
Abstract
Introduction
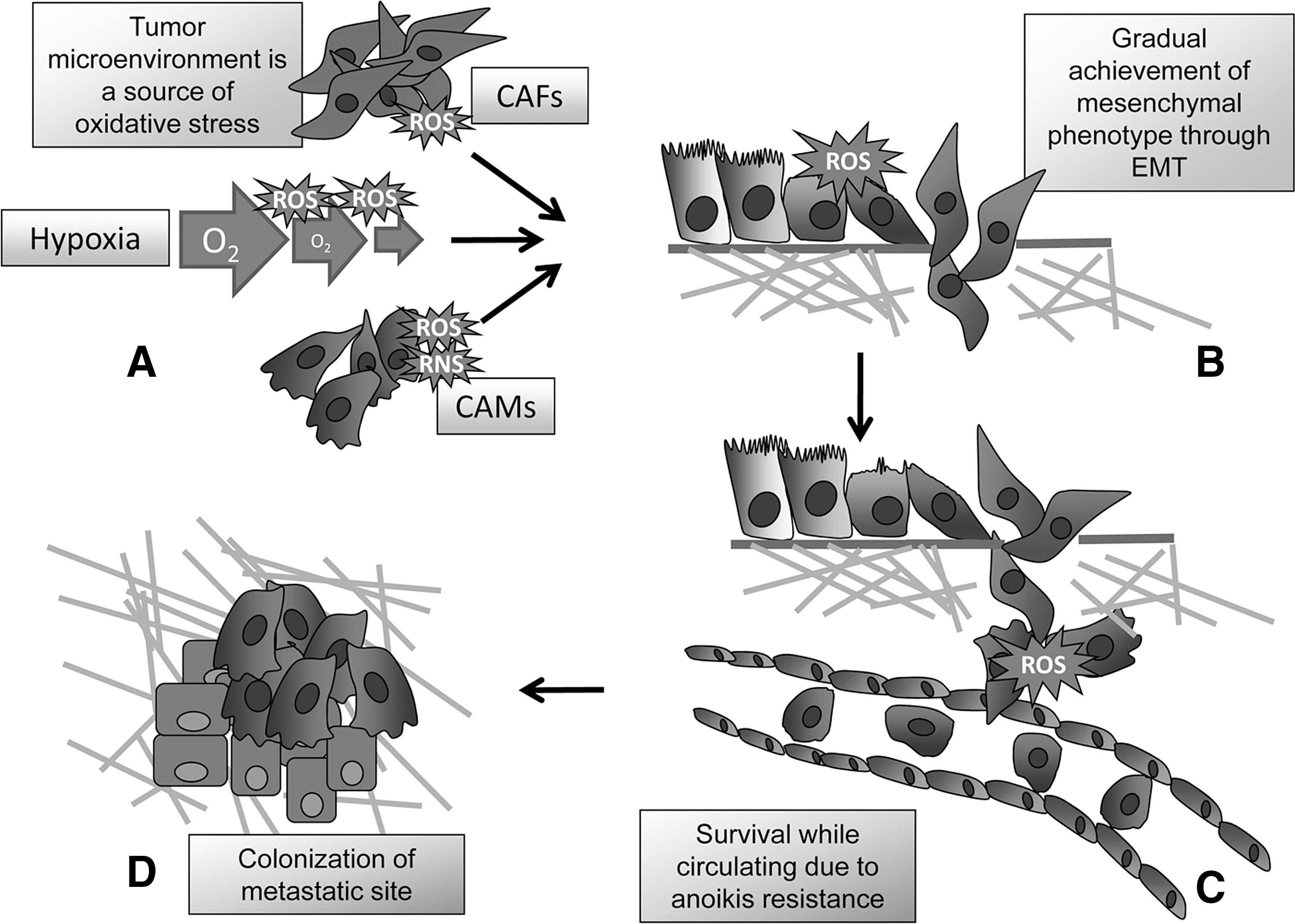
This review is focused on protein sensors of oxidative modifications engaged during tumor progression, a complex process characterized by morphological and behavioral changes of cancer cells, mainly due to accumulation of genetic and epigenetic changes. These lead to achievement of features, such as ability to withstand adverse environmental conditions, or to better respond to proliferative stimuli, which will be positively selected during tumor progression (Table 1) (14). With the progression of malignant disease, some cells acquire the ability to disengage from the primary tumor and colonize a distant organ. This process, called metastatic colonization, is a multistep phenomenon, which begins with the dissociation of tumor cells from surrounding cells by increasing their motility and invasive capacity, mainly due to concomitant factors of the tumor microenvironment, as cancer-associated macrophages or fibroblasts (CAMs or CAFs) or intratumoral hypoxia. These structural cellular factors are able to elicit an epigenetic program in cancer cells, called epithelial–mesenchymal transition (EMT), enhancing motility and the achievement of stem-like traits allowing these cells to proceed the long route of cancer metastasization. The process continues with tumor cell intravasation within the blood and/or lymphatic system, in which these cells have to survive to the mechanical insults of circulation, proceeds with extravasation, and terminates with the implantation and proliferation at new sites (Fig. 1) (31, 100, 103).
