
Abstract
Introduction
Eukaryotic organisms require extracellular signaling to survive. A variety of intracellular signaling pathways, including phosphatidylinsitol 3-kinase (PI3K)/Akt, Ras/mitogen-activated protein kinase (MAPK), and Jak/signal transducers and activators of transcription (STAT), mediate these extracellular signals. Binding of growth factors to their respective receptors results in the activation of a series of pathways resulting in a wide range of physiological events, including cell proliferation and cellular differentiation. Growth factor receptor activation results in complex patterns of downstream signaling events that are tightly regulated. These signal transduction pathways are rarely linear, with multiple pathways displaying cross-talk processes to produce highly dynamic signaling networks. In the previous decade, attempts to elucidate the intricacies of these networks have identified novel modes of signaling regulation. Redox regulation of receptor signaling is one such process.
Reactive oxygen species (ROS) were traditionally thought of as an unwanted byproduct of cellular respiration, but in recent years, there has been a renaissance of research into their role as mediators of intracellular signaling. Indeed, redox regulation of signaling events downstream of receptor activation has been recognized as critical for multiple cellular processes, and ROS such as hydrogen peroxide (H2O2) have been identified as second messengers in their own right [reviewed in (27, 28)]. Cellular H2O2 is generated in the mitochondrion, as a byproduct of oxidative phosphorylation at complex I or III in the electron transport chain (61). However, the major source of intracellular H2O2 that mediates signaling is generated by the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) family of enzymes, which have been associated with numerous growth factor receptors (see the ROS Generation for Signaling section). ROS regulate intracellular molecules typically through the oxidation of cysteine residues on proteins, which is known to alter protein function. Through such processes, ROS provide a critical link between extracellular growth factor signals and the complex intracellular networks that drive physiological processes such as proliferation, adhesion, migration, and survival. As an extension of this, redox signaling has implications in diseases such as cancer, degenerative disorder, and autoimmune diseases (see Fig. 1). A majority of cancer cell lines, for example, have demonstrated elevated endogenous levels of ROS (27). How this high level of ROS contributes to the tumorigenic phenotype remains elusive, however. Clearly, further study is needed to identify the specific processes involved in disparate redox-dependent diseases.
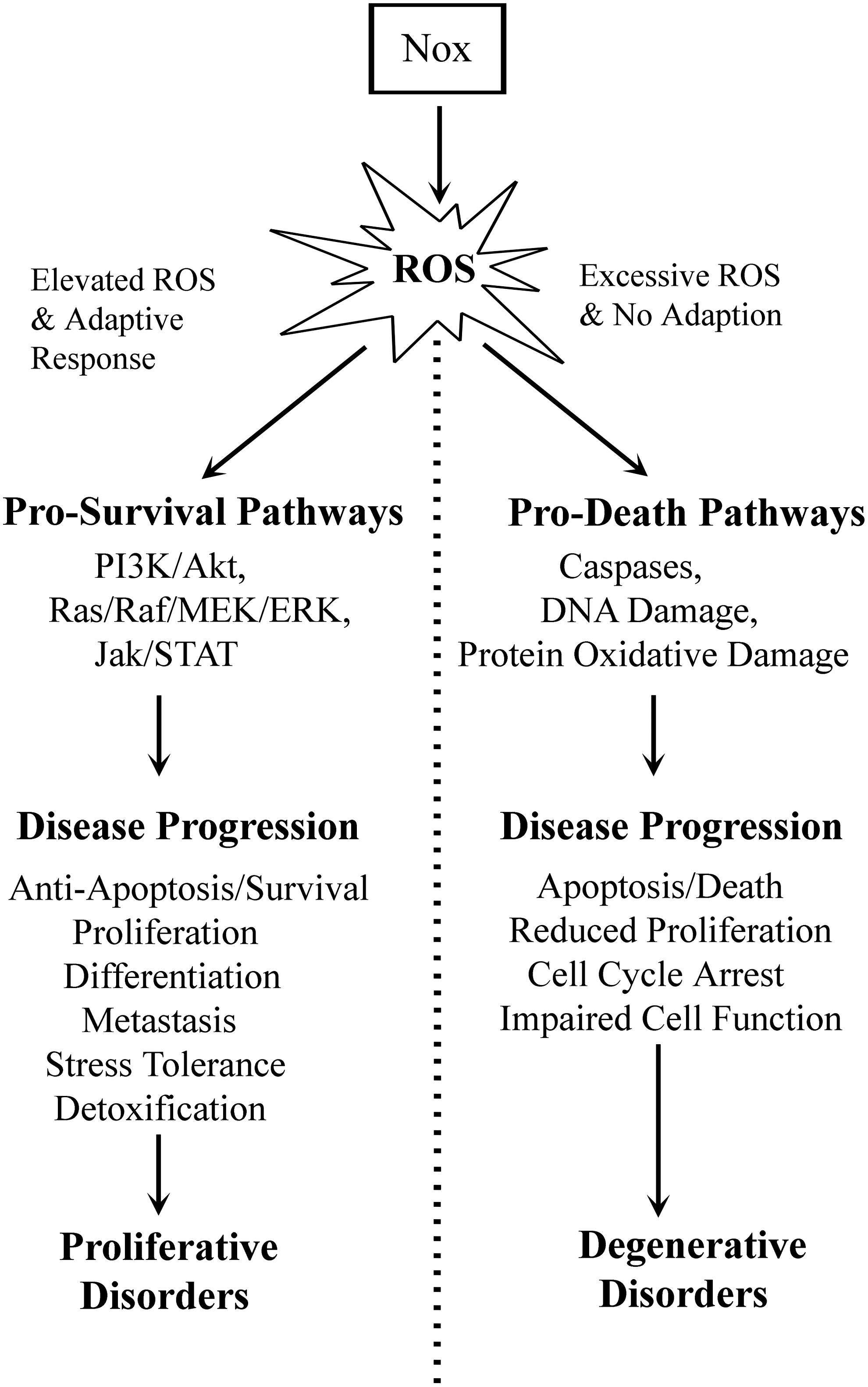
Overproduction of ROS can be accounted for by elevated expression and activity of Nox enzymes downstream from constitutively active growth factor receptors in a variety of tumors (27). Nox enzymes are inherent to processes synonymous with tumor phenotypes such as enhanced survival signaling, proliferation, angiogenesis, and metastasis owing to a novel oncogene–Nox relationship. Several oncogenes and constitutively active growth factor receptors are known to upregulate both Nox activity and expression, including Ras, Flt3, and BCR-ABL (42, 56, 74). The precise role that elevated ROS play in tumor development and progression, however, remains elusive. It is possible that elevated ROS facilitates increased prosurvival signaling downstream of oncogenic kinases