
Abstract
Introduction
Alzheimer Diagnosis and Staging
AD is clinically characterized by a decline in episodic memory that is often mistaken for normal cognitive deficiencies due to aging. Because the pathology remains hidden within the brain tissue, clinical diagnosis during the early stages remains inherently error prone and subjective, though advances have been made to aid a physician in making a correct diagnosis (100,167). To date, physicians have access to tools designed to help with diagnosis such as the Mini Mental State Evaluation (MMSE), which is used to track a patient's cognitive prowess (a 30-point scale is used with >25 being normal and <25 as probable AD), and also imaging alternatives such as magnetic resonance imaging (MRI) and positron emission tomography (PET) scans, which visualize both the potential hippocampal, sulci, and gyri degeneration and decreased glucose utilization (42,48,71,150).
Progression of typical AD can be stratified into four main stages: preclinical AD (PCAD), mild cognitive impairment (MCI), early AD (EAD), and late-stage AD (LAD). PCAD is defined as the potential stage of AD in which the patient presents as a fully functional individual in cognitive exams such as MMSE, yet the growing pathology within the brain tissue is present, but likely unknown precluding early death from a non-neurodegenerative means (141,144,166). MCI has been described as being the transition stage between normal cognition and EAD, and is subdivided into both amnestic MCI (aMCI) and non-amnestic MCI, the former of the two presenting with memory deficits and maintains a 10%–15% conversion rate per year to AD (123,124,150). Pathologically, each stage differs in that both amyloid plaques and NFTs increase in distribution and density from MCI to LAD, though non-demented individuals have also been known to possess both types of pathology while maintaining normal cognition (159). Through use of imaging techniques such as MRI, all stages of clinical AD described demonstrate varying degrees of degeneration, with MCI presenting relatively small degeneration affecting the hippocampus, sulci, and gyri, whereas a larger degree affects the same brain regions in LAD accompanied by additional atrophy of the frontal lobe and ventricular widening in EAD and LAD (45,47,71). Additionally, research conducted using PET scans concluded that regional glucose utilization within the brain, including the temporal lobe, was significantly reduced in AD patients, a trend that was shown to remain for possible PCAD and MCI patients, indicating a severe energy deficiency, as glucose is known to be the predominant source of energy for the brain (34,35,42,66,125).
APP Processing
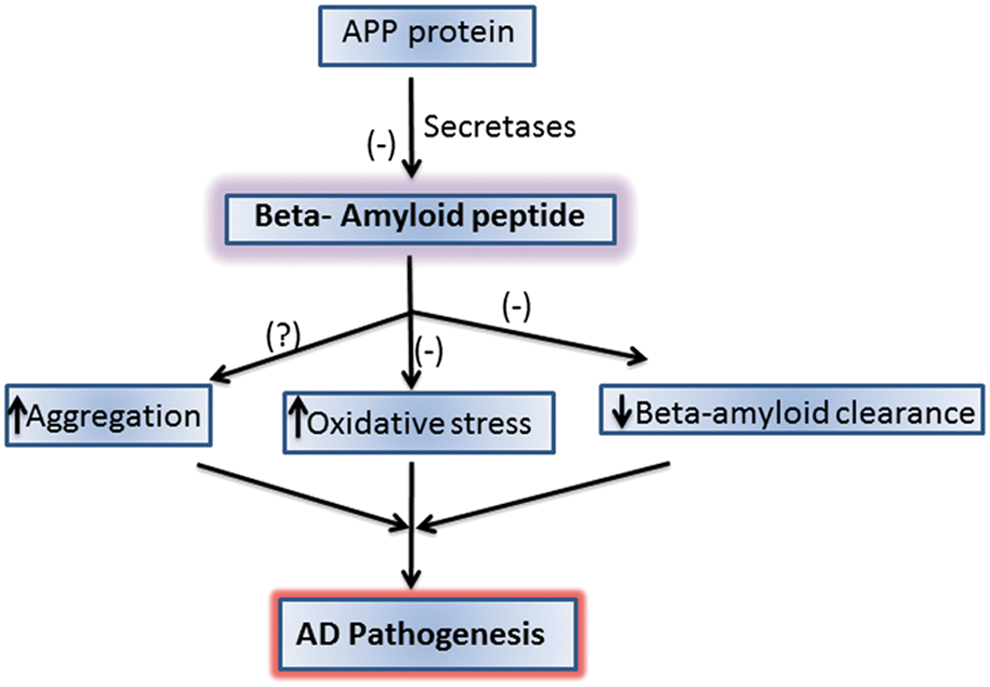
The main component of SPs is a 4-kDa protein called Aβ (61,99). Aβ is generated by the proteolytic cleavage of APP, a type I transmembrane protein suggested to play an important role in neurite outgrowth, neuronal protein trafficking, signal transduction, calcium metabolism, and others. (176). There are three major alternate splicing variants with APP770, APP751, and APP695.
