We examined the role of in vivo Gfrp overexpression on the de novo BH4 biosynthesis and observed a reduction in tissue BH4 levels in Gfrp overexpressing transgenic mice. The GFRP-mediated BH4 downregulation could be due to increased interaction of GFRP with GTPCH1 in transgenic mice. Indeed, our pull-down assay confirmed increased interaction of GFRP with GTPCH1 in different tissues of Gfrp+/Cre+ mice compared with control littermates.
Maintenance of BH4 homeostasis is critical for the preservation of cellular redox balance (26). Decreased BH4 bioavailability results in NOS uncoupling, leading to generation of more superoxide compared with NO, which in turn oxidizes BH4 to the catalytically incompetent BH2. This results in an imbalance in cellular redox homeostasis. The current study revealed reduced levels of BH4 and increased oxidative stress in Gfrp+/Cre+ mice, consistent with a role for BH4 in reducing oxidative stress. Moreover, oxidative and/or nitrosative stress have a profound effect on mitochondrial function (8). Assessing various aspects of mitochondrial function, we observed increased basal, ATP-linked, and proton-leaked OCR in primary thymocytes from Gfrp+/Cre+ mice, suggesting elevated oxidative stress in these animals compared with control littermates. Consistent with our observation, Dranka et al. (2011) also found that basal, ATP-linked and proton-leaked OCR was higher in neonatal rat ventricular astrocytes after hydrogen peroxide-induced oxidative stress (7). Moreover, in our study, we observed increased maximal OCR and reserve capacity in transgenic mice, suggesting that the mitochondria in thymocytes obtained from Gfrp+/Cre+ mice still maintain their membrane potential and might be responding to a stress-induced increase in energy demand. Notably, these changes occur without an increase in non-mitochondrial OCR, indicating that non-mitochondrial respiration is negligible in both groups of animals.
Oxidative stress, including radiation-induced oxidative stress, significantly alters GTPCH1 and GFRP expression resulting in alteration in BH4 bioavailability (14,16,17,22,29). We have recently shown that radiation causes a temporary, significant decrease in lung BH4 level (3), but the role of GFRP in this regard is unknown. The present study revealed a time-dependent increase in Gfrp mRNA expression in irradiated control mice, which may repress GTPCH1 activity and result in a relative BH4 deficiency. Like radiation-induced oxidative stress, oxidative stress induced by hydrogen peroxide has also been shown to induce Gfrp mRNA expression in a time-dependent manner in human endothelial cells (17). These in vivo and in vitro results clearly indicate that Gfrp expression is susceptible to oxidant signaling and that it negatively impacts BH4 bioavailability. On the other hand, Ishi et al. (16) reported decreased GFRP expression after oxidative stress induced by hydrogen peroxide in endothelial cells. The reason for these contradictory results may be the use of different cell types and different concentrations of hydrogen peroxide by the two groups.
Irradiation causes immediate production of ROS, with formation of superoxide being particularly detrimental because of its high reactive potential (24). Superoxide may readily react with available cellular NO, or NO produced after irradiation, to form the powerful toxic oxidant peroxynitrite. Recently, we showed that increased vascular peroxynitrite formation in irradiated CD2F1 mice was significantly reduced by gamma tocotrienol treatment, and by BH4 supplementation (3). Peroxynitrite formed after radiation exposure or radiation-induced ROS can oxidize BH4 to form BH2 resulting in decreased cellular BH4/BH2 ratio, which is critical for NOS uncoupling. Our data revealed significantly lower BH4/BH2 ratio and increased formation of peroxynitrite in irradiated Gfrp+/Cre+ mice compared with control littermates, consistent with the notion that irradiation induces more oxidative damage in transgenic mice.
Irradiation, by affecting the bone marrow, has a profound effect on the levels of circulating blood cells. It has been reported that WBC are more susceptible to radiation in mutant mouse models of oxidative stress. For example, p53 knockout mice exhibit a marked decrease in WBC counts compared with control mice after exposure to sublethal dose of IR (30). Similarly, we also observed a significant decrease in WBC counts in Gfrp overexpressing transgenic mice.
