
Abstract
Introduction
Here we consider the thiol redox control pathways of the single-celled eukaryote Saccharomyces cerevisiae, focusing on their functions in the different compartments of the cell, and on the known mechanisms that explain the extreme differences of thiol redox states in the environment of the mitochondrial matrix, cytosol and mitochondrial intermembrane space (IMS) that is very reducing, and the endoplasmic reticulum (ER) that is very oxidized. We also consider the interplay between the thiol-redox homeostatic controls of each compartment.
Cellular Distribution of Thiol Redox Control Components
Two complete TRX pathways are present in S. cerevisiae: one in the cytosol that consists of two apparently redundant TRX isoforms (Trx1 and Trx2) and a TRR (Trr1) (93, 94) (Fig. 1), and one in the mitochondrial matrix that consists of a TRX (Trx3) and a TRR (Trr2) (72) (Fig. 2). Similarly, two complete GSH pathways exist in this organism, one in the cytosol that consists of two dithiol GRXs encoded by GRX1 and GRX2 (Grx1, Grx2) (51), and a GLR encoded by GLR1 (Glr1) (15), and one in mitochondria consisting of activities also encoded by GLR1 and GRX2. Dual cytosolic and mitochondrial localization of both Glr1 and Grx2 is due to the presence of an in-frame translation start site upstream of the canonical one, which produces a N-terminal mitochondrial targeting presequence (69, 75, 78). The monothiol Grx6 and Grx7, which are ER and golgi luminal proteins anchored by N-terminal transmembrane domains to the membrane of these compartments, possess GSH-dependent oxidoreductase activity (36, 52, 58, 59), in contrast to the other monothiol Grxs, Grx3, Grx4, and Grx5, which are rather involved in iron metabolism and will therefore not be considered here. The dithiol cytosolic enzyme Grx8 carries a low oxidoreductase activity, but will not be further considered, because its function is still unknown (22). GSH is synthesized in the cytosol in yeast, as in mammals (30), by the sequential action of GSH1-encoded γ-glutamyl cysteine synthase, and GSH2-encoded glutathione synthase. GSH is found in all the yeast cell compartments, but how it distributes to these compartments is unknown. Cellular import of GSH and oxidized glutathione (GSSG) is operated by the plasma membrane proton-coupled oligopeptide transporter Hgt1 (or Opt1) that has a high affinity for GSH (50 μM) (7). HGT1 expression is stimulated under sulfur-free conditions, as well by all amino acids, except those containing sulfur (102). Degradation of GSH is also an important determinant of its homeostasis. Initially thought to be operated in the vacuole by γ-glutamyl transpeptidase (γ-GT) under conditions of nitrogen starvation (55), recent data indicate that the newly discovered Dug1/Dug2 glutamine amidotransferase, which cleaves GSH at the γ-glutamyl-cysteine bond (38), and Dug3, a Cys-Gly dipeptidase (39), form a complex that is responsible for all GSH-degrading activity of yeast cells under normal and stress conditions, including sulfur and nitrogen starvation and cadmium exposure (4, 23). Lastly, NADPH is the major proton donor for disulfide reduction. NADPH is primarily generated from NADP+ by the oxidation of pentose in the pentose phosphate shunt pathway in the cytosol (37, 66), and from nicotinamide adenine dinucleotide (NADH) by the NADH kinase Pos5 in mitochondria (68).

Cytosolic Thiol Redox Control
We will first address the respective functions of the TRX and GSH pathways in the cytosol, and then ask in which measure these pathways are functionally redundant.
The cytosolic functions of the TRX pathway
Functions of the cytosolic TRX pathway have been largely elucidated by genetics, and then refined by biochemical techniques that rely on the differential trapping of reduced versus oxidized Cys residues.
To observe a phenotype linked to cytosolic TRX inactivation, one must delete both TRX1 and TRX2 (Δtrx1Δtrx2), due to the redundant nature of the encoded proteins. The Δtrx1Δtrx2 strain has three remarkable phenotypes. The first one is a protracted cell cycle S phase (64), due to inefficient DNA synthesis by inefficient reduction of RNR (9, 40). Formal proof of the link between this phenotype and defective RNR reduction was provided by the presence in Δtrx1Δtrx2 of a significant decrease of dNTPs levels (9, 40), an accumulation of RNR in its disulfide form, and relief of these defects upon overexpressing RNR (9). The second phenotype is an auxotrophy for sulfur-containing amino acids (64), due to defective reduction of the catalytic disulfide of 3′-phosphoadenylsulfate (PAPS) reductase (Met16), an enzyme required for inorganic sulfate assimilation. A two-hybrid identification of Met16 using TRX as bait (101) further illustrated the role of TRX in PAPS reductase reduction. The third major phenotype is a profound decrease in tolerance to peroxides (24, 43), a consequence of multiple defects in peroxide metabolic pathways. TRX is indeed the exclusive reductase of the three cytosolic yeast peroxiredoxins (PRXs) Tsa1, Tsa2, and Ahp1 (11, 46, 100), of the three glutathione peroxidase (GPX)-like-enzymes Gpx1, Gpx2, and Gpx3 (19, 92), and of the two methionine sulfoxide reductases Mrx1 and Mrx2 (31, 101). TRX also regulates the Yap1 and Msn2/4 stress transcriptional regulators. In Δtrx1Δtrx2, the peroxide-stress regulator Yap1 is constitutively active and accumulates in its active disulfide form, which suggests that TRX negatively regulates this regulator by reduction (10, 18, 35). However, although Yap1 is constitutive in Δtrx1Δtrx2, the strain remains highly peroxide-sensitive, due to the concurrent peroxidase defect resulting from the lack of TRX. TRX is also required to activate the general stress regulators Msn2/4 and Maf1 in response to hydrogen peroxide (H2O2), through an elusive mechanism that requires TRX in its oxidized form (6). The major role of TRX in peroxide metabolism has been confirmed by the identification of Tsa1, Ahp1, and Met16 in a yeast two-hybrid assay using thioredoxin 2 as bait (101); Ahp1 was also purified from yeast lysates on the basis of its redox association with a plant TRX (100). The inability of Δtrx1Δtrx2 to use methionine sulfoxide as source of organic sulfate (62) is another proof of the exclusive role of TRX in assisting methionine sulfoxide reductase. Redox proteomics also showed that in Δtrx1Δtrx2, Tsa1, Tsa2, Ahp1, Prx1, Gpx2, and Mxr1 along many other proteins have a significant increase in Cys residues oxidation, which demonstrate the importance of TRX in controlling the redox state of these antioxidant proteins (45).