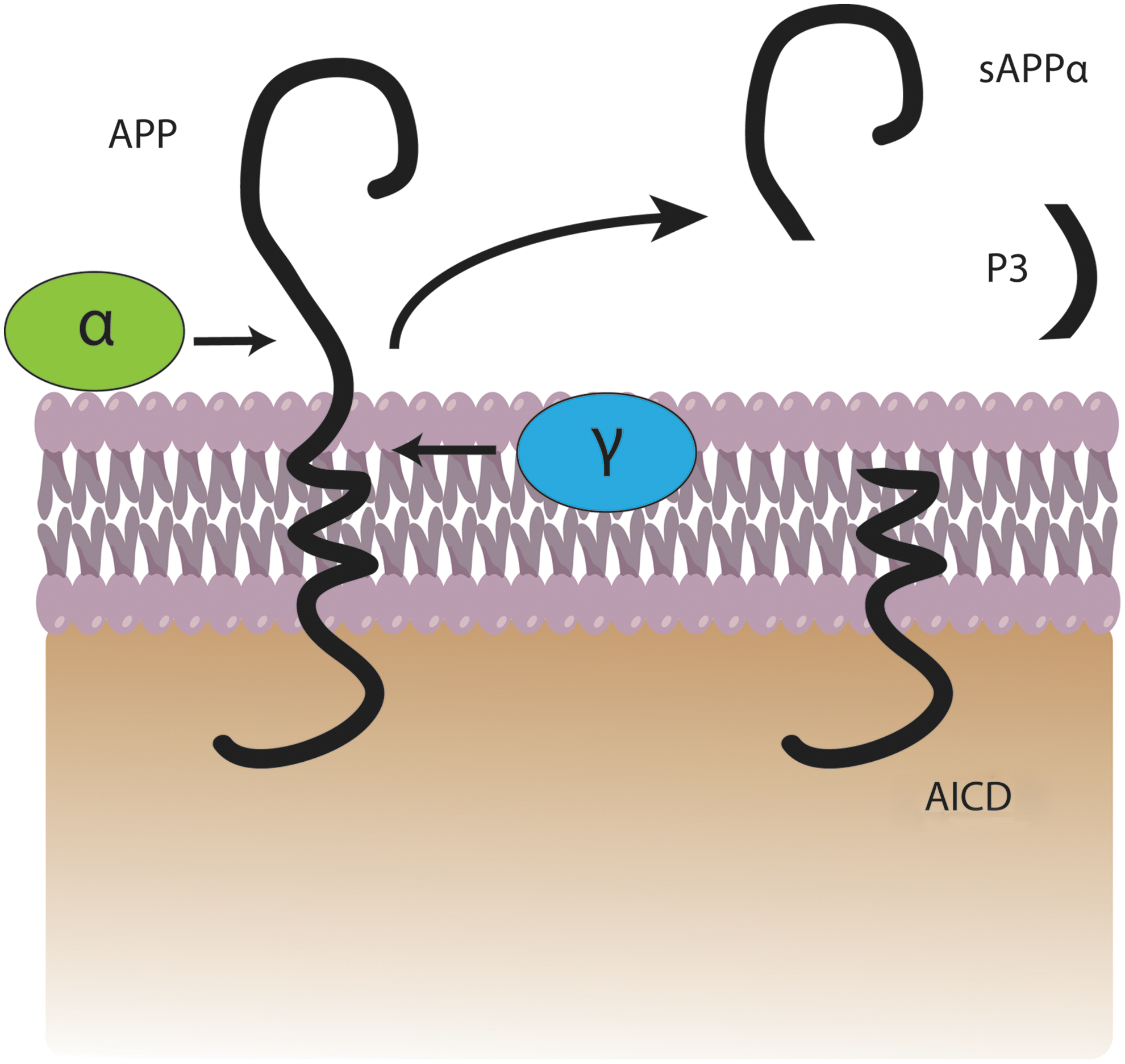
In the amyloidogenic pathway, APP is cleaved by β-secretase (also referred to as β-site APP-cleaving enzyme) (163) at position 671, resulting in the release of a large N-terminal derivative called β-secretase-cleaved soluble APP (β-sAPP) (Fig. 1). The β-sAPP differ from α-secretase-cleaved soluble APP (α-sAPP) (produced from non-amyloidogenic processing, Fig. 2) by lacking the Aβ(1 –16) regions at its C-terminus, but it has been reported to function as a death receptor 6 ligand and also mediate axonal pruning and neuronal cell death (114). The toxicity of C-terminal fragment (CTF) may possibly be mediated by the end products of γ- and/or caspase-cleavage including APP intracellular domain (AICD), C31, and Jcasp. Caspases can cleave APP at position Asp664 resulting in the formation of 31-amino-acid peptide of APP referred to as C31. C31 has been shown to induce cytotoxicity. Further, cleavage by γ-secretase generates JCasp (52,119); however, JCasp has been reported to play a minor role in cytotoxicity. In a transgenic mouse, mutation at caspase cleavage site in APP prevented AD-associated changes suggesting that caspase cleavage of APP might be crucial for Aβ-mediated neurotoxicity. In the next step, the 99-amino-acid CTF of APP (C99) is cleaved by the γ-secretase complex releasing free peptides ranging from 38 to 43 amino acids referred to as Aβ, P83 fragment, and AICD (Fig. 2). Hence, the γ- cleavage is critical for the amount and type of Aβ produced.