via negative regulation of phosphatases. Increased cellular ROS may also drive the evolution of cancer cell populations, generating heterogeneous tumors through DNA damage. These proliferative disorders would arise when the cells experiencing higher than normal levels of ROS produce an adaptive response (see Fig. 1) to alleviate or offset any deleterious effects of the species. This may manifest itself via changes in expression of antioxidant genes or reduced sensitivity to apoptosis, for example. On the other hand, a failure of cells to adapt to changes in endogenous ROS levels has been seen as a significant cause of degenerative disorders. This occurs when cells cannot deal with excess ROS, which in turn perturbs normal cellular signaling and reduces the viability of the cells in question. Failure of cells to adapt to altered ROS signaling can play a role in diseases such as atherosclerosis, cardiovascular disease, inflammatory disease, or diabetes, and we will discuss these further throughout this review.
In this review, we will discuss the regulation of events downstream of growth factor receptors by ROS. We focus on the most recent advancements in the field and describe redox-dependent signal transduction in a hierarchical manner describing ROS generation, signal propagation, and downstream targets of receptor activation. We have specifically focused on those areas that have developed rapidly in recent years.
ROS Generation in Signaling
ROS are generated in cells when oxygen is metabolized, a process that can occur by both endogenous and exogenous stimuli. There is an ever-growing body of literature demonstrating that receptor-mediated signaling can result in ROS production, which in turn can influence cellular signaling pathways. In eukaryotic cells, ROS are produced by a variety of sources. However, in contrast to the majority of these sources where ROS are produced as byproducts, the Nox family of proteins' primary function is to generate ROS. There are seven members of the Nox family. Nox2 (originally named gp91phox) was the first identified member and has been extensively studied in host defense for its involvement in ROS generation in phagocytic cells. Since its identification, six additional homologs of Nox2 have been described in nonphagocytic cells (Nox1, Nox3, Nox4, Nox5, Duox1, and Duox2), with their activity being involved in various cellular events, including survival, growth, differentiation, apoptosis, and immune responses. In 1995, Sundaresan et al. published one of the first demonstrations of growth factor receptor-mediated signaling-generated H2O2, which acted as a secondary messenger necessary for signal transduction (67). Subsequent studies demonstrated this ROS production to be dependent on Nox and their regulatory proteins (5, 8, 59). Further evidence has, in the interim, established a significant role for Nox-derived ROS in mediating signal transduction upon stimulation of a variety of growth factor receptors. Ligation of a growth factor to its receptor stimulates tyrosine kinase activity and phosphorylation of the carboxyterminal region of the receptor, which subsequently acts as a binding site for cytosolic signaling proteins, activating pathways (e.g., PI3K/AKT, phospholipase Cγ (PLCγ)/PKC, and Grb2/Sos/Ras/ERK), initiating a signal transduction cascade. This cascade can induce downstream effects, including the generation of ROS via activation of Nox proteins. Although regulation of ROS production via Nox has been extensively reviewed (8, 28, 41), some recent advancements merit discussion.