In addition to these three major phenotypes, Δtrx1Δtrx2 is paradoxically hypersensitive to the dithiol-reducing agent dithiothreitol (DTT) and resistant to the thiol oxidant diamide (95), which are phenotypes that have not been traced to any specific molecular defects. TRX was also biochemically identified as a factor required for vacuole inheritance, but this function is independent of the TRX Cys residues, and therefore not a redox one (104).
The phenotypes of the Δtrr1 strain should match those of Δtrx1Δtrx2, because of the epistatic relationship between TRR1, TRX1, and TRX2, but this is not so. Δtrr1 has a unique slow aerobic growth (44, 53, 96) that is significantly improved by anaerobiosis (94, 105), and a much poorer peroxide tolerance than Δtrx1Δtrx2. However, Δtrr1 does not carry the cell cycle and sulfate assimilation defects of Δtrx1Δtrx2. The distinct phenotypes of Δtrx1Δtrx2 and Δtrr1 could indicate that TRR has enzymatic functions independent of TRX, as shown for mammalian TRR (2). These differences may also be explained by a Δtrr1 gain of function caused by toxic accumulation of oxidized TRX leading to disulfide stress, as shown in E. coli (89). The latter hypothesis is supported by improvement of both growth and peroxide tolerance phenotypes by further deletion of TRX1 and TRX2 (Δtrr1Δtrx1Δtrx2) (17, 94, 105). Anyhow, lack of the cell cycle defect and organic sulfur auxotrophy of Δtrr1 also indicates that enough TRX activity remains in the cell to reduce RNR and PAPS reductase.
The Δtrr1 strain is also moderately DTT hypersensitive and exhibits constitutive activation of the unfolded protein response (UPR), which is physiologically triggered by ER stress (95, 96), and of the Aft1-dependent iron starvation response (44). The Δtrr1 constitutive UPR cannot be easily rationalized, but its cure upon GSH depletion (96) suggests that it might be linked to the compensatory increase in GSH levels in this strain (see below). Activation of Aft1 might be a consequence of GSH bearing the redox load increase caused by lack of Trr1, and therefore becoming limiting for its function in iron metabolism (see below). Δtrr1 has other complex defects in translation accuracy, ribosomal aggregation, and sensitivity to the translational inhibitors hygromycin and puromycin, which are to some extent shared with Δtrx1Δtrx2 and Δtsa1 (79, 98).
The cytosolic functions of the GSH pathway
Study of the yeast GSH pathway has been hampered by the essential requirement of GSH for viability. GSH1-null strains are unviable unless given exogenous GSH (26, 103), in contrast to those lacking either cytosolic GRXs (Δgrx1Δgrx2) or GLR (Δglr1), which indicates that GSH has functions not shared with the other components of the pathway. Δgrx1Δgrx2 and Δglr1 do not have any remarkable phenotypes under nonstress conditions, except for a strongly oxidized GSH pool in the latter mutant (Table 1) (25, 51). Δgrx2, Δglr1, and cells partially depleted of GSH are slightly peroxide-sensitive, whereas Δgrx1 is sensitive to the superoxide anion-generating drug menadione (25, 51, 65, 88). How can the peroxide phenotypes of GSH pathway mutants be rationalized? In mammals, GSH reduces GPxs, an important peroxide-scavenging family, but there are no bona fide GPx enzymes in yeast, and as mentioned above, the related enzymes, Gpx1, Gpx2, and Gpx3, are reduced by TRX and not GSH (19, 92). GSH might reduce protein-sulfenic acids (P-SOH) resulting from the reaction of thiols with H2O2, through formation of S-glutathione protein adducts, hence contributing to H2O2 scavenging. Oxidation of GSH and S-glutathionylation of several cytosolic proteins in response to H2O2 supports this notion (86). Grx1/2 was shown to possess peroxidase and glutathione S-transferase (GST) activities in the presence of GSH in vitro (13, 14), which if verified in vivo could also contribute to H2O2 scavenging. However, as GLR1 and GRX2 encode both the cytosolic and mitochondrial isoforms of these enzymes (see above), a mitochondrial peroxide scavenging defect should also be considered to explain the peroxide phenotypes of the corresponding mutants, which in fact prevails over a cytosolic defect (see below). GSH is also important for the resistance to heavy metals and electrophiles (76), a function relying on the chelating property of its Cys residue. Resistance to some electrophiles might also be linked to the GST-transferase activity of Grx1/2, a function involving GSH (13). Δgrx2, Δglr1, and GSH-partially depleted cells are paradoxically resistant to diamide, a phenotype shared with TRX mutants (see above) (51, 65).
From Ref. (44).
In the case of HGT1, measures were performed 1 h after adding the indicated amount of GSH. In the case of dgsh1, growth was initiated in the presence of the indicated amount of GSH, and measures were performed in the late exponential phase. In the case where GSH was not added, measures were performed after six divisions in medium not containing GSH.
EGSH was calculated according to the Nernst equation, assuming a pH=7.
GSH, glutathione; GSSG, oxidized glutathione.
Henceforth, the most salient phenotype of yeast GSH pathway mutants is the requirement of GSH for viability, which has been an enigma as to its molecular cause. The GSH auxotrophy of the Δgsh1 mutant cannot be rescued by alternative small thiol-reducing agents such as Cys, DTT, and N-acetyl cysteine, nor by anaerobiosis (47, 84, 88), which indicated that neither a general disulfide reduction defect nor lack of a reactive oxygen species (ROS) protective function can explain the inviability of GSH-depleted cells. Defective reduction of the essential RNR disulfide could not account for the inviability either, as this disulfide is preferentially reduced by TRX in yeast (9). Attempts at identifying yeast extragenic suppressors of GSH auxotrophy failed, only yielding mutations that, by diverting the proline biosynthetic pathway, restored synthesis of very small amounts of GSH (88). This and other studies thus indicate that minute amounts of the tripeptide suffice for full viability (47, 84, 88), excluding the idea that GSH is essential as a redox buffer. In fact, the essential nature of GSH is not linked to thiol reduction, but to a function in iron metabolism, a conclusion supported by the results of an analysis of GSH-depleted cells (44). Genome-wide mRNA profiling showed that the near-unique defect of these cells is an alteration of iron metabolism originating from a defect in extra-mitochondrial iron-sulfur cluster (ISC) maturation, a process that requires GSH (87). This requirement is possibly linked to an undefined role for GSH in iron trafficking and delivery that may, at least in part, involve the two cytosolic monothiols GRXs, Grx3 and Grx4, that require GSH to bind a [2Fe-2S] cluster (63) [for a recent review on this subject (48)]. The accumulation of iron in the mitochondria of GSH-depleted cells (87) is a consequence of the ISC assembly defect, and explains the high mitochondrial genome instability of these cells (3). The observation that both the ISC assembly defect and growth arrest characteristic of GSH-depleted cells are partially corrected by exogenous iron further establishes the link between the yeast GSH requirement for viability and its function in iron metabolism (44).