Aβ40 represents the most abundant form of Aβ in the brain, while Aβ42 shows a significant increase with certain forms of AD (112). Aβ42 has two extra hydrophobic amino acids compared to Aβ40, which promotes greater fibrillar formation in Aβ42 and is known to be more toxic (Fig. 3). The evidence of Aβ toxicity was provided by molecular pathology, human genetics, and discoveries from cell biology (13,16,38,169). The increased hydrophobicity of Aβ42 possibly allows this peptide to integrate within the lipid bilayer initiating the process of cell damage. Schmidt et al. using mass-per-length measurements and electron cryomicroscopy with 3-dimensional reconstruction on an Aβ(1 –42) amyloid fibril morphology showed that the Aβ(1 –42) fibril morphology has only one protofilament, in contrast to Aβ(1 –40) fibril forms two protofilaments. Further, Aβ(1 –42) showed pairs of β-sheets at the cores of the two protofilaments making up a fibril (135).

Once Aβ is produced, individual amyloid peptides (Aβ42 in particular) aggregate to form small assemblies of dimers, trimers, oligomers, protofibrils, and large insoluble fibrils. Studies showed poor correlation between plaque load and cognitive function (113). Recently, the role of Aβ has been amended to suggest that soluble Aβ oligomers are the more toxic species. Further research has indicated that the soluble oligomers and not the plaques correlate well with cognitive decline (44,53,54,117,165,168). Moreover, Aβ levels and temporal NFT density have been shown to be elevated to a higher degree in LAD when compared with MCI and EAD, which are likewise elevated compared with control (9,11,58,108,159). The relationship between Aβ-containing SPs and NFT formation has been debated, but recently Jin et al. reported that with the addition of soluble Aβ dimers, tau became hyperphosphorylated before cytoarchitectural disruption was observed, followed by subsequent neuritic degeneration. Interestingly, this process was exacerbated with the overexpression of human tau and prevented with the knockdown of human tau (74). Soluble Aβ has also been shown to modulate the pro-survival PI3K/AKT-GSK3β pathway, inhibiting various neurotrophin effects including that of α-sAPP (73). These lines of evidence provide insight into the progression of AD and a potential causal relationship between two known pathological hallmarks of this disease.
Genetic Evidences for Aβ Toxicity
The importance of APP and consequently Aβ in AD pathogenesis has emanated from genetic evidence of patients with familial AD (FAD) and Down syndrome (DS). After the cloning of the APP gene, a mutation causing FAD (autosomal dominant) was found at codon 717, close to the C-terminus of the Aβ domain of APP (55). Today, there are at least seven known APP mutations causing FAD (56,138). Interestingly, all APP mutations are located in or near the Aβ region of APP, close to the secretase sites. To date, 32 mutations in APP have been reported, and based on their locations they are grouped into three main classes: the Swedish mutation, located adjacent to the β-cleavage site of APP; London mutations, Flemish mutation, located near the γ-site of APP; the Arctic, Dutch, and the Iowa mutations, located within the Aβ sequence itself. All the APP mutations are found to alter the proteolytic processing of APP, resulting in either increased production of total Aβ or a selective increase in the 42-amino-acid form of Aβ (56,138). In addition to mutations of APP, 177 mutations in presenilin 1 (PS1) (392 families) and 14 mutations in PS2 (23 families) has been identified in FAD, which further support the role of altered APP metabolism in AD pathogenesis. The evidence of involvement of Aβ(1 –42) in AD pathogenesis is largely derived by observation of increased Aβ load and increased oxidative stress in FAD. Individuals with FAD mutations consistently show increases in the ratio of Aβ42/40, suggesting that elevated levels of Aβ42 is critical for AD pathogenesis (72,134).
DS patients have three copies of chromosome 21, and the APP gene is present on this chromosome; hence, if these patients live long enough they develop neuropathological features indistinguishable from AD. Further, DS patients had increased accumulation of intracellular Aβ preceding extracellular plaque formation, and the level of intraneuronal Aβ decreases as the extracellular Aβ plaques accumulate (59,109). Further, DS brain also has elevated oxidative stress (32,78,120,121).
Aβ and ROS/RNS in AD
In AD brain, increased levels of Aβ were found in the affected regions; however, Aβ42 is also the predominating form of Aβ in SPs (106), while shorter Aβ proteins predominate in both vascular amyloid and in cerebral spinal fluid (CSF) (106,164). Further, AD CSF showed reduced levels of Aβ42 compared with Aβ40 suggesting that the deposition of the protein in SPs in brain leads to reduced levels of Aβ in the CSF. In AD plasma, the levels of Aβ is controversial, one study found an increase in plasma Aβ42 (102). Most of the studies did not find any change in plasma Aβ42 between AD patients and age-matched controls (70,106). Current data do not provide clear-cut evidence that Aβ protein in plasma/CSF reflects the amount of Aβ deposited in the brain or that plasma/CSF Aβ42 has a potential as a biomarker for AD. Further studies are needed to develop biomarkers for AD diagnosis and therapeutic efficacy.