The profound alteration of mitochondrial activity caused by GFRP overexpression was a striking finding. The most likely explanation is that GFRP overexpression alters mitochondrial bioenergetics indirectly via redox processes and reduced levels of BH4. This is consistent with studies demonstrating that oxidative stress changes the mitochondrial bioenergetic profile (8) and that BH4 deficiency is linked to mitochondrial dysfunction (2). The first very simple explanation for why GFRP overexpression (accompanied by decreased BH4 bioavailability, NOS uncoupling, and decreased NO production) leads to changes in mitochondrial respiration is that NO reversibly binds to the oxygen-binding site of cytochrome oxidase (5). Hence, the amount of NO that competes with oxygen in complex IV is limited, thus likely causing an increase in mitochondrial respiration. Similar responses (increased oxygen consumption) was demonstrated when NO levels were decreased using L-NAME (a NOS inhibitor) in other studies (19). Another possibility to explain the altered mitochondrial function in GFRP overexpressing cells is a redox-mediated stress response. Hence, oxidative damage to proteins in the mitochondrial electron transport chain might lead to alterations in the rate of electron flux through electron transport chain and hence, changes in mitochondrial respiration.
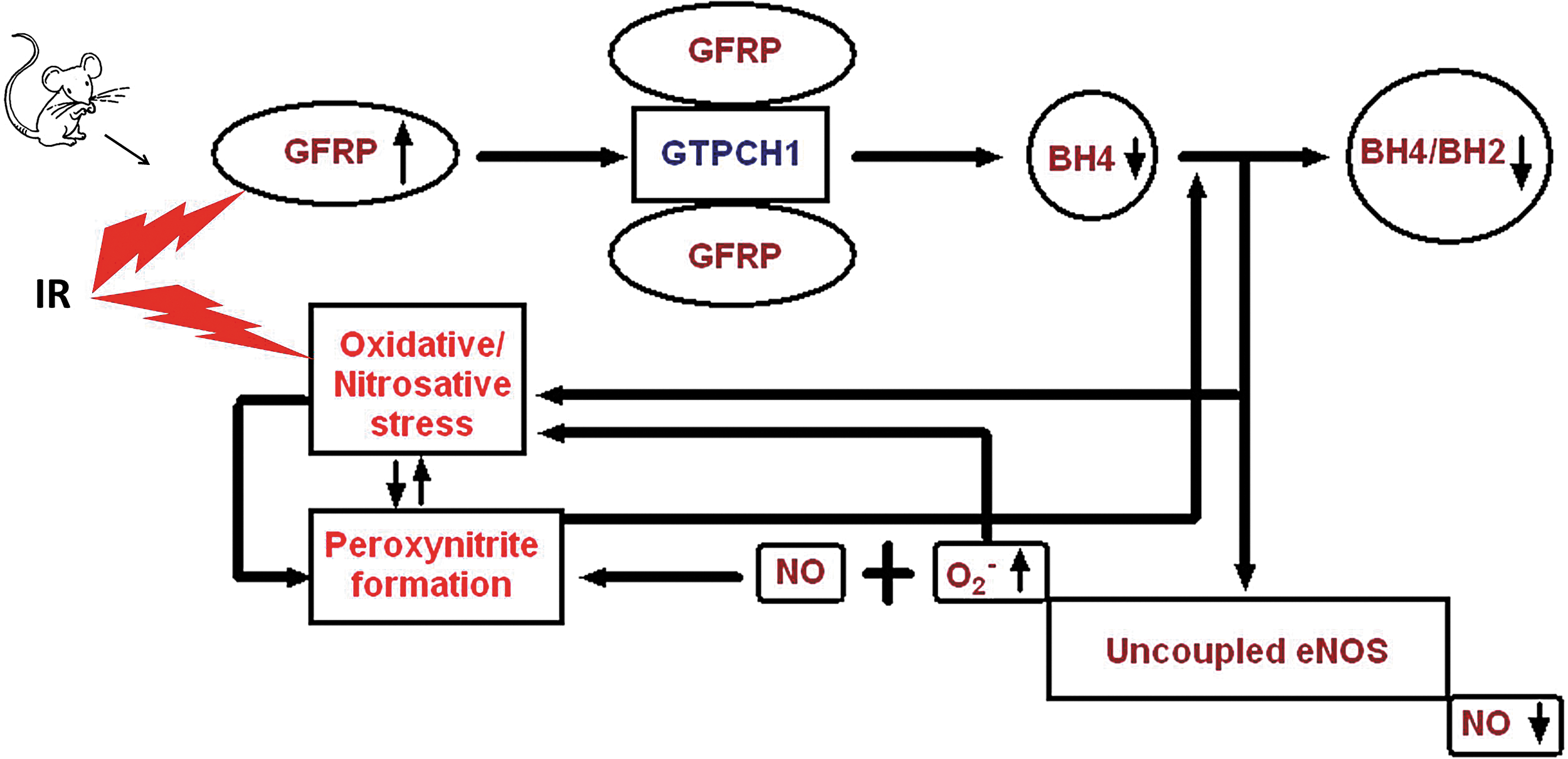
In conclusion, this is, to our knowledge, the first demonstration that in vivo Gfrp overexpression increases the interaction of GFRP with GTPCH1 to negatively regulate BH4 biosynthesis and increase the level of oxidative/nitrosative stress induced by IR. A schema summarizing the findings from the present study is shown in Figure 7. Further research is required to investigate the exact relationships among in vivo GFRP overexpression, NOS uncoupling, and oxidative/nitrosative stress. The novel Gfrp+/Cre+ transgenic mouse model presented here is a useful new tool for studies into the role of BH4 in health and disease and, because of the Cre-Lox technology used, provides opportunities for tissue-specific studies.