What are the other cytosolic functions of the GSH pathway (see Fig. 3)? Although this pathway, and GSH in particular, are classically described as important in cytosol thiol-redox maintenance, data obtained in yeast do not support this notion. It is first instructive to consider that no specific cytosolic thiol reduction functions have ever been attributed to GSH or to the GSH pathway in yeast, at least when TRX is present, and aside from the possible role of GRXs as peroxidases and/or GSH transferases (see above). In addition, the genome-wide mRNA profiles of GSH-depleted cells did not provide any hint of a thiol-redox maintenance defect (44), which is consistent with a redox proteomics study that also indicated that the redox state of protein-thiols is not altered in cells simultaneously depleted of both Glr1 and GSH, in contrast to those lacking both TRXs and TRR (see above) (45). When present at very high cellular levels up to 10-fold its wild-type concentration, GSH totally inhibits growth and causes cell death, a toxicity caused by a major ER reductive stress (see below) and impairment of ISC assembly, but not attributed to a defect in cytosolic thiol redox control (44).
The TRX pathway has the major contribution in cytosolic thiol redox control
It is clear from the data discussed above that the TRX pathway has a prominent, if not exclusive, role in cytosolic thiol redox control, in contrast to the GSH pathway that has only a limited role (see Figs. 1 and 3). Such a functional configuration would thus imply that these pathways do not overlap functionally, in contrast to their E. coli counterparts, either of which is able to support the thiol-redox duties of the cell in the absence of the other (94). However, although yeast thiol redox pathways are not redundant functionally, the GSH pathway can act as backup system when the TRX pathway is absent. RNR is essential as it provides the deoxyribonucleotides required for DNA synthesis and repair. RNR reduction, which thus constitutes a vital target for thiol redox control, is accomplished by the TRX pathway (see above). However, the viability of Δtrx1Δtrx2 indicates that the GSH pathway can substitute for the TRX pathway in this function, but only very inefficiently as evidenced by the cell cycle defects of this mutant. The synthetic lethality resulting from the simultaneous deletion of components in both pathways [e.g., Δglr1Δtrx1Δtrx2 (65), Δtrx1Δtrx2Δgrx1Δgrx2 (21), Δglr1Δtrr1, Δgsh1Δtrx1Δtrx2 (96), or Δgsh1Δtrr1 (44)] is probably caused by defective RNR reduction, which also supports the notion that GSH operates as a backup system in RNR reduction when TRX is absent. Another example of the inefficiency of the GSH pathway as a thiol-redox backup system involves redox control of PAPS reductase (64). The organic sulfur auxotrophy of Δtrx1Δtrx2 can be relieved by growth in liquid culture under low oxygen tension (21), which might indicate that Grx1 and/or Grx2 can substitute for TRX in PAPS reduction, but only when the cellular oxidative load is decreased. A putative alternate PAPS reductase other than GRX was also alluded to, as overexpressing GRX1 or GRX2 did not rescue the sulfur auxotrophy of Δtrx1Δtrx2 (21). The threefold increase of total cellular GSH in Δtrr1 and Δtrx1Δtrx2 (see Table 1) (96) probably compensates for the inefficiency of the GSH pathway in replacing the TRX pathway. Still, the very slow growth of Δtrr1 can be significantly improved by exogenous GSH, which indicates that, despite its compensatory increase, GSH remains extremely limiting in this strain (44). The activation of the iron starvation response observed in Δtrr1 is a probable consequence of the limiting amount of GSH in this strain, as it is also corrected by exogenous GSH.
In conclusion, the data examined here altogether indicate that in the cytosol the TRX pathway has the major, if not exclusive, role in thiol redox control. The GSH pathway in contrast has a very limited contribution to this control, serving as a backup system when the TRX pathway is absent, or when it is overwhelmed by severe redox stresses (44) (see Figs. 1 and 3). The limited contribution of the GSH pathway in cytosolic redox control is further underscored by data of two recent reports, one indicating that reduction of protein-S-glutathione adducts is essentially operated by TRX and not GRX (29), and the other indicating that the TRX pathway can function as an alternate system to reduce GSSG (91).