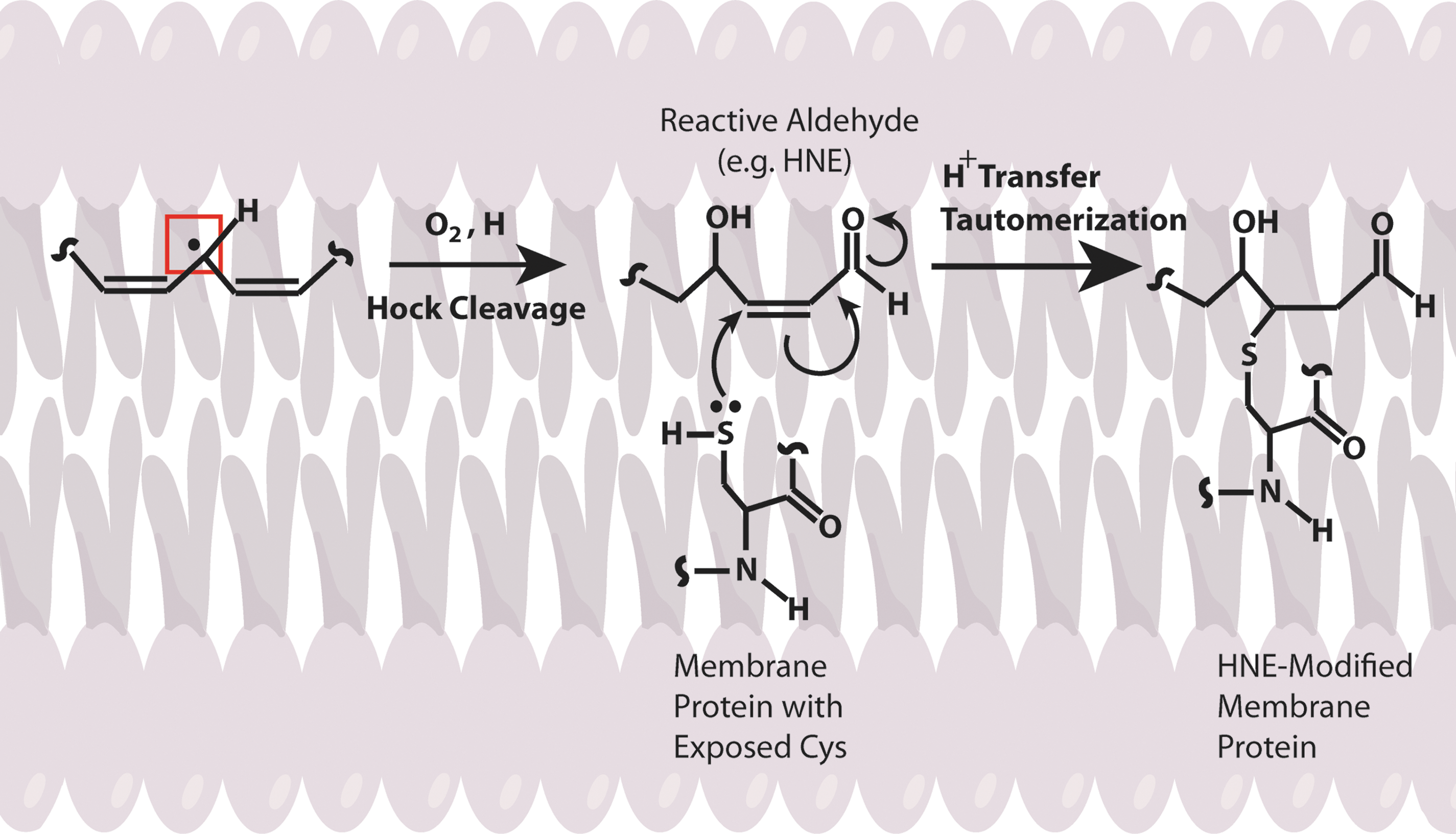
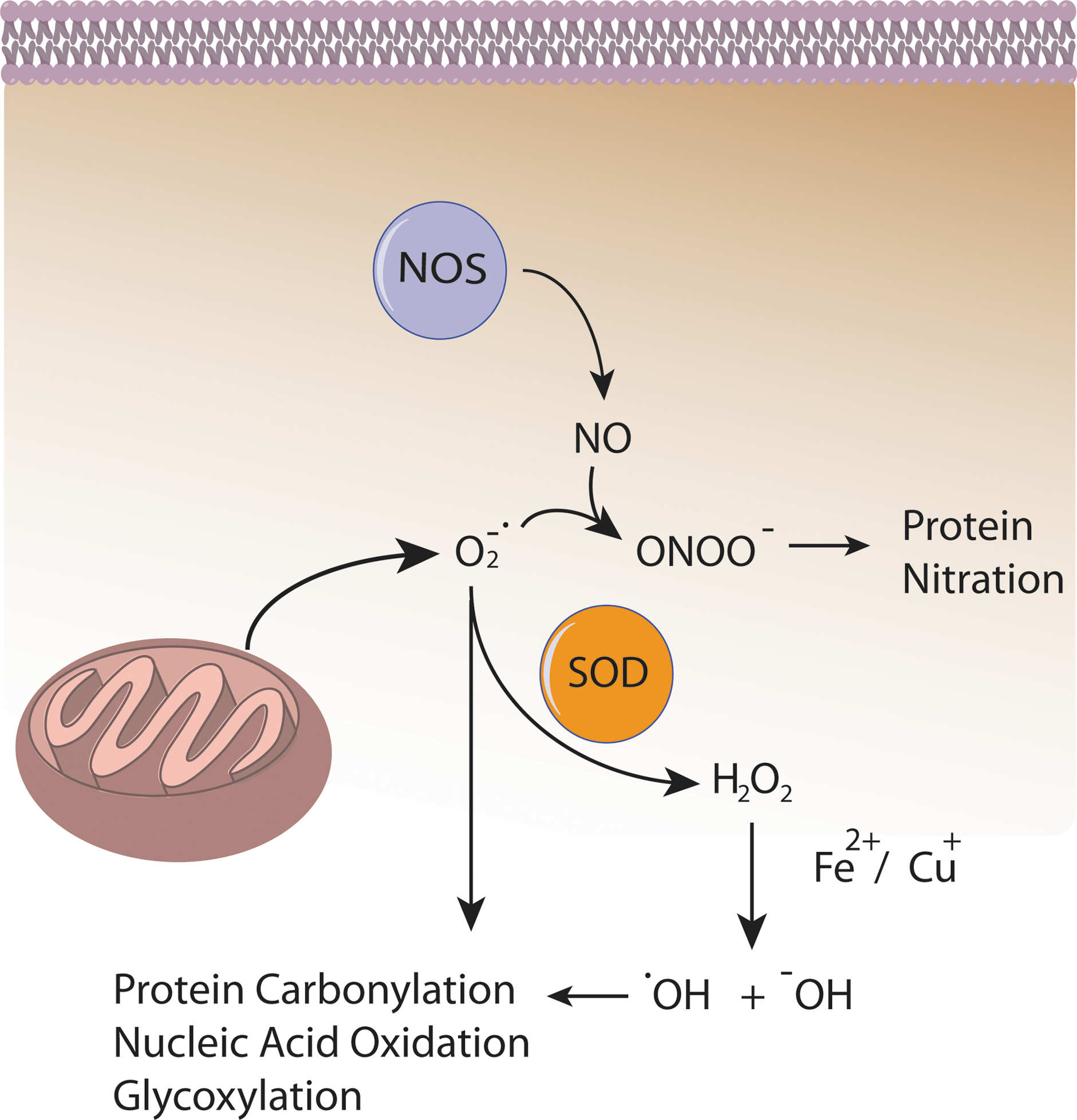
Several lines of evidence indicate that Aβ induces oxidative stress. Oxidative stress that occurs within the bilayer, hypothesized in the Aβ-induced oxidative stress hypothesis in which Aβ1–42 inserts as oligomers into the bilayer and serves as a source of ROS, has been shown to initiate lipid peroxidation (Figs. 4 and 5) (16,17,93,94,101). For a comprehensive review on oxidative/nitrosative stress in the cell, the reader is referred to the following articles (28,29,151).
Oxidative Stress at Different Stages of AD
Oxidative stress and its effects have been found as early as MCI in the progression toward AD. Studies conducted in our laboratory and others have found that oxidative stress markers for protein oxidation/nitration, such as protein carbonyls and 3-nitro-tyrosine, are elevated in brains from subjects with aMCI (6 –8,25,83). More recently, it has been shown that the phosphorylation profile of proteins such as heme-oxygenase-1 and biliverdin reductase A have been altered in MCI and AD indicating the possibility of aberrant signaling in at least this one critical antioxidant pathway. Increased levels of 8-OHdG, 8-OHG, 5-hydroxycytosine, 2,6-diamino-4-hydroxy-5-formamidopyrimidine, and 4,6-diamino-5-formamidopyrimidine, all markers of nucleic acid oxidation, were found in both mitochondrial DNA and nuclear DNA indicating nucleic acid oxidation in MCI (105,171). 8-OHdG (also found elevated in CSF of AD patients), 8-OHA, and 5-OHU were found in AD brain regions demonstrating that though DNA is more protected from oxidation than RNA, oxidation still occurs (2,50,104,150).
Significant RNA oxidation has been shown to exist in AD, as has been found in the earlier stages of the disease. A high percentage (30%–70%) of mRNA in the frontal cortex was shown to be oxidized in AD brain (139). In EAD brain, 8-OHG was found to be elevated in the cytoplasm of AD hippocampus, frontal, and occipital neocortex, which correlated with the β-amyloid load (89,115,116,140). Ribosomal RNA oxidation was observed in the superior middle gyri and inferior parietal lobule (IPL) of AD brain (43). 8-OHG levels decreased with increased Aβ and NFT levels, a finding that suggests that at the early stages of AD, oxidative damage to RNA may be an early event in AD progression (115).