There has been extensive study of the regulation of Nox in phagocytes. In this system, the enzyme consists of two membrane-bound subunits: the catalytic core Nox2 and p22phox (Fig. 2A). The p22phox protein is an integral partner for Nox2 as well as Nox1, Nox3, and Nox4, being essential for activity through stabilizing these proteins at the membrane (1). Activity of the enzyme is regulated by three cytosolic proteins, p47phox, p67phox, and p40phox, and the small Rho GTPase Rac2 (28, 41). After stimulation, p47phox undergoes a conformational change due to extensive phosphorylation at its autoinhibitory region mediated by the activity of various protein kinases (AKT, PKC, p38 MAPK, Erk, and PAK) (8, 33). This in turn facilitates translocation of the p47phox/p67phox/p40phox complex to the membrane where p47phox interacts with p22phox (28, 41). Concurrent activation then causes Rac2-GDP to be catalyzed to Rac2-GTP by guanine exchange factors, allowing Rac2-GTP to bind p67phox at the membrane resulting in the production of superoxide (

Regulation of platelet-derived growth factor (PDGF)-dependent H2O2 production demonstrated in smooth muscle cells gave one of the first insights into growth factor receptor-induced ROS production (67). PDGF-induced PKC phosphorylates p47phox, facilitating its translocation to the membrane, in turn activating Nox2 (4, 8). Further, PI3K activation and subsequent production of phospholipid phosphatidylinositol 3,4,5-triphosphate [PI(3,4,5)P3] downstream of the PDGF-receptor have been shown to be essential for Nox1-mediated H2O2 production in hepatocellular carcinoma cells (5). This is achieved through Rac1 activation, which results from PI(3,4,5)P3-induced phosphorylation and activation of the guanine exchange factor βPix (59). In more recent studies, the c-Src substrate proteins Tks4 and Tks5 have been shown to be functional members of a p47phox-related organizer superfamily (23). Src-mediated phosphorylation of Tks4 at Tyr508 and of NoxA1 at Tyr110 induces protein interaction via the SH3 domains of Tks4 and the proline-rich region of NoxA1, generating Nox1-dependent ROS (22, 24). Interestingly, Tks4 and Tks5 can mediate Nox1 and Nox3 ROS generation, but are unable to activate Nox2. This has important implications for Src regulation of redox signaling, mediating the Nox1 and Nox3 pathways specifically.
Unlike Nox1, Nox 2, or Nox3, Nox4 requires only p22phox for activity [Fig. 2B; (1)]. In addition to p22phox, polymerase delta-interacting protein (Poldip2) has been described as a novel regulator of Nox4 in smooth muscle cells, stimulating Nox4 activation through p22phox interaction (49). The molecular mechanism behind Poldip2 regulation of Nox4 still remains to be determined, however. Alternative evidence suggests that Nox4 is regulated at the level of messenger ribonucleic acid (mRNA) as opposed to post-translational protein modifications (63). Further, Nox4 is the only Nox family member demonstrating constitutive enzymatic activity, a property that seems to derive from a unique C-terminal region and distinct B-loop composition (69). Interestingly, insulin-like growth factor I (IGF-I), bone morphogenetic protein-2, transforming growth factor-β (TGFβ), and Toll-like receptor 4 are examples of some recently studied growth factor receptors that have been shown to activate Nox4 without engaging regulatory subunits (21, 43, 48, 50, 51). To date, growth factor-mediated activation of Nox4 has only been shown to occur through the transcriptional regulation of either Nox4 or p22phox. IGF-I receptor stimulation has been demonstrated to induce Nox4-dependent ROS production in pancreatic cancer cells, in turn playing an important prosurvival role (43). Activation of Nox4 occurred downstream of the IGF-I receptor through nuclear factor-kappa light-chain enhancer of activated B-cells (NF-κB)-mediated transcriptional upregulation of p22phox (21). Phosphorylation and activation of AKT after receptor ligand binding were integral in activating NF-κB in this system. The role that NF-κB plays in Nox regulation is not limited to p22phox, and its activation has also been shown to also be involved in the transcriptional upregulation of the Nox4 gene as well as Nox1 in smooth muscle cells downstream of tumor necrosis factor α (TNF-α), indicating a more widespread influence on Nox regulation (52).
Nox5, Duox1, and Duox2 are unlike the other Nox proteins in so far, as these proteins do not require p22phox for activity (Fig. 2C). Activation is dependent on intracellular calcium levels, which bind EF-hands present in the N-terminal domains of these proteins, inducing a conformational change and subsequent ROS production (2, 6). It is, therefore, possible that growth factor stimuli could potentially drive ROS by regulation of calcium-modulating pathways upstream of these Noxs. Interestingly, phorbol 12-myristate 13-acetate (PMA) exposure has been shown to increase calcium sensitivity of Nox5, mediated through PKC-dependent phosphorylation of Thr494 and Ser498 (34, 64). This increase in calcium sensitivity of Nox5 resulted in ROS production in cells with resting levels of calcium and introduced a novel method of Nox5 regulation. Data suggest that the MEK/ERK1/2 pathway is required for Nox5-derived
Peroxiredoxin Control of Redox Survival Signaling
H2O2 produced by Nox proteins downstream of growth factor stimulation must be tightly controlled, with multiple Noxs activated by multiple growth factors giving rise to a complex network of redox-dependent signaling pathways (see Table 1). Allowing the propagation of redox signaling while preventing unwanted protein damage is critical for proper cell function. Thus, a complex interplay of ROS generation (described above) and antioxidant systems determines how redox signals are transduced.