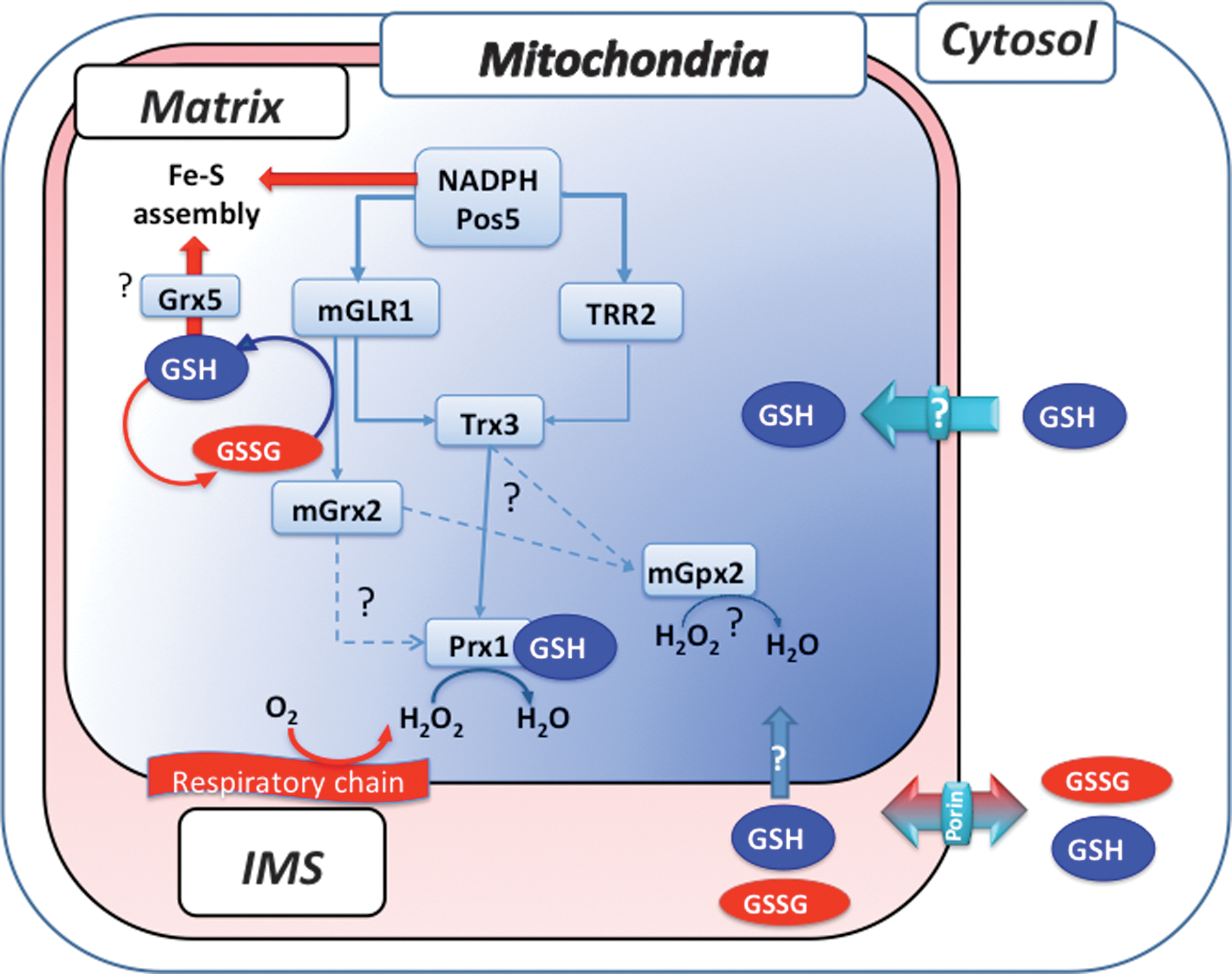
Mitochondrial Thiol Redox Control
As stated above, the yeast mitochondrion possesses the NADH kinase Pos5 as its own NADPH source and a complete set of both TRX and GSH pathways that are not functionally separated as the cytosolic ones (see Fig. 2). With the exception of Δpos5, phenotypes resulting from inactivating components of these pathways are only revealed under stress and respiring growth (glycerol) conditions (68, 72, 90, 97). However, it should be noted that due to our incapacity of depleting GSH from mitochondria with currently available experimental tools, the mitochondrial phenotypes of GSH are totally lacking.
The Δpos5 strain exhibits complex phenotypes, including slow growth in glucose medium that is exacerbated in respiratory medium, a mitochondrial ISC assembly defect and accompanying mitochondrial iron accumulation, and arginine auxotrophy (68, 90). The ISC assembly defect indicates the requirement for NADPH in this process, possibly at the level of the FAD and NADPH-dependent Arh1, an adrenodoxin reductase homolog, or for iron reduction to the biologically utile form Fe++, or for sulfite reduction into sulfur, or for another unknown process in mitochondrial iron metabolism (71). Decreased protection from respiratory-chain ROS is the other distinct defect underlying the lack of mitochondrial NADPH in Δpos5 (68), which combines with the ISC defect to engender the mitochondrial genome instability and protein carbonylation increase also seen in this mutant (90). The defective mitochondrial ROS protection of Δpos5 is evidenced by hyperoxic hypersensitivity that is partially alleviated by inactivating the respiratory chain via deletion of COQ1 encoding a coenzyme Q biosynthetic enzyme (68). The NADPH requirement for respiratory-chain ROS protection is at least in part linked to Glr1-dependent recycling of oxidized GSH, since GSH oxidation is significantly increased in Δpos5 and under hyperoxia, and since inactivation of the mitochondrial form of Glr1 also causes a hyperoxia phenotype (70). Such a ROS protective role for mitochondrial NADPH fits with the identification of POS5 in a screen of peroxide hypersensitive mutants (42).
The only demonstrated function of the mitochondrial TRX and GSH pathways is to assist the mitochondrial PRX Prx1, and possibly also Gpx2 that is also localized in this organelle (99), in protecting mitochondria against respiratory ROS as well as exogenous peroxide (see Fig. 2). GSH is suspected to fulfill important functions in mitochondrial iron metabolism, which should, at least in part, superimpose with those of NADPH (see above), but these functions have not been yet studied, due to the inability of mitochondrial GSH depletion. Inactivation of Trr2 alone causes no or only a very minor peroxide tolerance defect (72, 97), but the combined mutation of Trr2 and Glr1 (Δtrr2Δglr1) yields a synthetic interaction manifested by a severe respiratory growth defect not seen in either single mutant, and by the exacerbation of the Δglr1 minor peroxide tolerance defect (27, 97). Since as already discussed above GLR1 encodes both cytosolic and mitochondrial Glr1 isoforms (69), the synthetic interaction of GLR1 and TRR2 could either indicate a redox linkage between cytosol and matrix or an overlapping role for Trr2 and Glr1 within the matrix. Accumulation of oxidized Trx3 in Δtrr2Δglr1, but not in the corresponding single mutants (97), favors the latter hypothesis. Accumulation of oxidized Prx1 in either Δglr1 or Δtrr2, and loss in these mutants of the increased peroxide tolerance seen in wild-type cells when Prx1 is overexpressed (27) further support the functional redundancy of Trr2 and Glr1, and indicate that Prx1 is a common downstream substrate of these reductases. The identity of the Trr2 and Glr1 substrates that reduces the 1-Cys Prx Prx1 is differently appreciated. 2-Cys Prxs are almost exclusively reduced by TRX and TRR, but how 1-Cys Prxs are reduced is debated. Oxidation of Prx1 upon GSH depletion (27) indicates that the tripeptide contributes to Prx1 reduction, possibly as shown in vitro by formation of an S-glutathione adduct, which is then reduced by Grx2 (74) or Trx3 (28, 73), or less likely by Trr2 (27) or ascorbate (60). Although Δtrx3 is not peroxide hypersensitive, Trx3 clearly contributes to Prx1 reduction as indicated by the change of its redox state from fully oxidized in Δtrr2Δglr1 to almost fully reduced upon further inactivation of Prx1, and by the identification of a Trx3-Prx1 disulfide-linked complex using a Trx3-Cys trapping mutant (28). Interestingly, the latter study also showed that, similar to deletion of TRR1 that leads to toxic accumulation of oxidized cytosolic TRXs, accumulation of oxidized Trx3 is responsible for the respiratory growth defect of Δtrr2Δglr1, since further inactivation of either Trx3 or Prx3 in this strain rescues this growth phenotype. Grx2 might also contribute to Prx1 reduction in vivo, as suggests the peroxide hypersensitive phenotype of a strain lacking the mitochondrial but not the cytosolic form of the enzyme (77). Grx2 might also be involved in mitochondrial iron metabolism, based on the synthetic lethality of Δgrx2Δgrx5 and the fact that Grx5 plays a role in Fe–S cluster biogenesis (82).