Increased protein-bound 4-hydroxy-nonenal (HNE) and free HNE, TBARS, and MDA were found, and a higher isoprostane (F2isoP) level in plasma, urine, and CSF in MCI when compared with healthy controls (83,96,173). There have been high levels of free and protein-bound HNE found in AD brain (16,23,86,88,97,103,122). In addition to lipid peroxidation, protein carbonyls were found to be increased in regions of the brain heavily associated with AD, including the hippocampus and parietal cortex, while leaving the cerebellum relatively untouched (64). Moreover, another index of protein oxidation, protein nitration, was also found to be increased in the CSF and AD brain in regions such as the IPL, neocortical regions, and the hippocampus (7,31,65,143,155). Increased protein nitration and protein-bound HNE were found in brains of subjects with EAD (130,131). Inversely correlated to the increase in oxidation observed was the activity of antioxidant systems (both enzymatic and nonenzymatic) found by Sultana et al. and Guidi et al. while no changes in total protein levels were observed, which may be a result of, and contribute to, the observed increase in free radicals during the progression of AD (57,153).
Redox Proteomics Studies of MCI, EAD, and AD
Redox proteomics is a method of identification of oxidatively modified proteins pioneered by our laboratory that employs redox-specific antibodies, two-dimensional polyacrylamide gel electrophoresis, and tandem-mass spectrometry (MS/MS) with the identification of specific proteins based on their tryptic peptide amino acid sequence after interrogation of protein databases such as SwissProt (41,67,68). Our laboratory has identified proteins in MCI, EAD, and AD brain that are vital to cellular function as being oxidatively modified and dysfunctional; however, for the sake of this review only a select few will be discussed. For a discussion of oxidatively modified proteins discovered by our laboratory using redox proteomics, the reader is referred to articles cited here (4,24,27,41,150).
Sultana et al. found that the important protein regulator Pin1 is oxidized and activity decreased (148). Recently, there has been much interest in the area of regulation via the phosphorylation specific peptidyl-prolyl cis-trans isomerase (PPIase), Pin1, and its role in neuronal cell cycle checkpoints and cellular phosphorylation status in diseases such as AD and cancer (5,14,46,82). Pin1 recognizes the specific motif of phosphorylated serine or threonine on the amino-terminal side of an adjacent proline (pSer/Thr-Pro) and catalyzes the isomerization of the peptide bond (90,128). This regulation has been shown to be important in the phosphorylation status of both APP and Tau, and some kinases and phosphatases that act on those target proteins, giving Pin1 both a direct and indirect regulation of two key pathological hallmarks of AD (14,85,87,92).
Another link between the stages of AD is the presence of oxidatively modified proteins important to cellular energy production (150). Three enzymes, α-enolase, adenosine-triphosphate-synthase, and lactate dehydrogenase were implicated as being oxidatively modified in brains of subjects with aMCI and AD, while α-enolase in particular was found to modified in EAD as well (25,30,31,122,129,149,152,155,156). Additionally, enolase was identified by redox proteomics as oxidatively modified in brains of subjects with FAD (19).
The activity of α-enolase as a glycolytic protein is well understood. Consequently, the oxidative modification and subsequent loss of activity may significantly hinder energy production (150). α-Enolase however, possesses nonglycolytic activities in signaling pathways important to cell survival and in Aβ clearance (22). Evidence also suggests that α-enolase may be a neurotrophic factor, play a role in hypoxic stress regulation, and have transcription factor capabilities (1,62,147,158).
The examples of Pin1 and α-enolase were selected to demonstrate the power of redox proteomics in identifying specific links in cell signaling pathways that are damaged and dysfunctional as opposed to global tissue oxidation, and free radical induced oxidative stress of enzymatic proteins with multifunctional roles that may have far-reaching effects. In using redox proteomics, researchers may identify proteins that are more susceptible to oxidative modification and from this information garner insight regarding the progression and possibly even potential treatment for diseases such as AD.
Role of Methionine in Aβ-Induced Oxidative Stress
Studies from our laboratory and others showed Met-35 of Aβ peptides is critical for Aβ-associated toxicity and oxidative stress (15,160,174,175). Met can undergo two-electron oxidation to form methionine sulfoxide (MetSOx) (127,137). Oxidation of Met to the sulfoxide might play an important role in the regulation of protein function or cellular defense mechanism (145). Further, the presence of methionine sulfoxide reductase (MSR), which catalyzes the conversion of MetSOx to Met (91,98,146), suggests that MSR might play an antioxidant role. Interestingly, in AD brain the activity of MSR is less, and a significant fraction of SP-resident Aβ peptide has Met in the form of MetSOx (112), suggesting that Met oxidation might play an important role AD progression and pathogenesis (49). However, in vitro studies showed that Aβ with MetSOx is less toxic at a shorter incubation time (160), this could possibly be related to altered production of toxic Aβ oligomers (75,107).