Redox Molecular Machines Involved in Tumor Progression Toward Malignancy
PTK, protein tyrosine kinase; PTEN, 3-phosphatase and tensin homolog; SHP-2, Src homology-2 domain–containing phosphatase 2; FAK, focal adhesion kinase; LWM-PTP, Low molecular weight-protein tyrosine phosphatase; NF-κB, nuclear factor-κB; IKK, Inhibitor of κB kinase; FoxO, forkhead box class-O; TF, transcription factor; HIF, hypoxia inducible factor; PHD, prolyl hydroxylase; Nrf2, nuclear respiratory factor-2; PKM2, pyruvate kinase M2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PPP, phospho-pentose pathway; AMPK, adenylate monophosphate kinase; ATM, ataxia telangiectasia mutated; G6PD, glucose-6-phosphate dehydrogenase; ADP, adenosine-diphosphate; ATP, adenosine-triphosphate; NEMO, NF-kappa-B essential modulator; PKA, protein kinase A; RTKs, receptor tyrosine kinases.
For an efficient engraftment both at the primary tumor site and at the metastatic location, tumor cells must have the ability to adhere to various extracellular matrix (ECM) components through specific cell receptors that belong to the family of integrins. Indeed, cell adhesion to the ECM is an essential condition to allow cells to survive and proliferate (25). However, during cancer progression, some cells may acquire the ability to grow and proliferate in the absence of anchorage to the ECM, thereby becoming resistant to anoikis, the natural apoptotic program that normal cells undergo in the absence of ECM contact (125). In particular, anoikis-resistant cells may obtain a real advantage in terms of successful metastatic colonies, due to their ability to survive when circulating within the bloodstream or the lymph system in suspended conditions (Fig. 1) (47, 125).
Tumors progression is the result of the sequential acquisition of genetic and epigenetic alterations promoting cell's ability to adapt to changes in their microenvironment and to remodel their surroundings. In addition, many cytotoxic anticancer agents can induce oxidative stress. Extensive research has unveiled the mechanism by which continued oncogenic, oxidative, and environmental stress can mediate cancer progression toward malignancy. Conversely to non-neoplastic cells for which oxidative stress usually leads to extensive damage, in cancer cells, oxidative stress-related pathways can activate a variety of proteins and/or transcription factors (TFs) responsible for multiple epigenetic adjustments of transcriptome, leading to adaptive responses used by cancer cells to increase their resistance to the stressful environment, engaging modifications of cell metabolism, de novo angiogenesis, stem cell survival, and spread through activation of motogen escaping plans. Studies in several cancers have clearly established that oxidative stress players are expressed aberrantly in cancer and positively affect several of the above-mentioned mandatory steps.
The term reactive oxygen species (ROS) covers a range of partially reduced metabolites of oxygen (e.g., superoxide anions, hydrogen peroxide, and hydroxyl radicals) possessing higher reactivity than molecular oxygen. Inside cells, they are by-products of normal aerobic metabolism, or second messengers in various signal transduction pathways, including growth factors and integrin signaling. ROS affects cell proliferation and anchorage-independent cell growth, causes insensitivity to apoptosis, sustains de novo angiogenesis, and alters the migration/invasion program through metabolic and epigenetic mechanisms (37, 103, 104). The consequences of the production of oxygen radicals on cancer biology are pleiotropic and complex. In fact, beside being directly involved in mutagenesis and genomic instability, ROS also contribute epigenetically to cancer development and progression, by acting as signaling intermediates downstream of mitogen receptors and adhesion molecules and as inducers of genetic programs leading to cell invasion and malignancy as mesenchymal–epithelial transition (45, 50). Furthermore, oxidation of cell constituents is a general cause of cell stress, also due to tumor microenvironment as CAFs or CAMs producing ROS and/or reactive nitrogen species (23, 24, 50, 130), and promotes spontaneous and therapy-induced tumor cell death by making cells more vulnerable (82).
The primary effect that ROS exert as signal transduction messengers is the reversible regulation of proteins, adjusting protein functions through their oxidation of low pKa cysteines. Growing evidence indicates that thiol groups act as redox-sensitive switches, thereby providing a common trigger for a wide range of ROS-mediated signaling events. Oxidation of cysteines forms reactive sulfenic acid that can form disulfide bonds with other near cysteines, or undergo further oxidation (sulfinic or sulfonic acid) (15, 114). In case of presence of close nitrogen residues, the sulfenic acid may react to form a sulfenamide. Glutathionylation of proteins, which occurs through the formation of a mixed disulfide between one cysteine of glutathione and one cysteine of the other protein, constitutes an efficient mechanism to protect proteins from irreversible modifications, and also seems to play an important role in cell signaling. All these oxidative modifications always result in structural or functional changes of the redox sensor protein (113). Protein oxidation can be reversed by thiol donors such as glutathione, glutaredoxin, peroxiredoxins, and thioredoxin. In particular, peroxiredoxins are members of a newly discovered family of peroxidases able to efficiently decrease growth factor-induced hydrogen peroxide (116). To regenerate S-thiolated proteins, these systems act as reducing agents, rather than as free radical scavengers. The enzymes that catalyze protein thiol disulfide redox reactions have a CXXC motif in the active site and oxidoreduction of these cysteines, by either mono- or di-thiol mechanisms, is part of their catalytic cycle. While thioredoxin is able to drive reduction of sulfenic acids (SOH) and disulfides (S-S), glutaredoxin can support the reversibility of mixed disulfides with reduced glutathione (GSH) and S-nitrosothiols, while only sulfiredoxin can reverse the formation of sulfinic acids (SO2H). Finally, sulfonic acids (SO3H) are currently believed to be irreversible terminal products of overoxidation.