Nicotinamide Adenine Dinucleotide Phosphate Oxidase-Regulated Growth Factor Survival Signaling
In the previous decade, it has become increasingly apparent that the Nox family of proteins play a vital role in the regulation of redox-sensitive growth factor signal transduction pathways. Multiple Noxs have been implicated as mediators of signaling downstream of growth factors, and Noxs are known to be downstream of multiple growth factors. In addition, there is a wide range of prosurvival effectors activated in Nox-dependent pathways. These data highlight the complexity of Nox prosurvival signaling, and suggest an as-yet poorly understood regulation of redox-dependent signaling networks.
Peroxiredoxins (Prx) have recently been established as a significant cellular enzyme responsible for the reduction of H2O2 in eukaryotes (71), with a very high rate constant (in the order of ∼107 M−1s−1) (30). The enzymatic action of Prxs relies on two conserved cysteine residues, a peroxidative and a resolving cysteine. The peroxidative cysteine reduces the peroxide substrate and is oxidized, leading to a conformational change in the active site. This allows for reactivation of the enzyme wherein the resolving cysteine forms a disulfide bridge with the peroxidative cysteine. The initial step in catalysis (peroxidation) is the reaction of the peroxidative cysteine with the peroxide substrate, forming an enzyme-bound sulfenic acid and water, at the active site (60). This active site contains four conserved residues (the peroxidative cysteine, arginine, threonine, and proline), which serve to protect the active site from water, lowering the pK a for the peroxidative cysteine. The form of the enzyme carrying out the initial reaction is fully folded, with the peroxidative and resolving cysteines separated. The second step in the reaction (resolution) is facilitated by the resolving cysteine attacking the sulfenic acid–peroxidative cysteine complex to release water, forming a disulfide bond. For this bond formation, the active site containing the peroxidative cysteine and the C-terminal region containing the resolving cysteine must locally unfold. Upon recycling of this disulfide bond, typically by a thioredoxin-like molecule, the enzymatic reaction is complete, regenerating the free thiol forms of the peroxidative and resolving cysteine. In addition to transformational changes associated with the enzymatic action of Prxs, they undergo oligomerization, forming decamers, octamers, and dodecamer during catalysis to enhance protein stability (3).
It has recently become apparent that Prxs play important roles in H2O2 signaling downstream of receptor activation, and indeed are often critical for signal propagation to downstream targets. Given the importance of H2O2 as a second messenger in survival signal transduction, alongside its potential to damage a myriad of biological molecules, there is great interest not only in the mechanisms of its generation in the cell but also in how it is controlled at physiologically safe levels. Nox proteins are responsible for generating H2O2 upon receptor activation, and data suggest that the primary control mechanism for this process is via Prxs. Recent evidence suggests that PrxIV is a principal signaling mediator for the granulocyte colony-stimulating factor receptor (G-CSFR) in myeloid precursors (57). PrxIV was shown to be the only Prx that interacted with the CSF-R receptor C-terminal domain, in a manner independent of the redox status of PrxIV's cysteine core. Interestingly, PrxIV appears to only interact with endocytosed G-CSFR in the endoplasmic reticulum (ER), and not at the plasma membrane, where it associates with protein tyrosine phosphatase 1B (PTP1B), which has the ability to maintain PrxIV's activity via dephosphorylation. This elegant study by Palande et al. demonstrates a potential model for Prx control of growth factor signaling, whereby PrxIV attenuates the level of Nox-derived ROS in the ER, thus allowing PTP1B activity, in turn inhibiting G-CSFR signaling. Further, the ability of PTP1B to activate PrxIV through dephosphorylation was also demonstrated, highlighting the intricate redox control of this receptor.
Inactivation of the receptor tyrosine kinase (RTK) vascular endothelial growth factor receptor 2 (VEGFR2) via oxidation at a conserved cysteine by H2O2 is prevented by PrxII in human aortic endothelial cells (36). PrxII was identified as the only peroxiredoxin capable of regulating VEGFR2-dependent signaling, where knockdown of PrxII inhibits global tyrosine phosphorylation downstream of the receptor, reduced eNOS and ERK signaling, as well as reduced migration and proliferation in response to VEGF. PrxII prevents oxidation of a conserved cysteine in VEGFR2, which normally leads to the formation of an intramolecular disulfide bond preventing activation of the receptor through a hitherto undetermined conformational change at the active site. Thus, in contrast to G-CSF regulation, PrxII demonstrates positive control of H2O2-dependent growth factor survival signaling.