In summary, apart from the suspected involvement of GSH in mitochondrial iron metabolism, the mitochondrial TRX and GSH pathways have one established and redundant function, which is to assist Prx1 in scavenging respiratory ROS and exogenous H2O2 (see Fig. 2). Further, based on phenotypes, and in contrast to the cytosol, the GSH pathway appears as the dominant thiol redox control system in this organelle.
Thiol Redox Control of the ER
The ER is the first stage for proteins that are destined for secretion. These proteins are co-translationally translocated into the ER, where they are glycosylated, folded, and acquire their mature conformation, before they are transported, via the Golgi, to the plasma membrane and the extracellular space. For many of them, folding requires formation of one or more disulfides [for a review see ref. (54)]. To attain their native arrangement, formed disulfides must be isomerized and reduced. Hence, opposing reactions—the oxidation and reduction of Cys residues—coexist in the ER. Disequilibrium between these two processes impairs oxidative folding and leads to protein misfolding and ER stress.
The yeast machinery catalyzing disulfide bond formation is relatively well understood, involving a redox relay made of the ER oxidase Ero1 that generates disulfides, which are transmitted to protein disulfide isomerase (Pdi1), and then to substrates (8) (see Figs. 4 and 5). This relay is feedback regulated by the inactivation of Ero1 through negative regulatory disulfide formation when the ER becomes hyperoxidizing (83).
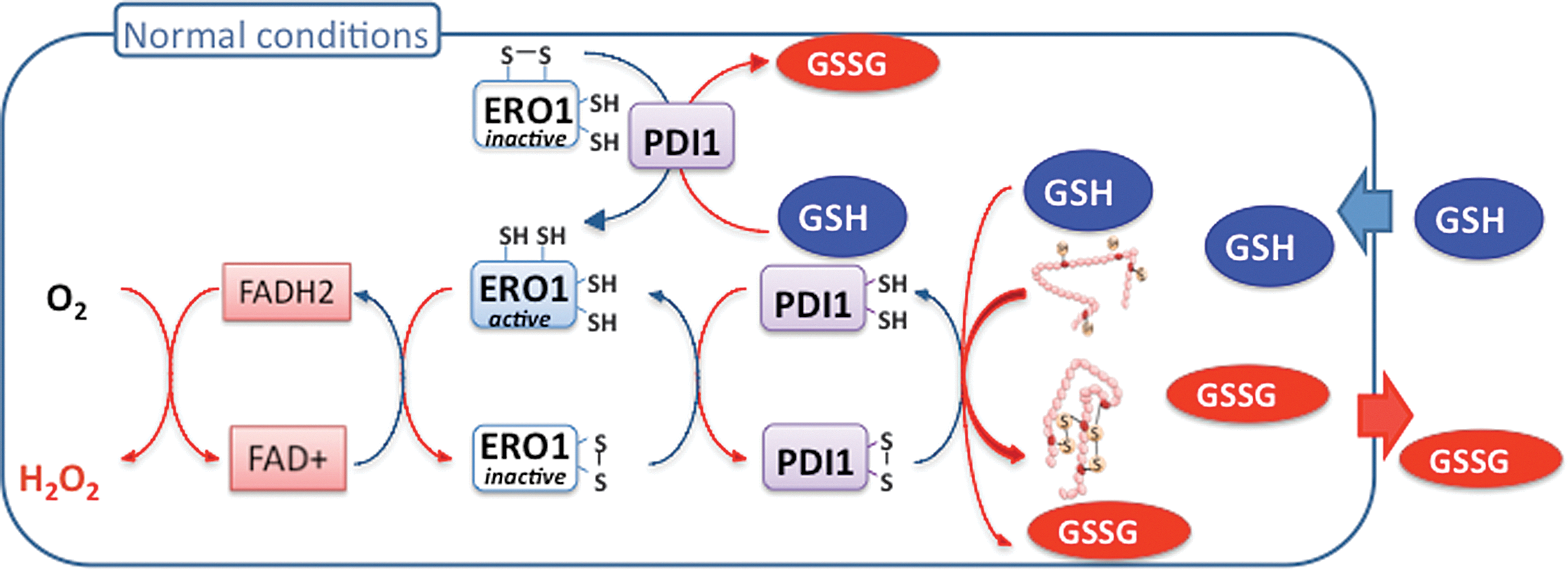

The reducing system of the ER is, however, poorly understood (12). Both isomerization and reduction steps are operated by reduced Pdi1, but how this enzyme is recycled to the reduced form is unclear, especially considering the very oxidized GSSG/2GSH ratio of the ER (see Table 2 and Fig. 6) that is unfavorable for thiol reduction. What then is the reducing source for the ER? Newly synthesized proteins entering the ER certainly constitute a net source of reduced thiols. However, whether such reducing source is sufficient to balance the ER oxidative load is questionable, even when considering the decrease of this load by Ero1 feedback inhibition. With the exception of GSH, Grx6, and Grx7, none of the disulfide reduction catalysts of the other cell compartments, or homologues thereof, are present in the yeast ER. Although induced as part of the UPR, Grx6 and Grx7 do not participate, however, to ER oxidative protein folding (36, 59). Grx6 possess both GSH-dependent oxidoreductase and glutathione transferase activity, or alternatively can assemble an iron–sulfur complex together with GSH. This protein could therefore sense the GSH redox state of GSSG/2GSH redox couple or another ER redox signal and keep in the reduced state specific Cys residues that in enzymes coordinate prosthetic groups or constitute catalytic centers (52, 58).