Reversible cysteine oxidation plays a pivotal role in redox signaling cascades, major molecular targets being protein tyrosine phosphatase (PTP) or lipid phosphatases, proteases, signaling adaptors, and TFs. The effect of cysteine oxidation varies among redox-regulated proteins, from inactivation of PTPs and some TFs, to activation of protein tyrosine kinases (PTKs) (either receptor or cytosolic kinases), small GTPases, and a small group of TFs. Cysteine oxidation may be either inhibitory for proteases of the caspase family, exerting an antiapoptotic role (94), or activatory for matrix metalloproteases (MMPs), through the disruption of the inhibitory interaction between the prodomain and the catalytic site (57). Reversible cysteine oxidation of TFs affects DNA binding and transcriptional activity of several nuclear factors, including activating protein-1, nuclear factor-κB (NF-κB), p53, and hypoxia inducible factor (HIF) (90). In most cases, oxidation interferes with DNA binding and inhibits transcriptional activity that is reactivated upon cysteine reduction assisted by the redox factor-1 (140).
Beside directly targeting enzyme activities, cysteine oxidation can modulate signal transduction by interrupting protein–protein interactions. One example of this kind of regulation is the nuclear translocation of nuclear respiratory factor-2 (Nrf-2) following disruption of its interaction with the adaptor Keap-1, upon oxidation of the latter on cysteines 273 and 288. Free Nrf-2 migrates to the nucleus, where it activates the antioxidant and detoxifying response through an antioxidant response element (ARE)-dependent gene transcription (30, 90).
Beside cysteine oxidation, ferrous iron containing proteins may also be targeted by ROS, though oxidation of ferrous to ferric iron and the consequent disruption of protein function. Among this group of proteins, at least for what the purpose of this review is concerned, we can mainly focus on 2-oxoglutarate (2OG) dioxygenases. These are ubiquitous 2OG-dependent oxygenases that couple substrate oxidation to the conversion of 2OG to succinate and carbon dioxide and their role includes collagen biosynthesis, DNA repair, chromatin modifications, and hypoxic sensing (13, 95). Fe++/2-OG-dependent dioxygenases catalyze a range of oxidative reactions, possibly the widest of any enzyme family, but they are sensitive to oxidative functional inhibition. Among this group of enzymes, prolyl hydroxylases (PHDs) and JmjC histone demethylases have been reported to play important roles during cancer progression (69, 97, 120).
Non proteins substrates
Beside proteins, oxidants may also target DNA or lipids eliciting the formation of molecules endowed with signaling abilities. For example, the formation of 8-oxo-guanine upon oxidative injury of DNA has been reported to play a role in transcriptional activation downstream to estrogens receptor and Myc oncogene (2, 107). Beside, several lipid peroxides take a role as signaling molecules downstream to lipoperoxidases or cycloxygenases metabolism of arachidonic acid. In addition, nonenzymatic lipid peroxidation alters the noncovalent interactions within the membrane bilayer, contributing to local membrane destabilization (58).
Redox Machines Involved in Cell Adhesion and Cytoskeleton Remodeling
Several molecular targets of redox balance belong to cell cytoskeleton machinery, thereby deeply affecting the ability of cancer cells to adhere to the ECM or other cells, as well as to move and invade surrounding tissues, allowing tumor cells to metastatize. These two characteristics of cell motility have been included within the hallmarks of cancer, thereby accounting for the great impact that redox regulation of cytoskelon components have within cancer malignancy (63).
This class of redox machines includes both structural components of cytoskeleton as β-actin, or proteins regulating cytoskeleton dynamics, as the tyrosine kinase Src or receptor tyrosine kinases, as well as PTPs (Fig. 2).

β-actin
The movement of cancer cells escaping the primary tumor, invading surrounding tissue and reaching and colonizing other organs in which they metastatize, is driven by actin polymerization at the leading edge of the moving cells, an event that is under redox control. Indeed, ROS can act directly on structural cytoskeleton proteins thereby affecting cell spreading and the overall organization of the cytoskeleton architecture. A proteomic screen for cysteine glutathionylated proteins in T cells exposed to oxidant agents diamide or hydrogen peroxide identified several cytoskeleton components (actin, vimentin, myosin, tropomyosin, cofilin, and profilin) (41). Hydrogen peroxide produced by integrins upon ECM contact has been involved in in vivo β-actin redox regulation. Actin oxidation takes place via the formation of a mixed disulfide between cysteine 374 and reduced glutathione. The impairment of actin glutathionylation, through either GSH depletion or expression of the C374A redox insensitive mutant, significantly affects the formation of stress fibers and consequently cell spreading (38, 39). The redox regulation of actin has serious implications for cell contractility and movement as glutathionylation of β-actin causes the disassembly of the actinomyosin complex. Although a high-resolution structure of the actomyosin interface is not yet available, it is likely that the redox modification to which actin undergoes during cell spreading impedes actin binding to myosin, thus exerting a crucial effect on assembly/disassembly dynamics of the contractile machinery (36, 39).