Materials and Methods
Animals
Male mice with an average body weight of 23–26 g were used. The experimental protocols were reviewed and approved by the Central Arkansas Veterans Healthcare System Institutional Animal Care and Use Committee (IACUC) and by the IACUC at the University of Arkansas for Medical Sciences.
C57BL/6J (stock No. 000664) and EIIa Cre (stock No. 003724) mice were obtained from The Jackson Laboratory. The Gfrp transgenic mice were developed on a C57BL/6 background in the Transgenic Core Facility of University of Arkansas for Medical Sciences.
Plasmid construction for transgenic mice
Briefly, total cDNA was generated from mRNA of CD2F1 bone marrow cells by RT-PCR using the oligo dT primers (Universal Riboclone cDNA synthesis system; Promega). The Gfrp gene specific primers (pQFLNL-gfrp forward primer sequence 5′-GAATTCCAGCCACGCACCATGCCCTATC-3′ and pQFLNL -gfrp reverse primer sequence 5′-GCGGCCGC GCTCAAGCAGCTCATTC-3′) were used to amplify a 300 bp fragment to incorporate EcoR I and Not I sites at 5′ and 3′ ends respectively from the total cDNA. This product was gel purified, subcloned in vector, and verified by sequencing. The GFP gene in pCALNL-GFP was replaced with the 0.3 kb Gfrp cDNA by digesting with Bcl I/BstB I. In the pCALNL-Gfrp construct, the Gfrp cDNA was flanked by loxP sites on either sides of a stop codon, downstream of a CAG enhancer in the pCALNL vector (Addgene). Cre-mediated excision of the stop codon will allow for the expression of the mouse Gfrp transgene. The resulting plasmid, pQFLNL-Gfrp was again confirmed by DNA sequencing using standard techniques and used to create the Gfrp transgenic founder lines.
Breeding strategy
Gfrp positive male founders were crossed with female C57BL/6J mice to maintain the founder lines. To generate Gfrp overexpressing transgenic animals (Gfrp+/Cre+), founders were crossed with EIIa Cre females and pups were genotyped to determine the presence of the Gfrp transgene. The littermates that had not received the Gfrp transgene after crossing with EIIa Cre were used as control mice (Gfrp−/Cre+). All experiments were carried out with the mice obtained in first filial generation. The Cre was expressed in all the first filial generation mice, irrespective of their Gfrp status.
Genotyping
The presence of the Gfrp transgene was determined with PCR on tail DNA. Mouse tail was lysed in DirectPCR (Tail) lysis reagent (Viagen Biotech) with Proteinase-K (Applied Biosystems) and incubated overnight at 55°C. Two microliters of lysate was used for PCR.
The forward primer (5′GTTATCTCGAGTCGCTCGGTACG 3′) in the vector construct and the reverse primer (5′CATGGTGGGGCCCACCTCCATAC 3′) in the transgene were designed to particularly amplify transgene. Gfrp PCR amplification was performed in a 25 μl reaction mixture containing 1× Green Go Taq reaction buffer (Promega), 1.25 mM magnesium chloride (MgCl2; Invitrogen), 200 μM deoxynucleoside triphosphate (Thermo Scientific), and 0.04 U of Go Taq DNA polymerase (Promega). The thermal conditions for Gfrp PCRs were as follows: initial denaturation for 5 min at 94°C, followed by 30 cycles of 94°C for 1 min, 58°C for 45 s, and 72°C for 1 min, and finally 1 extension cycle of 10 min at 72°C.