Refined measures obtained using GSH redox probes explain the important difference from preliminary estimates.
ER, endoplasmic reticulum; IMS, mitochondrial intermembrane space; NA, non-available.
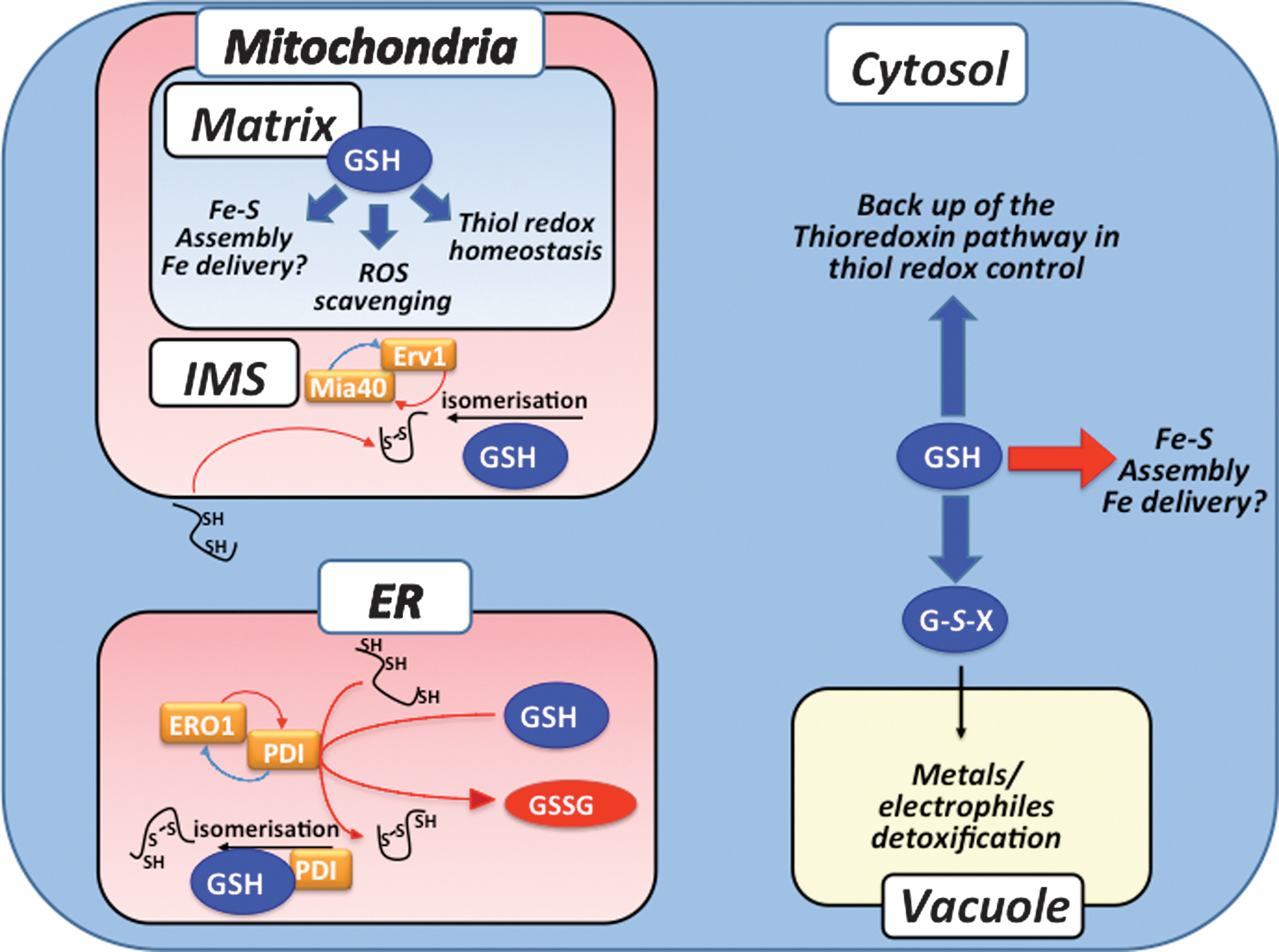
Although apparently dispensable for protein secretion in yeast (16), the role of GSH in ER redox homeostasis is, nevertheless, important, but still obscure (Figs. 3, 5, and 6). GSH was initially proposed to act as the ER source of oxidizing equivalents because of the high GSSG/2GSH ratio reported for this compartment (34) (Table 2), but is now thought to influence the ER oxidative machinery by acting in a competing pathway for disulfide reduction (16). Accordingly, the function of GSH in the ER should be to buffer excess Ero1-dependent oxidation. Still, how could GSH act as a redox buffer if no mechanisms are present to recycle its reduced form? This opened question requires further study.
Thiol Redox Control Mechanisms in the IMS
The IMS houses the mitochondrial disulfide relay that is composed of the sulfhydryl oxidase Erv1 and of the oxidoreductase Mia40 (Figs. 1 and 3). This relay couples the oxidative folding of a set of proteins with characteristic twin CX3C and twin CX9C motifs to their import into the IMS [for a review see ref. (32)]. Unfolded substrates are translocated, most likely co-translationally, in the reduced form in the IMS where they are recognized by and interact with the oxidized form of the IMS import receptor Mia40, which leads to formation of transient Mia40-substrate mixed disulfides. Mixed disulfides are then transposed into intramolecular substrate disulfides that prevent protein back translocation to the cytosol by promoting protein folding. Released, reduced Mia40 is re-oxidized by Erv1 via transfer of electrons to cytochrome c and O2. Except for GSH, the IMS is not known to carry any classical components of thiol redox control pathways (Grxs, Trxs, GSH or Trx reductases). In the IMS, GSH was proposed to increase the efficiency of Mia40-mediated substrate oxidation in an in vitro reconstituted system (5). In the absence of GSH, Mia40 accumulated into kinetically trapped mixed disulfide intermediates, which suggested that GSH might favor isomerization of non-native disulfide intermediates by reduction. Initial EGSH measurements using the redox sensor rxYFP expressed in the IMS showed this value to be very oxidizing (∼−255 mV, see Table 2 and Fig. 4) (33). However, a more recent study that used Grx1-roGFP2 indicated the IMS to be much more reducing (EGSH ∼−302 mV), and also provided data suggestive of redox linkage between Mia40 and GSH (41). The discrepancy between these two IMS redox measurements may be due to differences in the rate of equilibration of each sensor with the local GSH:GSSG environment and argues against a native Grx in this compartment to catalyze equilibration of the green fluorescent protein-based sensors with the IMS GSH:GSSG pool. In any case, as already argued upon discussing ER redox homeostasis, if GSH indeed acts as a reducing buffer in the IMS, what is the mechanism that recycles its reduced form?