Src kinase
The tyrosine kinase Src, a major effector in integrin signaling, is transiently oxidized and activated upon cell adhesion to the substrate (71). Src-family kinases are critically involved in the control of cytoskeleton organization and in the generation of integrin-dependent signaling responses, inducing tyrosine phosphorylation of numerous signaling and cytoskeletal proteins. Beside the well-known phosphorylation/dephosphorylation circuitry involving Tyr416 and Tyr527 of c-Src kinase, cysteine oxidation has been recently reported as a further mechanism of enzyme activation. A first direct proof of a redox chemical modification of Src kinase has been achieved from in vitro experiments on mercuric chloride and NO-releasing agents (1, 98). Beside in vitro evidence, Src is also regulated via oxidation during anchorage-dependent growth (48). During ECM contact, Src kinase undergoes biphasic activation in response to integrin receptor activation: there is a slight early activation in concomitance with focal contact formation and a late stronger activation of the kinase, driving cytoskeleton organization and cell spreading. The kinase undergoes oxidation/activation in response to the formation of an S-S bond between Cys245 and Cys487, respectively, located in the SH2 and in the kinase domain of the Src molecule (48). Conservative mutation of the two cysteines to alanine gives rise to redox-insensitive kinases, unable to become activated during ECM adhesion. Mounting evidence describes Src activation via its redox regulation as a key outcome in several circumstances, including growth factor and cytokines signaling, integrin-mediated cell adhesion and motility, membrane receptor cross talk as well in cell transformation and tumor progression (15, 51). Finally, Src oxidation has also been involved in survival to hypoxia due to mitochondrial delivery of ROS and regulation of NF-κB TF (91).
Beside c-Src, activation via oxidation is mandatory also for the oncoproteins v-Src and SrcY527F, which are oxidized by exogenous oxidative bursts or during anchorage-independent cell growth. The redox regulation of v-Src or SrcY527F greatly affects the oncoproperties of transformed cells, as indicated by the susceptibility to antioxidants or cell-permeable catalase of MMP expression, as well as their serum- and anchorage-independent growth or solid tumor formation (48). In keeping with a key role of redox regulation of oncogenic Src kinases, v-Src activity as well as its oncogenic potential is reduced by curcumin, a powerful dietetic antioxidant (87).
Src oxidation has also been reported for molecular cross talk between membrane receptors, as receptor tyrosine kinases, integrin receptors, and G-protein-coupled receptors (15, 51, 91, 111). For example, Src oxidation occurs during anoikis protection by ECM contact in nontransformed cells (47). ROS developed upon integrin engagement lead to Src oxidation/activation, which in turn sustain, via ligand-independent phosphorylation and activation of the epidermal growth factor receptor, a prosurvival pathway inhibiting anoikis death. Indeed, the activated epidermal growth factor receptor leads to activation of Akt kinase, phosphorylation of the proapoptotic protein Bim, and suppression of apoptotic death (47). In metastatic carcinoma cells, naturally resistant to anoikis, Src oxidation and a sustained prosurvival signal has been acknowledged as mandatory to resist apoptosis during metastatic spread and obtain successful metastases (49).
Protein tyrosine phosphatases
PTPs are a large group of hydrolases, encoded by about 100 different genes. They regulate the tyrosine phosphorylation of several signaling proteins and exert multifaceted functions in cell proliferation, survival, and motility. Although members of the PTP family differ in their intrinsic susceptibility to oxidation, they invariably contain a catalytic Cys residue, which confer redox sensitivity. PTP active-site cysteine is targeted by various oxidant species, such as H2O2, superoxide or nitric oxide, and this modification can be reversed by incubation with thiol compounds (16). In vivo the reversibility of PTP inactivation is guaranteed by specific blocks of excessive oxidation of the SH group, through formation of a mixed disulfide with glutathione, an intramolecular S-S bridge among cysteines, or a sulfenyl-amide intermediate (16, 114). Numerous PTPs undergo redox-dependent inactivation in cells in response to various growth factors, thereby obtaining a shift of PTP/PTK activity favoring protein phosphorylation. Redox-regulated PTPs include PTP1B, Low Molecular weight PTP (LMW-PTP), 3-phosphatase and tensin homolog (PTEN), Src homology-2 domain–containing phosphatase 2 (SHP2), and many others. Several of these PTPs behave as regulators of cytoskeleton dynamics during cell motility, adhesion, and proliferation and their regulation via redox-based mechanisms plays a mandatory role in such a context.
Redox regulation of the lipid phosphatidyl-inositol (PI) PTEN acts on adhesion and motility mainly through its indirect regulation of the downstream kinase Akt (85). Indeed, PTEN metabolizes cellular PtdIns(3,4,5)P3 and acts in direct opposition to PI3-kinase, thus playing a main role in the inhibition of PI3-kinase-dependent downstream signaling, leading to inactivation of Akt kinase and mammalian target of rapamycin signaling. PTEN is sensitive to rapid inactivation by hydrogen peroxide or by S-nitrosothiols, forming a stable intramolecular disulfide bond between the active site Cys124 and the proximal Cys71 (80). PTEN oxidation/inhibition leads to inability to contrast PI3-kinase and to sustain Akt activation, acknowledged to favor proliferation, adhesion, spreading, and survival to stresses. Furthermore, oxidation/inactivation of PTEN seems to be regulated by thioredoxin-interacting protein or peroxiredoxin 1, both acting through reduction of the PTEN disulfide. PTEN is a tumor suppressor gene localized to the human chromosome 10q23.31, a genomic region frequently lost in glioblastoma and prostate cancer. In addition, studies of both humans and mice show that even partial loss of PTEN function is sufficient to promote some cancer types, particularly in the breast. The PTEN protein undergoes oxidation in response to growth factors stimulation, correlating with ROS-dependent activation of downstream Akt phosphorylation (86). PTEN oxidation has been implicated in the development of T-cell acute lymphoblastic leukemia, as well as in the multiorgan tumorigenesis in mice lacking peroxirexoxin-1 (11, 123).