The prevailing model for Prx control of signaling in eukaryotes has been the Floodgate model [Fig. 3A; (73)]. This hypothesis purports that these enzymes act to rapidly eliminate cellular H2O2 under normal circumstances and prevent oxidation of target proteins. However, elevated levels of H2O2 would lead to inactivation of the Prx itself via oxidation, for example, during a Nox-mediated ROS burst upon receptor activation, which would facilitate H2O2 signaling. This has been suggested as an acquired evolutionarily ability of eukaryotic Prxs, selected for in higher organisms to allow enhanced control of multiple complex signaling pathways, given the observation that prokaryotic Prxs are not susceptible to inactivation via hyperoxidation. The sulfenic form of the peroxidative cysteine formed through oxidation during the normal enzymatic cycle of Prx may further react with H2O2, giving rise to higher oxidation products, typically sulfinic acid. Cellular antioxidants such as glutathione or thioredoxin cannot reduce protein sulfinic acids, thus producing an irreversible inactivation shuttle in the Prx cycle in circumstances where excessive levels of H2O2 prevail. In accordance with this model, recent data suggest that PrxII is inactivated in exactly this manner to allow signal propagation, while PrxI is inactivated via phosphorylation to a similar end (72). The phosphorylation-dependent inactivation of PrxI is mediated by Src kinase downstream of both PDGF, epidermal growth factor (EGF) stimulation, as well as both T- and B-cell receptors. PrxI inactivation in both keratinocytes and hepatocellular carcinoma cells was shown to be driven by Nox1-derived H2O2, which inactivates PTPs, promoting further H2O2 accumulation. This differential regulation of PrxI and PrxII at the membrane has been suggested as a safeguard against the rapid amplification of H2O2 levels through positive feedback loops upon receptor stimulation (72).
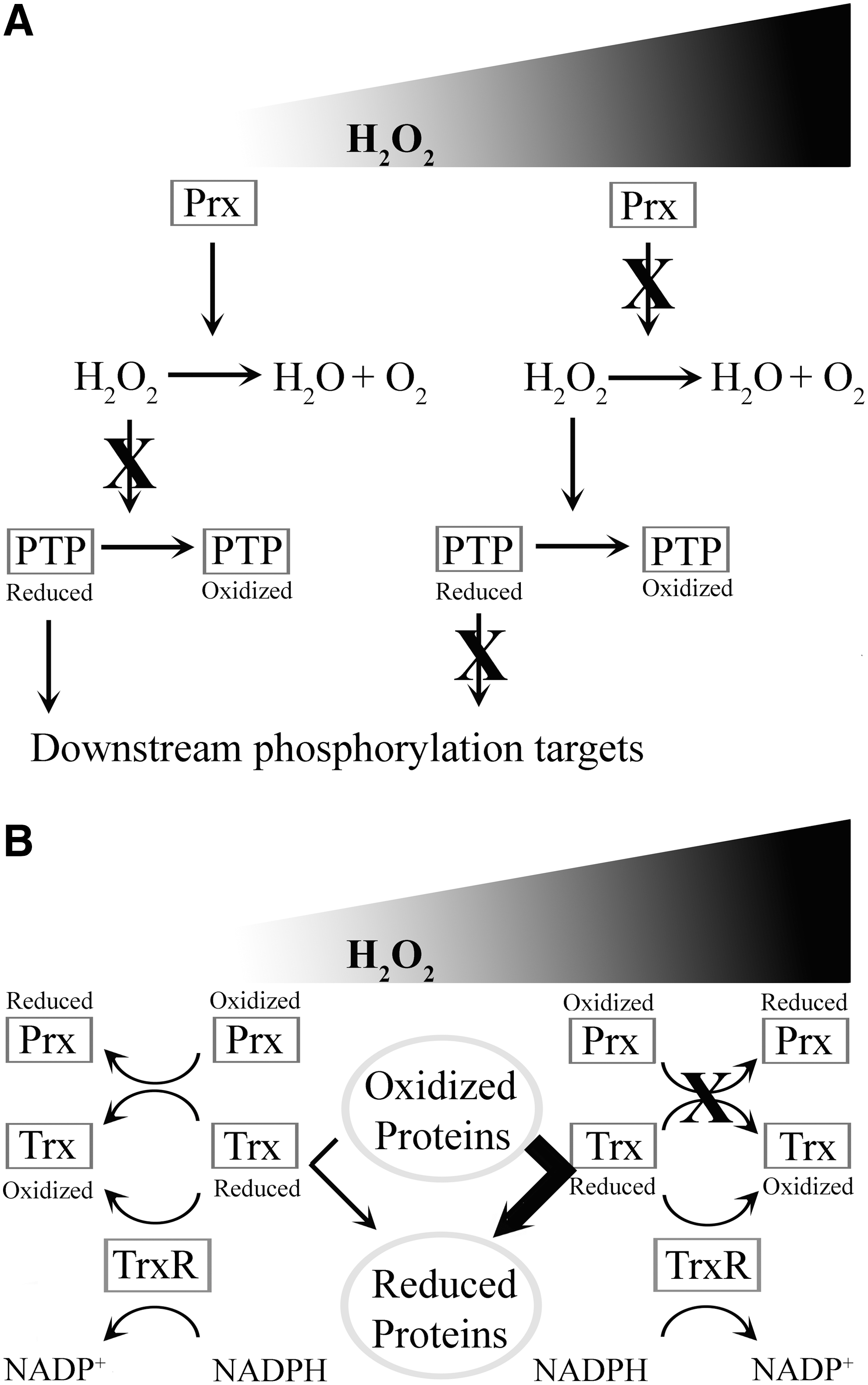
In contrast to the Floodgate model, recent evidence suggests a different explanation for the hyperoxidative inactivation potential of eukaryotic Prxs. In this case, it appears that Prx hyperoxidative inactivation allows the oxidoreductase activity of a thioredoxin to target other more critical downstream targets [Fig. 3B; (18)]. These data suggest that the primary responsibility of Prxs is to maintain ROS homeostasis, and their oxidation is an adaptive mechanism to protect the cell from oxidative stress. It should be noted that these data come from studies on yeast, and the argument could be made that redundancy in the multiple Prxs in higher eukarya has evolved to support complex signaling networks. It should also be noted that individual Prxs downstream of receptors may have evolved distinct roles, as seen with PrxI and Prx II (72), with relaxed selection constraints. However, while the precise mechanism of Prx regulation of signal transduction remains elusive, recent data highlight the importance of these enzymes in signal propagation downstream of H2O2 originating from receptor-activated Noxs.
Redox-Regulated Phosphatases
Signal transduction further downstream of receptor activation is also maintained by a host of redox-modulated proteins. Perhaps, the most widely studied of these are the PTPs, which represent a form of regulation of protein tyrosine kinase (PTK) signaling by tempering and terminating phosphorylation cascades. PTPs are themselves regulated by the dual processes of phosphorylation and oxidation (15), resulting in a complex coordination of PTP activity (see Fig. 4).