The concentration of MgCl2 was 1.5 mM for Cre PCR. The forward primer sequence was 5′GCG GTC TGG CAG TAA AAA CTA TC 3′, while the reverse primer sequence was 5′GTG AAA CAG CAT TGC TGT CAC TT 3′. The Cre amplification parameters were as follows: after an initial denaturation at 94°C for 3 min, amplification was performed for 35 cycles at 94°C for 30 s, 51°C for 1 min, and 72°C for 1 min, followed by a final extension step at 72°C for 2 min. All the primers were obtained from Integrated DNA Technologies, Inc.
Isolation of genomic DNA from mouse tails
DNA was extracted from mouse tail biopsies using a DNeasy Blood & Tissue Kit (Qiagen) as per manufacturer's protocol. Genomic DNA samples were quantified by NanoDrop (Thermo Scientific).
Custom-made TaqMan-based Copy Number Assay
Custom plus TaqMan® Copy Number Assay was designed using online software (Applied Biosystems) to determine copy number as previously described (15). The amplicon was designed spanning the vector construct and the transgene to avoid endogenous Gfrp amplification. Each PCR mix contained 10 μl of 2×TaqMan® Universal Master Mix, 1 μl of the TaqMan Copy Number target assay (RP-GFRP), and 1 μl of the TaqMan Copy Number reference assay (Tfrc), which is known to exist only in two copies in a diploid genome, 4 μl of Nuclease-free water, and 4 μl of tail DNA (20 ng). All the reagents were procured from Applied Biosystems. The reactions were processed in an ABI 7500 Fast Real-Time PCR System. The copy number of transgene was calculated using the standard curve method as previously described (15).
RNA extraction and qRT-PCR
Total RNA was purified from frozen tissue using RNeasy Plus Mini Kit (Qiagen) as instructed by manufacturer after homogenizing the samples in TRIzol® Reagent (Life Technologies). cDNA was synthesized using a cDNA reverse transcription kit (Applied Biosystems) after treating with RQ-DNase I (Promega). Predesigned TaqMan assay (Applied Biosystems) for mouse gene: Gfrp, Mm00622819_m1; Gtpch1, Mm01322973_m1; and 18s rRNA, Hs99999901_s1 was used. The mRNA levels were normalized to eukaryotic 18s rRNA and calculated relative to control mice, using the standard ΔΔ Ct method.
Western blotting analysis
Standard method was followed as previously described (1). GFRP (Proteintech), GTPCH1 (Santa Cruz Biotechnology, Inc.), and β-actin (Cell Signaling Technology) was used at 1:1000 ratio, while secondary antibody was used at a ratio of 1:20,000.
Immunohistochemistry
Immunohistochemical staining for GFRP was performed with the same antibody used for western blotting as described elsewhere (25). Images were captured at 20× magnification.
BH4 and BH2 estimation
Frozen tissue was homogenized for 1 min in 500 μl of homogenizing buffer, consisted of Tris-HCl (pH 7.3), 10 mM dithiothreitol, 1 mM ethylenediaminetetraacetic acid (EDTA) solution, and 166 ng/ml internal standard mix [(5-15N)-biopterin, (5-15N)-dihydrobiopterin, and (5-15N)-tetrahydrobiopterin]. Biopterins were purchased from Schircks Laboratories. The homogenate was centrifuged at 14,000g for 5 min at 4°C. An aliquot (250 μl) of supernatant was added to 100 μl of a mixture containing ascorbic acid (5 mM), gallic acid (2.5 mM), hydroquinone (2.5 mM), EDTA (1.0 mM), and epigallocatechin gallate (1.0 mM) and gently mixed. Perchloric acid (38 μl) was added and incubated on ice for 5 min; the samples were then centrifuged. The supernatant (250 μl) was added to 25 μl of a 2.5 M potassium bicarbonate and mixed. Samples were placed on ice for 10 min and then centrifuged. The supernatant (240 μl) was added to 30 μl of an ammonium formate buffer (500 mM, pH 2.7). The samples were transferred to autosampler tubes and analyzed by LC-MS/MS system.