Redox regulation of SHP-2, a widely expressed positive regulator in signaling pathways mediated by several growth factors and cytokines, has also been correlated with adhesion and motility (6, 126). Upon integrin receptor engagement by ECM proteins, SHP2 undergoes a reversible oxidization/inactivation to which mitochondrial and 5-lipoxygenase-derived ROS contribute differentially. Indeed, the first wave of oxidation of SHP-2 is mainly driven by mitochondrial delivery of ROS, while the second wave is driven by cytosolic ROS. The final effect of reversible oxidation/inhibition of SHP-2 is to prevent the dephosphorylation and inactivation of its substrates p125 focal adhesion kinase (FAK) and SHP2-substrate-1, thereby enabling the continued propagation of signals arising by integrin engagement. SPH-2 has also been found oxidized in response to hypoxia, a common feature of several growing cancers (118, 129).
LMW-PTP is a redox molecular machine acting in multiple points during cell proliferation and adhesion. First, during PDGF receptor signaling, it undergoes reversible inhibition due to oxidation of its catalytic Cys12 and nearby Cys17, thereby maintaining receptor activation through tyrosine phosphorylation and allowing signal transduction toward the nucleus (18). Second, its oxidation plays a mandatory role during anchorage to the ECM and cell motility. Indeed, the transient and reversible inhibition of LMW-PTP is instrumental for activation of FAK in response to integrin engagement (20). Beside FAK activation, LMW-PTP oxidation is involved in the antagonistic cross talk between Rac-1 and RhoA small GTPases (16, 17), a crucial event for cellular dynamics, the former promoting membrane protrusion, cell polarity and spreading, the second cytoplasm contractility and tail retraction (35). Rac-1 and RhoA have been acknowledged to orchestrate two different and mutually exclusive motility styles in invading malignant cells: Rac-1 is mandatory for polarized and proteolytic mesenchymal motility style, while RhoA has been involved in cytoskeleton contractility and amoeboid motility, an ancestral motility style used by lymphocytes and numerous cancer cells to invade the ECM independently from its proteolytic degradation (35, 42). Rac1-3 small GTPases have been reported to be upstream regulators of ROS production thorough activation of NADPH oxidases, as well as other membrane-bound or mitochondrial ROS sources (16, 114). During polarized motility Rac-1-driven ROS, by inactivating LMW-PTP, causes hyperphosphorylation of the PTP substrate p190Rho-GTPase Activating Protein (GAP), thereby increasing its inhibitory activity on RhoA (17). On the contrary, during amoeboid motility in metastatic carcinoma cells, there is a reduction of Rac-1 activity owing to attenuated generation of ROS, causing LMW-PTP activation, p190RhoGAP dephosphorylation, and to an increase of Rho signaling (8). Hence, LMW-PTP takes a central role in the decision of the most useful motility style to be engaged for cancer cells. Of note, the key event driving cancer cells toward polarized or amoeboid motility is switched by the LMW-PTP redox engine.
Redox Machines Involved in Control of Gene Expression
The activity of TFs is modulated by redox cascades either in a direct or indirect fashion, that is, through the regulation of their phosphorylation, acetylation, hydroxylation. Among the plethora of redox-sensitive TFs, we will focus only on NF-κB, Nrf-2, forkhead box class-O (FoxO), p53 and HIFs, owing to their mandatory role in cancer progression and antioxidant response.
Nuclear factor-κB
NF-κB was the first TF recognized to be sensitive to redox homeostasis in eukaryotic cells (124). NF-κB is the mandatory TF, originally involved in the inflammatory response orchestrated by a plethora of cells. More recently, NF-κB has been involved in several steps of tumor progression, as the activation of CAMs and their polarization toward M2 phenotype, reactivity of CAFs, the recruitment of endothelial precursor cells to instruct de novo angiogenesis and vasculogenesis, as well as in the motogenic epigenetic transcriptional programs of metastatic cancer cells, leading these tumor cells to enhance their motility through EMT and their stem cell traits. All these events, driven by NF-κB activation, concur to the successful metastatic spread of tumor cells (33, 52, 139).
NF-κB forms homo- or heterodimers composed by Rel or NF-κB subfamily members, which interact with the inhibitory proteins IκBs, preventing translocation of the TF to the nucleus and its binding to DNA. In the absence of proinflammatory stimuli, NF-κB is kept latent in the cytosol bound to its inhibitors IκBs. Prototypical activators of NF-κB, as tumor necrosis factor α, lead to phosphorylation of IκBs by IκB-kinase (Inhibitor of κB kinase [IKK] complex), rapidly leading to proteosomal degradation of IκBs. Consequently, free NF-κB migrates to the nucleus and activates transcription of target genes. Redox regulation of NF-κB embraces both cytoplasmic and nuclear steps in NF-κB activation, including degradation of its inhibitor IκBα, regulation of DNA binding ability, and transcriptional activity, as well as chromatin remodeling (52, 76).