Oxidation of PTPs appears to be essential for the functioning of certain intracellular signaling pathways. For example, the redox-mediated inactivation of suppressor of high-copy PP1 (SHP1) has recently been implicated in the macrophage colony-stimulating factor (M-CSF)-induced proliferation of hematopoietic progenitors (16). Transient oxidation of SHP1 affects the PI3K/AKT pathway, but not the MAPK survival pathway to stimulate bone marrow monocyte-/macrophage-lineage cell proliferation. This demonstrates how the specificity and integrity of the signal initiated by M-CSF binding its receptor c-Fms and subsequently transmitted via ROS can be maintained. Interestingly, SHP2 was not oxidized in their system, even upon exogenous addition of peroxide, despite its accepted status as redox-sensitive phosphatase in other systems (54, 70). Oxidation of PTP's in a signaling context requires the specificity of an enzyme–substrate reaction to maintain the integrity of the propagated signal. The sensitivity of individual PTP's to different oxidant species is one mechanism whereby this differential regulation is ensured. Similarly, data suggest that PTP1B is inhibited by H2O2 in vivo, but not by
Alongside oxidative regulation of PTPs, another method of ensuring the specificity of redox signaling is the localization of both the oxidant-producing enzyme and the target of the oxidant to a particular subcellular compartment. Recent evidence points to spatial regulation that occurs in the ER of multiple cell types, whereby ER-resident PTP1B, which acts within the ER to attenuate endocytosed EGFR signaling, is itself inactivated by H2O2 produced by another ER protein, Nox4 (11). The specificity of the redox signal was highlighted by the failure of Nox4 to oxidize a cytosolic mutant form of the phosphatase.
The list of PTPs that are redox-regulated in response to growth factor signaling continues to grow, and among the most recent additions to this list is the tumor suppressor PTP density-enhanced protein tyrosine phosphatase-1 (DEP-1), which was shown to be oxidatively inactivated in response to aberrant redox signaling driven by the oncogenic RTK FLT3-ITD (26). Transformation of mouse hematopoietic progenitor cells by FLT3-ITD is so dependent on the redox regulation of DEP1 that blocking the ROS burst induced by FLT3-ITD was sufficient to halt transformation in vivo and in vitro. This effect was reversed upon knockdown of DEP1, both establishing the powerful tumor suppressive role of this phosphatase in leukemia, as well as uncovering further details of the relatively obscure FLT3-ITD-signaling pathway.
Much of the focus on the source of GF-stimulated ROS production centers on the Nox family of enzymes, the oxidizing mediators produced by 12/15-lipoxygenase, which can act as second messengers by inactivating PTPs to amplify and prolong PDGF-β receptor signaling (17). The signaling molecules in question were shown to be peroxidized arachidonic acid-derived lipids that induced oxidation of SHP1, PTP-H1, and TC-PTP in vitro. This previously unrecognized class of oxidizing mediators introduces another dimension to the already-complex situation of redox regulation, and they may also help to account for features of redox signaling that cannot be attributed to diffusible ROS.