Extracted biopterins were separated on a Betabasic 3 μm C8 analytical column (150 mm×2.1 mm), which was maintained at 45°C. The linear binary gradient consisted of solvent A; 5 mM ammonium formate buffer (pH 2.7), 0.1% perfluoroheptanoic acid, and 10% acetonitrile and solvent B; 99% acetonitrile, 5 mM ammonium formate buffer (pH 2.7), and 0.1% perfluoroheptanoic acid. The sample (8 μl) was injected onto a 20-μl loop. The initial flow rate and %B was 0.3 ml/min and 10%. The %B was increased over the next 4 min to 30%. From 4 to 4.5 min the gradient was increased to 90% B and the flow rate increased to 0.5 ml/min. The gradient was held at 90% for 2 min and then returned to initial conditions (6.5–7.0 min). The total run time was 10 min.
Positive ions were generated using electrospray ionization at a capillary voltage of 2.5 kV. The cone voltage (35 V) was adjusted to optimize the precursor ion for each compound. The mass analyzer, a Quattro Premier triple quadrupole (Waters) was operated in multiple-reaction-monitoring mode using argon at a pressure of 3.9×10−3 bar. Biopterin, (5-15N)-biopterin, dihydrobiopterin, (5-15N)-dihydrobiopterin, tetrahydrobiopterin, and (5-15N)-tetrahydrobiopterin, were detected using transitions 237.9>219.9, 238.9>220.9, 239.9>167.9, 240.9>165.9, 241.9>165.9, 243.3>167.0, respectively.
IP assay
Frozen lung and liver tissue was rapidly homogenized in 500 μl buffer (50 mM Tris-HCl, 150 mM sodium chloride, 1 mM ethyleneglycoltetraacetic acid (EGTA), 1 mM EDTA, and 1%Triton×100). One milligram of protein was incubated with 5 μg antibody (GCH-1 or GFRP) or 5 μg of normal IgG for 1 h on a rotary shaker at 4°C. After incubation, protein G-magnetic beads (Life Technologies) were added for an additional hour, and then washed with fresh buffer, and the immunoprecipitates were eluted, boiled for 5 min in laemmli sodium dodecyl sulfate (SDS) sample buffer, and frozen until used for western blotting.
GSH-GSSG measurement
Blood samples were collected from the retro-orbital plexus, immediately lysed in 5% sulfosalicylic acid and centrifuged. The 5,5′-dithiobis-2-nitrobenzoic acid recycling assay was performed to measure GSH and GSSG levels in the supernatants, as described previously (12,23). The data were normalized to protein content, determined by the bicinchoninic acid (BCA) protein assay, as per the manufacturer's instructions (Pierce Biotechnology).
Measurement of mitochondrial function in primary thymocytes using the XF96-extracellular flux analyzer
OCR was measured at 37°C using an XF96 extracellular analyzer (Seahorse Bioscience) as previously described (10). Briefly, 175,000 freshly isolated thymocytes per well were plated in CellTak-coated plates, changed to unbuffered Dulbecco's Modified Eagle Medium (DMEM) supplemented with 4 mM glutamate and incubated in a non-CO2 incubator for 1 h at 37°C. Four baseline measurements were acquired before injection of mitochondrial inhibitors or uncouplers. Readings were taken after sequential addition of oligomycin (4 μM), FCCP (4 μM), and rotenone/antimycin A (10 μM). OCR was calculated by the Seahorse XF-96 software and represents an average of 40–80 measurements on four different days (10–20 wells per mouse per day). The OCR was normalized to cell number.
Irradiation
TBI was performed as described before (4). The average dose rate was 1.21 Gy/min.
Blood cell counts
At 24 h and 3.5 day after exposure to 8.5 Gy of TBI, whole blood was collected into EDTA-coated tubes (Fisher Scientific). Mouse peripheral blood cell counts were obtained using a veterinary hemocytometer (Hematrue System; Heska Corporation) according to the manufacturer's instructions.
Peroxynitrite assay
Vascular peroxynitrite production was measured at 3.5 day after 8.5 Gy of TBI as described earlier (3). Protein concentration in the supernatant was measured using BCA protein assay kit. Fluorescence was expressed per mg protein.
Statistical analysis
Results were expressed as mean±standard error of mean. Data were analyzed using Prism software (version 4.0; GraphPad). Comparison among multiple means was performed by analysis of variance and pairwise comparison of means with the Student's t-test. A two-sided value of p<0.05 was considered statistically significant.
Footnotes
Acknowledgments
We would like to thank Dr. Charles A. O'Brien of the University of Arkansas for Medical Sciences Transgenic Mouse Facility for helping in generation of Gfrp overexpressing transgenic mice. Financial support was received from the National Institutes of Health (AI67798 and CA71382) and the Veterans Administration.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.