First, NF-κB activation following tumor necrosis factor-α stimulation is mediated through ROS-mediated signaling, leading to protein kinase A-mediated NF-κB/RelA Ser phosphorylation and critical modification for its transcriptional activity (73). Second, H2O2 directly targets NEMO, an essential regulatory component of the IKK complex, which controls activation of the NF-κB signaling pathway. Indeed, hydrogen peroxide induces formation of NEMO dimers through disulfide bonds formed between Cys54 and Cys347, leading to inhibition of the IKK complex, a clear requirement for a correct NF-κB activation (68). In addition, tyrosine phosphorylation of IκB-α is also under indirect redox control. For example, during hypoxia exposure of cancer cells, a very common feature of cancer microenvironment characterized by mitochondrial delivery of ROS due to incomplete oxygen reduction (13), IκB-α is phosphorylated by oxidized/activated c-Src (91). Hence, the activation of NF-κB through c-Src-mediated phosphorylation is indirectly due to oxidation of the tyrosine kinase and not to components of the TF, but has been reported to promote carcinogenesis through maintenance of cell survival and tumor progression. H2O2 also inhibits the nuclear import of IκB-a, thereby facilitating its proteasome degradation, and concurring to maintain NF-κB in the nucleus (52). NF-κB is also sensitive to oxidation of Cys62 in its p50 subunit, essential for DNA binding (96). Of note, NF-κB activation and nuclear translocation are stimulated by oxidizing conditions, common within cytoplasms receiving proinflammatory or motile stimuli, while DNA binding is inhibited by Cys62 oxidation. Indeed, once in the nucleus, Cys62 of p50 needs to be re-reduced by Ref-1 to enable NF-κB transactivation of the DNA (108).
Although NF-κB is regulated by cellular redox homeostasis at multiple levels, the role of this control in the numerous environmental settings in which it occurs, as proinflammatory, promigratory, or prosurvival stimuli, is still not completely understood. One attractive idea is that H2O2 could behave as a synergistic player with canonical activation of the TF by prototypical activators, thereby leading to upregulation of genes containing low-affinity NF-κB sequences (102).
Forkhead box class-O
FoxOs are a family of TFs, including four members called FoxO1, FoxO3a, FoxO4, and FoxO6, involved in crucial cellular process, including cell cycle regulation, apoptosis, and resistance to oxidative stress (29, 136). In addition, several members of the family have been involved in cancer progression and metastatic spread. Oxidative stress-mediated activation of FoxOs was shown to regulate a variety of genes, including genes that promote cell motility as MMPs, stress resistance proteins as superoxide dismutase and peroxiredoxins, stemness and self renewal regulators as p27 or p21 cyclin inhibitors. (99).
FoxOs can be either directly or regulated via redox mechanisms. Indirect redox regulation of FoxOs is mainly mediated by the control of their phosphorylation. FoxOs are regulated by several kinases, leading to opposing effects on TF activity. Generally, an increase in intracellular ROS facilitates the localization of FoxOs to the nucleus where they are transcriptionally active (34). Indeed, an increase in intracellular reactive ROS induces the activation of the c-Jun-N-terminal kinase, as well as the acetylating enzyme complex p300. FoxO4 could be phosphorylated through the Jun N-terminal kinase pathway, while FoxO3a and FoxO1 are targeted by mammalian sterile 20-like kinase signaling (34, 84). Of note, redox-dependent phosphorylation of FoxO can also produce opposite effects, as reported for Akt-mediated phosphorylation of the TFs. FoxOs are directly phosphorylated by Akt on three conserved residues and this correlates with nuclear export, cytoplasmic retention, and inhibition of transcriptional activity of FoxOs (29). ROS can also act on this pathway, as upon PTEN oxidation/inhibition, Akt is largely activated, leading to FoxO phosphorylation and to nuclear exclusion of the factor (7). However, several post-translational modifications are needed to exert the full spectrum of FoxO-mediated responses to ROS and multiple phosphorylation (activatory or inhibitory), acetylation, and monoubiquitination of FoxO concur to regulate the transcription of specific subsets of genes that regulate cellular detoxification, survival, cell cycle arrest, or apoptosis.
FoxO activity can also be regulated via direct oxidation. Following hydrogen peroxide treatment or glucose deprivation-induced mitochondrial ROS production, cysteines within FoxO4 are subject to oxidation, disulfide formation, but avoiding protein dimerization. Indeed, an intramolecula disulfide can be formed between FoxO and the histone acetylase p300. The covalent binding of p300 with FoxO leads to acetylation of the TF, driving a shift in transcriptional targets (28). This acetylation-dependent inhibition can be reversed by the NAD-dependent deacetylase SirT-1, thereby rescuing classic FoxO transcriptional activity (131).
Hypoxia inducible factors
The HIF family, composed by three members HIF-1/2/3, plays a key role in the reprogramming of cancer metabolism, motility, and survival to stress and chemotherapeutic agents. HIFs consist of an O2-regulated HIF-α subunit and a constitutively expressed HIF-β subunit (120, 121). At least for the best known HIF-1 member, in well-oxygenated cells, HIF-1α is hydroxylated on proline residue 402 and Pro-564 by PHDs, which use O2 and 2-OG as substrates (77). Prolyl-hydroxylated HIF-1α targets HIF-1α for proteasomal degradation. In oxygenated cells, beside hydroxylation of prolines, Asp803 is hydroxylated by factor inhibiting HIF-1 (FIH-1), leading to inhibition of binding of histone acetylase coactivators, as p300 (121). Severe hypoxia due to O2 deprivation leads to inhibition of both PHD- and FIH-mediated hydroxylation, thereby causing HIF-1 stabilization and enhancing its transcriptional activity.