Although PTPs are the prototypical redox-modified molecules, they are the only one arm of the RTK phosphorylation signal transduction cascade, and recent evidence suggests that their signaling counterpoints, the PTKs, can be similarly redox regulated if they contain a cysteine residue vulnerable to oxidation. Particular focus has been applied to the Src family kinases as potential targets of ROS because of their prominent role in both physiological and oncogenic signaling, and a recent comprehensive review by Giannoni et al. (25) discusses the mechanism and effects of the redox regulation of Src. The oxidation of PTKs appears to render the enzymes more active, thus amplifying the phosphorylation signal already facilitated by the transient oxidative inactivation of PTP's.
Further to the Giannoni review and expanding upon the pathological significance of Src redox regulation, Xi et al. (75) more recently provided direct evidence for a role for Src oxidation in response to IGF-1 stimulation in a mouse model of diabetes. Similarly Lee et al. (44) demonstrated that VEGF-driven ROS production resulted in the thiol oxidation of both c-Src and the VEGFR in endothelial cells. These modifications prompted a physical interaction between the two molecules, stimulating the Src/PI3K/AKT pathway downstream of VEGFR. Interestingly, the PLCγ-1/ERK1/2 pathway, which is also downstream of VEGFR, was not activated upon oxidation of the receptor, again suggesting that redox signaling can discriminate between potential targets and tailor a distinct response.
Aside from PTPs and redox-sensitive PTKs, there are many other direct targets of growth factor-driven ROS production. Included in these are the GTPases, particularly Ras, whose redox sensitivity has implications for a range of pathologies (31). In addition, transcription factors (TFs) such as STAT3, which was shown to be directly sensitive to intracellular peroxide (45), are crucial for translating the oxidative signal into adaptive changes at the level of gene expression. Guttmann and Ghoshal recently reviewed the redox regulation of thiol proteases, including calpains and caspases (29), whereas Koch et al. (40) offered a new concept of matrix metalloprotease (MMP) regulation via the oxidative inactivation of an MMP-1-bound inhibitory-acting thiol. In addition to these, the protein disulfide isomerase (PDI) class of chaperones such as ER protein 72 kDa (ERp72) was shown by Chen et al. (12), to be redox-modified by Nox 1-derived ROS in response to EGF signaling at the plasma membrane.
Redox-Regulated Effector TFs
Oxidation-sensitive Prxs and phosphatases are controlled directly downstream of growth factor-stimulated ROS production, thereby allowing proper activation of effector molecules further along the signaling cascade. At the furthest end of redox transduction pathways are TFs. There are at least 14 mammalian TFs known to be redox regulated [reviewed in (7, 10)]. Nox-generated ROS have been shown to affect the activity of NF-κB and activating protein-1 (AP-1) [reviewed in (7)], but since that very thorough review covering all aspects of Noxs in 2007, more recent studies have delved deeper into the link between ROS produced by Noxs, TFs, and survival signaling.
While there are a myriad of articles linking the ROS produced by Nox family members to NF-κB activation, very few delineate the signaling mechanisms involved. Many publications assume that this mechanism involves ROS directly modifying the cysteines found in TFs [e.g. (38)], but this is not necessarily the case (10). Brigelius-Flohe and Flohe (10) argue that changes in the activity of TFs are much more likely to be due to protein thiol oxidation of their interacting partners, as there is little evidence in the literature that any oxidant signal directly modifies a mammalian TF. In the case of NF-κB, this is seen through ROS influencing the cytosolic complex where NF-κB is held in an inactive state through its interaction with inhibitor of κB (IκB). If IκB kinase (IKK) is activated, it phosphorylates IκB, leading to its degradation and freeing NF-κB to translocate to the nucleus. Li et al. (46) demonstrated that this is the case for Nox-derived ROS, through studying the production of H2O2 triggered by TNFα. H2O2, produced at endosomes by Nox2, facilitates increased TNF receptor-associated factor 2 (TRAF2) recruitment, thus enabling activation of IKK and hence release of NF-κB from its inhibitory complex. Similarly, in an interleukin-1 (IL-1) system, ROS produced by Nox2 were linked to the recruitment of the adapter protein, TRAF6, to the IL-1 receptor, which determined the degree of activation of IKK and subsequent NF-κB activation (47). There is no doubt that the ROS produced by Nox enzymes alter the activity of NF-κB, but recent evidence makes it clear that this is through NF-κB's signaling partners, rather than ROS acting on NF-κB itself.