Thiol Redox Control in the Remaining Compartments
This survey of the compartment-specific mechanisms that control thiol-redox homeostasis has not yet considered the vacuole, peroxisomes, and the nucleus (see Figs. 1 and 4).
The peroxisome is probably an important organelle with regard to redox homeostasis because of the production of H2O2 and other ROS during the β-oxidation of long-chains and polyunsaturated fatty acids and other metabolic processes (1). However, except for the presence of the catalase Cta1 and possibly also of the atypical 2-Cys Prx Ahp1 that carries a peroxisomal targeting sequence (46), it is not yet known whether thiol redox control enzymes and GSH are present in this organelle.
Similarly, the yeast vacuolar compartment hosts an important redox metabolism, principally operated by GSH that has not been yet really studied (see Figs. 1, 3, and 4). The ATP-binding cassette transporter yeast cadmium factor (Ycf1) is present at the vacuolar membrane where it mediates the ATP-dependent transport of GSH conjugates with metals such as cadmium (49, 50), and electrophiles. In the vacuole, these GSH S-conjugates are presumably degraded by γ-GT and a dipeptidase (56). Ycf1 is also proposed to mediate transport of GSH into the vacuole with low affinity (80), and could possibly also transport GSSG.
In the context of thiol redox control, the nucleus and cytosol can be considered a single compartment, based on the lack of a nucleocytoplasmic diffusion barrier for proteins <40 kDa, and on the knowledge that, with the exception of Glr1, all thiol redox pathways components have masses <40 kDa. Such an assumption is borne out by the observation that the GSH redox potential (EGSH) is similar in the cytosol and nucleus (17). Only in cells lacking both cytosolic TRXs was EGSH slightly more reduced in the nucleus compared to the cytosol.
Redox Linkage Between Compartments
We have so far considered thiol redox control mechanisms in each of the yeast cell compartments. It is now relevant to evaluate whether and how these mechanisms are linked to each other (Fig. 6).
Independence of cytosolic and mitochondrial matrix redox control
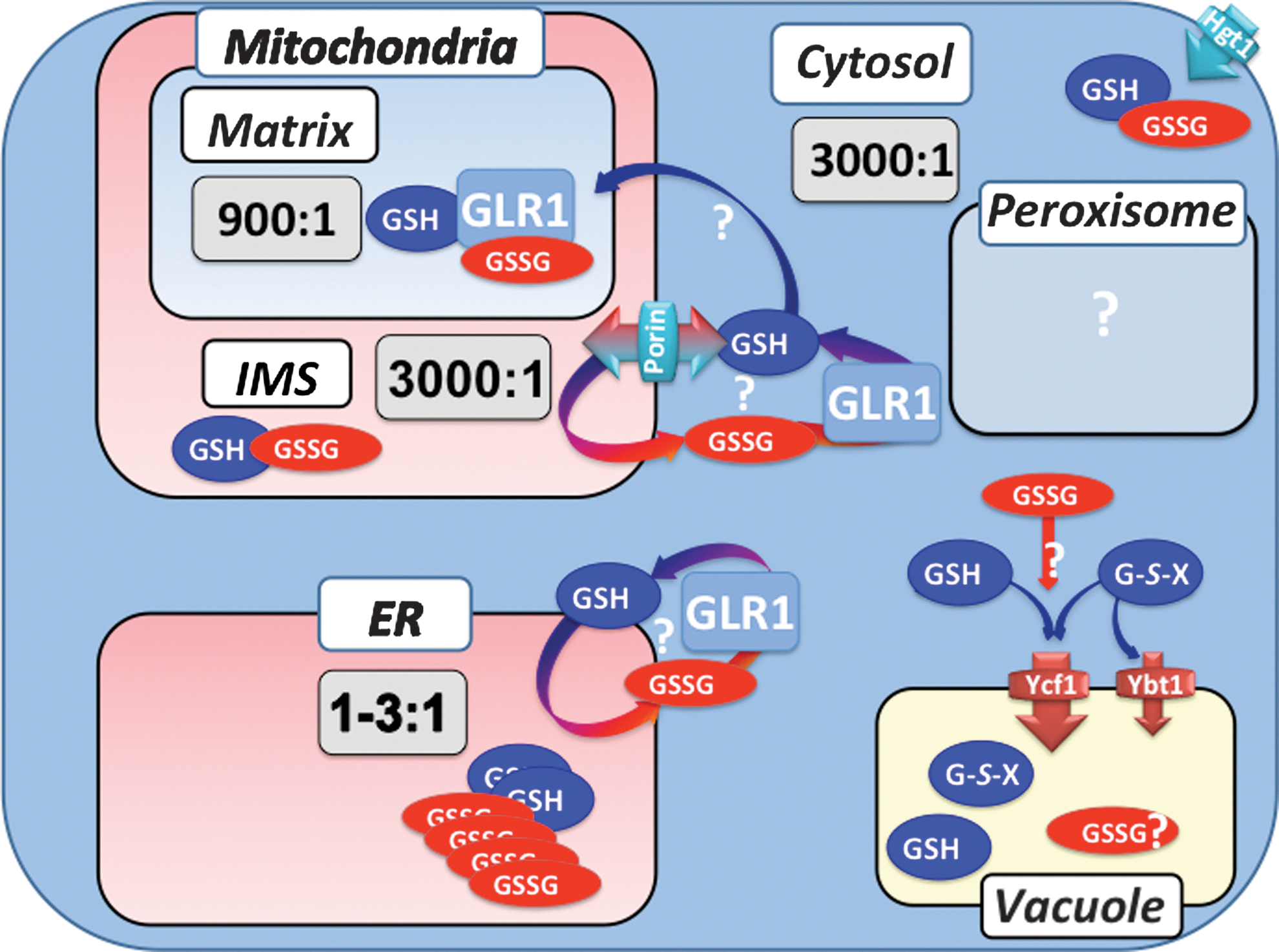
The nucleo-cytosol and mitochondrial matrix should have autonomous thiol redox homeostasis, as each carry entire sets of the TRX and GSH pathways, together with their own NADPH recycling system (see Figs. 1 and 2).