In addition, Aβ-resident Met in the lipid bilayer can undergo one-electron oxidation forming sulfuranyl free radical [MetS+]. Since, Aβ is generated from cleavage of APP, a transmembrane protein as discussed above, we proposed that Aβ once produced can insert as small oligomers into the lipid bilayer adopting an α-helical conformation (9,175). According to the α-helix conformation rule of i+4 rule, that is, every fourth amino acid interacts; hence, the Met-35 S-atom would interact with carbonyl oxygen of Ile-31 (79,80,137) (Fig. 6). Since oxygen of Ile31 is more electronegative than sulfur it will pull the electron density toward it, making the S-atom in Met-35 more vulnerable to one-electron oxidation to form sulfuranyl free radical [MetS+] on Met (79,136,160) (Fig. 7). The substitution of Ile-31 by proline, an α-helix breaker, abrogates the oxidative stress and neurotoxicity associated with Aβ(1 –42) (80), suggesting that the secondary structure of Aβ(1 –42) contributes to reactivity of the neurotoxic peptide. However, until now the source of oxidant that triggers this event largely remains unknown. It is proposed that either molecular oxygen or Cu2+ might be key in the oxidation of Met to the sulfuranyl radical. In the absence of oxygen, Aβs cannot lead to free radical production (161). Prior studies showed that Aβ(1 –42) has Cu/Zn SOD-like properties (39), and that amyloid plaques had high levels of copper (33). In vitro studies showed that Aβ(1 –42) can promote the reduction of peptide-bound Cu2+ to Cu+ and form hydrogen peroxide (H2O2). Further Cu+, can react with the H2O2 to form highly reactive, hydroxyl free radicals (69,76). Further, chelation of copper by clioquinol (CQ, 5-chloro-7-iodoquinolin-8-ol), hydroxyquinoline antibiotic that has nanomolar affinity for Cu2+ (118), reduced the formation of H2O2 by Aβ (12,142,172). In vivo studies showed that oral administration of the clioquinol in Tg2576 mice reduced amyloid levels. Further, Phase 2 clinical trial showed that CQ slowed the rate of cognitive decline and reduced the plasma Aβ42 levels in moderately severe AD patients (132). The importance of copper in Aβ-induced toxicity is suggested by a study where Met35 was substituted by Val that showed to increase the toxicity (36), suggesting that this substitution might lead to a change in the conformation of Aβ from α-helix to a mixture of α-helical and β-sheet conformations, thereby increasing the binding of Cu+2 and subsequently its associated toxicity. Further, substitution of His 6,13,14 in Aβ(1 –42) by Tyr, which binds Cu2+ with less affinity than His, showed that it did not affect the oxidative stress and neurotoxicity further emphasizing the importance of Met-35 in the Aβ-induced toxicity and oxidative stress (10,160). Further research is needed to understand the role of copper in Aβ.

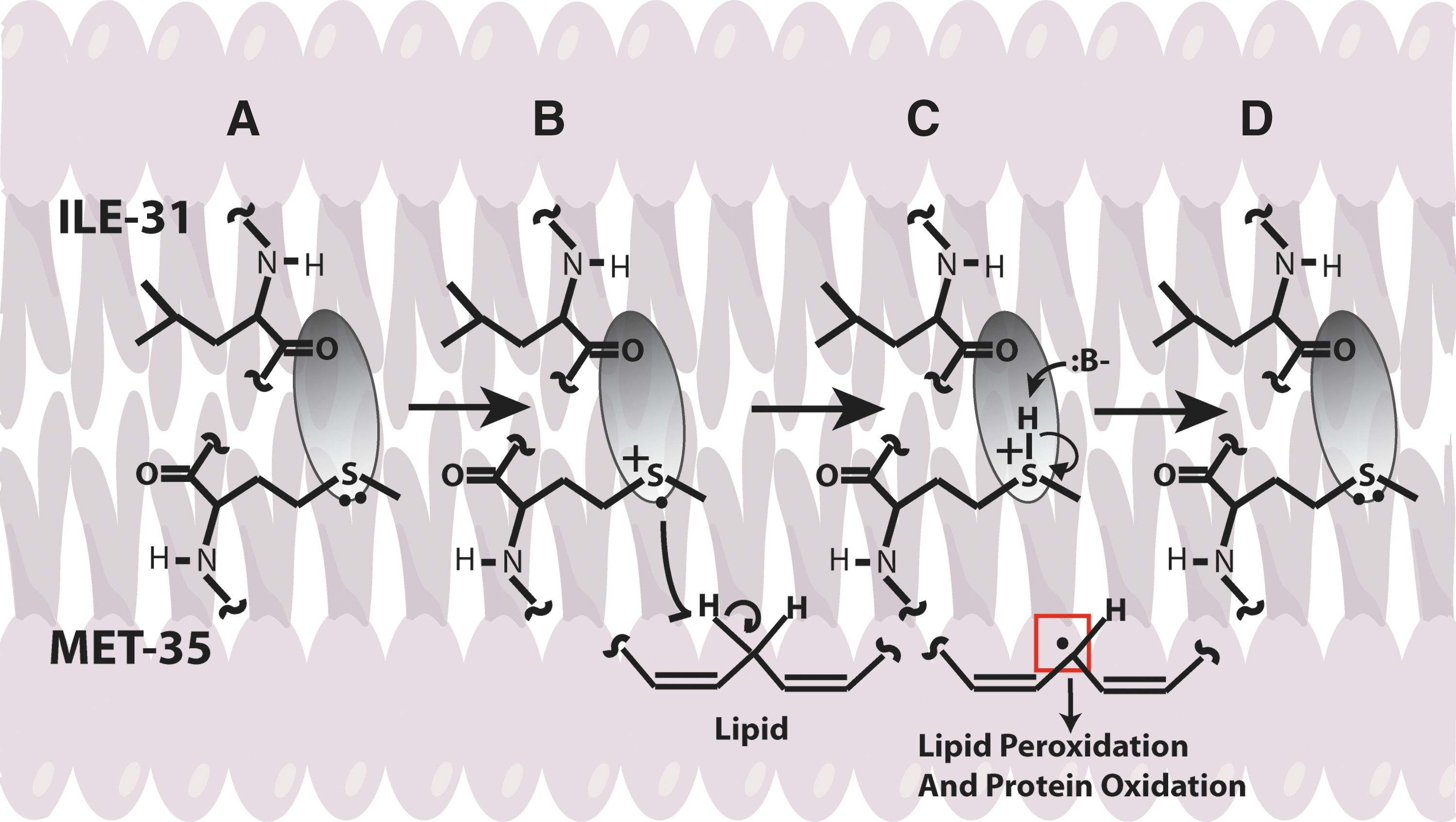
Once MetS+ radical is generated it can abstract allylic H atoms from the acyl chains of unsaturated fatty acids in the lipid bilayer to initiate the process of lipid peroxidation (60), and consequently affect the lipid bilayer. The products of oxidation further diffuse through the membrane affecting other cellular compartments, greatly amplifying the effect of the original Aβ-centered free radical, eventually leading to cell loss and AD. Consistent with this model, we substituted Gly at residue 37 of Aβ(1 –42) by aspartic acid. The effect of this negatively charged amino acid was to remove the Met-35 residue from the bilayer, and no oxidative stress was observed in neuronal cultures (93). Vitamin E, a chain-breaking antioxidant blocks the chain reaction in the mechanism of lipid peroxidation, preventing oxidative stress to neurons (80). However, clinical trials conducted using vitamin E for the most part did not show beneficial effects in AD, which could be due to experimental design (77).