Nuclear factor (erythroid-derived 2)-related factor 2 (Nrf2) is another redox-sensitive TF and interacts with an inhibitory protein, namely, Kelch-like ECH-associated protein 1 (Keap1), maintaining its cytoplasmic localization in unstimuated conditions (10, 38). Recently, Nrf2 was linked conclusively to Nox4. Overexpression of Nox4 in cardiac myocytes led to a significant upregulation in antioxidant and detoxifying genes, which were not found in Nrf2-null mice with the same overexpression system (9). This study established that Nox4 increases enzymes such as thioredoxin reductase 1 and the levels of glutathione, both of which contribute to the removal of excess ROS, and so are considered to increase the survival of cells. A second study demonstrated that AP-1 can also work in conjunction with Nrf2 in response to Nox2-generated ROS to upregulate the protein sulfiredoxin, which is known to be protective in cells (39). This study also demonstrated that the effects of ROS on Nrf2 may be through multiple, but all indirect, pathways (39). These indirect pathways may be the result of changes in JNK signaling, or changes in Keap1 expression (10). This is because Keap1 is a redox sensor, with murine Keap1 having 25 cysteines, 4 of which are highly susceptible to redox regulation. Any changes in these cysteines result in alterations of Keap1 activity and hence Nrf2 activity also (20). Therefore, any ROS, generated by Noxs or other enzymes, are likely to have an effect on Nrf2 transcription activity, but this is most likely to be via indirect mechanisms.
The link between Noxs and STATs is developing briskly with numerous studies identifying Nox enzymes as intermediates in growth factor-stimulated Jak/STAT pathways. IL-13 leads to the activation of Duox1 in keratinocytes, which inhibits PTP1B, leading to the sustained phosphorylation and hence activation of STAT6 (32). Another family member, STAT3, is downstream of EGFR (65). Activation of EGFR by 15(S)-hydroxyeicosatetraenoic acid results in the generation of ROS through Nox. These ROS contribute to the phosphorylation and activation of the Src/JAK2/STAT3 pathway and ultimately to the upregulation of monocyte chemoattractant protein-1, increasing survival of cells by permitting the remodeling of vascular walls. Although the JAK-STAT signaling pathway is considered to be relatively simple, with these two proteins forming the main constituents, other proteins can also modify this pathway, for example, Src (65). Therefore, even though there has been a recent discovery of redox-sensitive cysteines in JAK2, which result in JAK2 inhibition when oxidized (66), similar to phosphatases, this does not mean that all ROS produced will lead to the inhibition of STAT. These studies by Hirakawa et al. (32) and Singh et al. (65) clearly demonstrate that the ROS generated by Noxs can lead to an increase in the phosphorylation of STAT. The generation of ROS alone, therefore, does not automatically imply that STATs will be activated, as their effect on STAT signaling can vary depending on many factors. Recent data also showed a link between Nox and STAT signaling in leukemia (62). Work from our group has added further weight to this link and identified STAT5 as a downstream target of p22phox-associated Noxs, regulating the expression of the proto-oncogene Pim-1 (74). There are a wide range of other TFs that are redox sensitive and linked to cell survival, all of which cannot be mentioned here due to space limitations. One of the more recent and unusual TFs to be connected with Nox-derived ROS is the serum response factor (SRF). In human aortic smooth muscle cells, this TF was shown to be the link between the TGFβ signal to differentiate and the expression of smooth muscle α-actin. However, SRF was not directly affected by ROS, but its phosphorylation at serine 103 proved to be crucial in this signaling pathway (53), implicating the involvement of a redox-sensitive phosphatase.
Overall, the known downstream gene targets that have been linked directly to growth factor-induced ROS produced by Nox and activation of a specific TF are very limited (Table 2). From Table 2, it is clear that the identified downstream targets play a role in cell motility/contractility (MMPs, MCP-1, and smooth muscle α-actin) and also in the further production of ROS [Nox4 and Duox2; (19)]. Other studies have focused on individual components of this pathway rather than looking at the pathway in its entirety. There are numerous other targets of the TFs examined in the preceding paragraphs, but there has been no conclusive link to date with their specific targets and Nox-derived ROS stimulated by growth factors. These other targets are known to modify diverse cellular functions, ranging from apoptosis to inflammation to antioxidant gene expression, resulting in them playing roles in various diseases from cancer to Parkinson's disease.
Downstream Targets of Growth Factor-Stimulated Nox-Derived Reactive Oxygen Species
Examples given in this table consist only of those pathways where four components are known: (i) the growth factor, (ii) a link to Nox, (iii) transcription factor, and (iv) downstream targets. Many other publications have shown a link between two or three of these, but not all four.
Conclusion
Regulation by ROS downstream of growth factor receptors occurs at multiple steps, from the point of ROS generation and homeostasis control at the receptor initially to the signaling partner phosphatases and eventually the effector TFs themselves (see Fig. 5). Clearly, much further work is needed to tease out the intricacies of these redox-mediated processes, but in recent years, a clearer picture has developed in this field. Acknowledging the importance of ROS molecules, such as H2O2, in signaling opens up not only new avenues of research, but greatly enhances our knowledge of cellular signaling and expands disease therapeutic strategies.

Footnotes
Acknowledgments
This work was supported by the Science Foundation Ireland, Irish Cancer Society, and Children's Leukemia Research Project.