Compelling evidence indicates that also HIFs are subdued to multiple mechanisms of redox regulation. S-nitrosylation of HIF1 at Cys-800 has been revealed to increase protein stability and transcriptional activity, by promoting HIF binding to p300 histone acetylase through a disulfide with p300 Cys-388 or 393 (141). In parallel, hydrogen peroxide indirectly promotes HIF stabilization by oxidizing the catalytic Fe++ of PHDs and inhibiting their activity (44). This second mechanism may be implicated in physiological stabilization of HIFs under mild (1%–3%), but not deep, hypoxia, a circumstance accompanied by production of ROS both from mitochondria and from membrane-bound NADPH oxidases (60, 119). In keeping with a key role exerted by redox regulation of HIF-1, it has been recently reported in three different tumorigenesis mouse models that antioxidants exert their antitumoral effect mainly acting on the HIF-1α level (43). The regulation at a redox level of HIFs plays a particularly important role in normoxic circumstances (46, 62, 106). Indeed, several reports have indicated a role of the HIF-1 transcriptional response in circumstances in which oxygen is not limiting, as growth factor supply or treatment with chemotherapeutic drugs. PHD inhibition by ROS delivery, common to both growth factor signaling and citotoxic drug sensing, may thus explain the normoxic accumulation of HIFs in cancer cells (137). ROS also mediate oncogene-driven accumulation of HIF-1 in normoxia, a phenomenon crucial for the establishment of the Warburg effect (see Metabolic Reprogramming Due to Redox Molecular Machines). For example, H-Ras stabilizes the hypoxic factor through the generation of superoxide anion (137).
p53
p53 is a key redox-sensitive TF, that controls a wide variety of target genes and regulates numerous cellular functions in response to stresses that lead to genomic instability (79). Upon activation by a multiplicity of stress-related signals (DNA damaging agents, hypoxia, heat shock, etc.), p53 transactivates genes responsible for cell cycle arrest, apoptosis, and DNA repair. Recent studies present evidence that ROS function upstream of p53 in some model systems, while in others ROS production could be a downstream effect of p53 activation (89).
Oxidative modification of the cysteine residues of p53 causes its conformational changes affecting its transcriptional activity and biological responses (93). First, p53 is glutathionylated on its Cys141 under oxidative stress, with a consequent inhibition of p53 dimerization and association with DNA (134). Thioredoxins, a family of small redox proteins that undergo NADPH-dependent reduction by thioredoxin reductases and reduce oxidized cysteines on proteins, might play a direct role in regulating the redox status of p53. For example, oxidative stress due to GSH depletion significantly reduces p53 DNA binding and activation of its specific reporter genes (117). Indeed, p53 may also be oxidized during oxidative stress forming an intramolecular disulfide, which can be re-reduced by thioredoxins to facilitate the DNA binding activity of p53 (93). Finally, NO or ONOO− can also modulate the redox status of p53 via nitration of critical tyrosine residues present in its DNA binding domain, resulting in p53 aggregate formation, and loss in DNA binding ability (22). Of course, these redox modifications shielding of reactive cysteines contributes to a negative regulation for human p53 and may represent an acute defensive response with major consequences for oncogenesis.
Nuclear respiratory factor-2
To prevent oxidative stress, the cell must respond to ROS by mounting an antioxidant response. The primary TF involved in activation of AREs under oxidative stress conditions, is Nrf2, a cap ‘n’ collar-b-zip TF (72). Nrf2 is commonly segregated into the cytoplasm by association with Keap1, which serves for proteasomal degradation of Nrf2 (135). Keap1 Cys151, Cys273, and Cys288 behave as redox sensors, and their oxidation results in the dissociation of Nrf2 from Keap1, allowing Nrf2 stabilization and translocation into the nucleus, dimerization with Maf proteins, binding to the ARE enhancers and activation of transcription of target genes with antioxidants and detoxification properties (135, 143). In addition to this regulatory mechanism, Nrf2 Cys-514, located in the DNA binding domain, or Cys119, located in the transactivation domain, undergo Ref-1-mediated control and their mutations affect binding to ARE and decrease transcription (5).
To highlight the importance of the Nrf2 antioxidant response pathway as a determinant of susceptibility to carcinogenesis, multiple studies reported a correlation between Nrf2 disorders and enhanced incidence and tumor burden in several models of colon, bladder, lung, mammary, and skin cancers (127). In many human cancers, missense mutations in Keap1 and Nrf2 genes have been identified, always having the effect to disrupt the Keap1/Nrf2 interaction. This causes constitutive activation of Nrf2 and an increased antioxidant and detoxification response, with a clear advantage in terms of stress resistance and cell proliferation in normal and cancer cells (66, 127).
Chromatin remodeling
Modification of chromatin remodeling factors like histone acetylases/deacetylases, or directly to DNA, is rising as a further mechanism of redox-based transcriptional regulation. For example, nitrosylation of histone deacetylase-2 (HDAC2) upon neurotrophin stimulation of neurons promotes transcription by the cAMP-responsive element binding factor, through the release of the HDAC2 from chromatin. This leads to an increase in acetylation of histones surrounding neurotrophin-dependent gene promoters and promotes transcription (101). Furthermore, it has been reported that oxidative stress correlated with inflammatory diseases, could lead to peroxynitrite formation, which impairs the HDAC2 activity through nitration of critical tyrosine residues (4).
DNA may also be the molecular machine sensing oxidative stress and eliciting a cellular response (27). Demethylation of H3K4 by lysine-specific demethylase-1 (LSD1) produces hydrogen peroxide, which nearby oxidizes guanine to 8-oxoguanine and induces the recruitment of DNA repair enzymes, as glycosylase-1 and topoisomerase-IIβ, triggering chromatin and DNA conformational changes that are essential for transcription (2, 107). LSD1 demethylation, DNA oxidation, and chromatin looping have been reported as a mandatory mechanism for transcription driven by estrogens, Myc, and Snail TFs, likely affecting Myc-driven tumorigenesis and tumor invasion associated with Snail-mediated EMT, two hallmarks of cancer aggressiveness (2, 78, 88, 107).