Combined mutations in genes encoding both cytosolic and mitochondrial thiol redox components (97) showed only minimal if any linkage in the redox control of these compartments. The synthetic interaction resulting from the simultaneous mutation of Trr2 and Glr1, as already discussed above, indicated the overlapping role of these enzymes in mitochondrial matrix thiol redox maintenance. In fact, only additional loss of Trr2 in Δtrr1 produced a synthetic phenotype, manifested by an aggravation of the respiratory growth and peroxide tolerance defects already existing in the latter, which indicates that to some extent mitochondrial and cytosolic TRRs might compensate for each other. Other observations unambiguously indicate the independence of the cytosol and matrix redox controls. The exclusive mitochondrial genome instability phenotype of the POS5 null mutation is a probable outcome of an exclusive matrix peroxide-scavenging defect (90). Another consequence of the Δpos5 mutation is an exclusive matrix increase of GSSG; reciprocally, mutation of ZWF1, which encode the first enzyme of the oxidative part of the pentose pathway, results in the exclusive cytosolic increase of GSSG (70). Using rxYFP, Hu et al. (33) further demonstrated that the GSH redox state of the cytosol and matrix are kept independently at different values (see Table 2). Re-expressing the cytosolic form of Glr1 in Δglr1 corrected the GSH redox state of the cytosol but not the matrix, and reciprocally, with the mitochondrial form of Glr1. The latter condition also caused some correction of EGSH in the cytosol, due to inefficient targeting of Glr1 in the matrix. These results have recently been reproduced by another group, also concluding to the independence of the redox control of GSH in the cytosol and matrix (41).
Are ER and IMS compartments in redox linkage with the cytosol?
The ER and IMS share unique redox homeostatic features not shared with other compartments. In both of them, oxidative protein folding involves thiol oxidation by two similar redox relays (Figs. 3, 4, and 5). Furthermore, thiol reduction that is needed during oxidative protein folding is probably provided by GSH in both cases; however, there are no enzymes to recycle GSSG in either the ER or IMS. The mechanisms by which reduced GSH is replenished in the ER and IMS are still unknown. Whether such replenishment is a consequence of GSH import, and possibly also of GSSG egress, into and out of the lumen of these organelles is highly suspected (12) (Figs. 4 –6).
In the ER, real-time EGSH measurements upon tunicamycin-induced ER stress indicated a switch from very oxidizing to very reducing values that lasted hours (57). Could such reduction of the ER GSH pool be linked to a regulated GSH ER import? Existence of ER facilitated GSH transporters is suggested in another study, which examined the consequences of a 10-fold abrupt increase of total cellular GSH (44). A potent and long-lasting UPR was observed at the genome-wide level, along with a major alteration of iron metabolism (discussed above). Secretion of a disulfide-containing folding substrate was eventually arrested. Under these conditions, the ER became profoundly reduced, as manifested by the major and partial reduction of Ero1 and Pdi1, respectively (Fig. 5). These ER manifestations were interpreted as a consequence of an import of GSH into the ER that perpetuated Ero1 activation by keeping the enzyme in the reduced, active conformation, and by providing reduced thiol-substrates in unlimited amounts. The fast and enduring reduction of the ER upon increase of GSH levels suggested a facilitated import of GSH into and trapping within this organelle. A regulated ER ingress of GSH is also suggested by the mechanism of Ero1C150SC295S unable to form one of the enzyme's negative regulatory disulfides. Overexpression of Ero1C150SC295S causes a marked growth inhibition and massive cell death that is explained by constitutive enzyme cycling (83). However, for enzyme cycling to perpetuate, increased supply of reduced-thiol substrates is needed, and the remaining Ero1-negative regulatory Cys residues must stay reduced. These two conditions could be fulfilled by continual ER ingress of GSH, an idea fully supported by the finding that GSH depletion both alleviates the growth defect of Ero1C150SC295S and switches the enzyme from its fully reduced to its fully oxidized form (83).
EGSH measurement in the IMS showed values that followed those of the cytosol but not the matrix (41). For instance, inactivating the cytosolic form of Glr1, which results in oxidation of the cytosolic GSH pool, also resulted in the oxidation of EGSH in the IMS, whereas inactivating Pos5, which specifically oxidizes the matrix GSH pool, did not affect EGSH in the IMS. This study also showed that inactivating POR1, which encodes the outer mitochondrial membrane channel porin, resulted in oxidation of the IMS GSH pool, suggesting that IMS ingress of reduced GSH through the porin channel is a mechanism ensuring the tripeptide reducing function in the IMS (see Fig. 6).
Conclusions and Perspectives
The TRX and GSH pathways are the two main and universally conserved disulfide reduction systems, and are therefore engaged in a diverse array of cellular functions that involve reversible disulfide formation. Survey of the data on these pathways in S. cerevisiae has highlighted several principles of their functional organization and compartmentation that can be now enumerated. In this organism, the TRX and GSH pathways are not functionally redundant, in contrast to E. coli. The pathways nonredundant functions differ with the compartment: in the cytosol, the TRX pathway exerts the dominant thiol redox control function, whereas the GSH pathway appears as backup of the former; in mitochondria, the GSH pathway seems to dominate over the TRX pathway. The mitochondrial matrix has an entire set of both pathways that exert a totally independent thiol redox control insulated from influences from other compartments. One main function of the matrix thiol redox system is to assist respiratory ROS scavenging. The ER and IMS are small organelles that lack thiol reductase systems, except for GSH. They host an intense oxidative thiol metabolism and therefore must generate GSSG. How GSH is replenished is unknown, but could involve fluxes of GSH and GSSG between these organelles and the cytosol (see Fig. 6). How these principles apply to higher eukaryotes is unknown, although they should help explore thiol redox control in mammals and plants.
Footnotes
Acknowledgments
This work was supported by grants from ANR (ERRed), FRM, and INCA to M.B.T., and from VLM to A.D.-M.