The earliest study using transgenic Caenorhabditis elegans expressing human Aβ(1 –42) showed increased oxidation that correlated with the phenotypic expression (e.g., paralysis) of the worm (170,175), which was confirmed by others (44). However, when the Met-35 was substituted by Cys no oxidative stress was found, but the deposition of modified Aβ(1 –42) was not altered (175). Consistent with the role of Met, an in vitro study demonstrated that when the sulfur atom of methionine in Aβ(1 –42) was substituted by a methylene moiety [Aβ(1 –42)M35NLE] that has the same side chain length and hydrophobicity as Met (175), Aβ loses its associated free radical formation, oxidative stress, and toxicity (37,40,111). In contrast, some studies suggested that the 33–35 region of Aβ (25 –35) is critical for the aggregation and neurotoxic properties of Aβ peptide, but substitution of Met by norleucine did not reduce the toxicity associated with this peptide (126). However, the chemistry of C-terminal Met is entirely different than Met within the peptide chain.
A recent study from our laboratory used for the first time an in vivo mammalian model to show that Aβ-resident Met-35 is critical to oxidative stress and neurotoxicity (18). In this study the PDAPP mouse, with Swedish and Indiana familial mutations of APP, has a third mutation introduced: substitution of leucine in APP at M631, corresponding to Met-35 of Aβ(1 –42) (18). These mice were referred to as PDAPPM631L mice. In contrast, to the brain from PDAPP mice, which demonstrate oxidative stress, brain from PDAPPM631L mice showed no in vivo oxidative stress. In addition, punctate deposits of Aβ(1 –42) were found in the latter brain compared to frank amyloid deposits in the brain of PDAPP mice, suggesting that Aβ(1 –42)-resident Met not only affects in vivo oxidative stress but also affects plaque formation. Interestingly, Met substitution in Aβ(1 –42) did not rescue spatial learning and memory deficits at 6 months of age as assessed by the Morris water maze test. Given that APP is processed to produce toxic sAPPβ and other toxic fragments of APP, this result may not be surprising. Other, more sensitive cognitive tests are needed to better understand the effect of the loss of Met on learning and memory. Proteomics analysis on brain from PDAPPM631L mice showed reduced oxidation of key proteins that are critical in regulating cellular pathways such as energy metabolism, cellular defense, protein degradation, and pH regulation compared to PDAPP mice (133,157). The decreased oxidation in general and reduced oxidation of key proteins like Pin1 (Pin1, discussed earlier) might play an important role in preventing AD pathogenesis (157).
Conclusion
The overproduction and accumulation of Aβ are key to the progression and pathogenesis of AD. Hence, the use of treatments to reduce Aβ formation or the downstream oxidative stress associated with Aβ could be helpful in preventing, treating, or delaying the progression of AD (51). There are various approaches that could be potential candidates to reduce Aβ levels: inhibiting Aβ production (by inhibiting secretase enzymes) or increasing the clearance of Aβ or using compounds that bind Aβ to impair aggregation (Fig. 8). Dissolving the extant SP may not be a good idea to combat this devastating disease, since oligomeric Aβ, the likely main toxic species of this peptide would be elevated by simple equilibrium considerations. Studies are in progress in our laboratory and others to further delineate the mechanism of Aβ-associated toxicity and develop a regimen to treat, slow, or hopefully one day prevent AD.
Footnotes
Acknowledgment
This research was supported by NIH grants to D.A.B. [AG-05119].