Metabolic Reprogramming Due to Redox Molecular Machines
Cancer cells undergo profound changes in their own intrinsic metabolism, including the well-known Warburg metabolic reprogramming, characterized by an increased activity in aerobic glycolysis, as well as lipid metabolism deregulation and shift toward phospho-pentose pathways (PPPs). These complex metabolic reprogramming events have been reported to causally participate to tumor progression toward malignancy and in resistance to stressful and hostile environmental or pharmacological insults (63, 138). Indeed, one of the main differences observed among cancer and normal cells is their glucose metabolism. Indeed, cancer cells mainly use glucose by aerobic glycolysis, producing lactate (the so-called Warburg effect), while normal cells completely catabolize glucose by oxidative phosphorylation. The Warburg effect, coupled with an increased glucose uptake due to incomplete glucose oxidation, facilitates in cancer/proliferating cells the efficient anabolism of macromolecules needed to construct a new cell from glicolytic intermediates supporting the anabolic requirements of cell growth: mainly lipid, protein, and nucleotide synthesis (biomass). Excess carbon is secreted as lactate. Cancer cells also use strategies to decrease their ATP production, while increasing their ATP consumption, thereby maintaining the ADP:ATP ratio necessary to sustain their glycolytic flux. The redox balance has been claimed to explain this complex metabolic reconfiguration of cancer cell glycolysis, mainly due to the glycolytic enzyme pyruvate kinase (PK). Several key players of the metabolic reprogramming of cancer cells are redox molecular machines and deserve our interest in this forum review. These include metabolic enzymes, which are directly targeted by ROS, as PK, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), adenylate monophosphate kinase (AMPK), or enzymes, which can affect metabolism due to their redox regulation as ataxia telangiectasia mutated (ATM) (Fig. 3).

Pyruvate kinase M2
PK is the final enzyme in glycolysis, converting phosphoenolpyruvate (PEP) to pyruvate with the production of ATP. Glicolytic flux in cancer cells is greatly determined by expression, in virtually every cancer cell type so far analyzed, of the isoform M2 of PK (PKM2) (21). Conversely, to its splice variant PKM1, which is expressed in several adult tissues, PKM2 is activated in a feed-forward regulatory loop by fructose-1,6-bisphosphate and is susceptible to several kind of inhibitory post-translation cues, as tyrosine phosphorylation by growth factor signaling, lysine acetylation in response to high glucose environment, or cysteine oxidation in response to oxidative stress (3, 92, 138). It has been shown that oxidation/inhibition of PKM2 in cancer cells, due to exposure to oxidative stress or to hypoxia, allows proliferating cells to divert glucose into anabolic pathways starting from glycolysis intermediates to other metabolic pathways, the most important of which is PPP. PPP is acknowledged to convert glucose into penthoses or glycolysis intermediates, mandatory molecules for anabolism of amino acids and nucleotides, as well as NADPH, the key masterpiece of cellular reducing activity, driving re-reduction of intracellular glutathione. The concerted inhibition of glycolysis and activation of PPP, leads to increased anabolism and allows growing cells to meet the increased biosynthetic demands of proliferation (132, 133).
Although PK orthologs from bacteria to humans have been reported to be inhibited by oxidants, more recently, the Cantley group reported oxidative inhibition of PKM2 in human lung cancer cells, owing to an acute increase in intracellular concentrations of ROS (3). PKM2 is therefore a redox molecular machine, sensing intracellular oxidative stress through oxidation of its Cys358 and inhibition of its enzymatic activity on PEP, able in this way to divert glucose flux into the pentose PPP, and thereby generate NADPH for detoxification of ROS. In keeping, replacement of endogenous PKM2 with a redox-insensitive C358S mutant increases sensitivity to oxidative stress of cancer cells and impairs xenograft tumor formation. Of note, oxidation of PKM2 has also been reported in hypoxic conditions, known to increase the Warburg metabolism and to signal through ROS delivery by mitochondria or cytosolic NADPH oxidase (3). Whatever the source of oxidative stress within the cells, treatment with reducing agents as dithiothreitol restored PKM2 activity, confirming that PKM2 undergoes a reversible redox regulation in proliferating cancer cells due to oxidative stress.
The adaptation of proliferating cells to gain resistance to oxidants, following oxidative block of PKM2, is attributable to accumulation of its substrate PEP, acting as a feedback inhibitor of the glycolytic enzyme triosephosphate isomerase. This latter enzyme is directly responsible for the stimulation of PPP, leading to increased antioxidant metabolism, and to a general reconfiguration of central carbon metabolism (56).
Of note, an increased Warburg metabolism accompanied by activation of PPP is implicated in chemoresistance of cancer cells (81, 115). Beside the role of ATM in the regulation of PPP, PPP can be engaged in cancer cells due to the interaction between PKM2 and CD44, an acknowledged cancer stem cells marker. This interaction has been reported either following p53-deficiency or hypoxia exposure. CD44 ablation resulted in a depletion of GSH, an increase in oxidative stress, and an enhancement of the effect of chemotherapeutic drugs (128).
Glyceraldehyde-3-phosphate dehydrogenase
GAPDH enzymes are a family of richly expressed oxidoreductases known for their involvement in glycolysis, catalyzing the reversible phosphorylation of glyceraldehyde-3-phosphate to 1,3-bisphosphoglycerate using NAD+ as a cofactor. Its catalytic mechanism involves Cys-152, resulting in the formation of a hemiacetal. Seminal studies revealed that this glycolytic enzyme is a redox sensing protein, mainly acting through oxidation of Cys-152 (9). Interestigly, S-nitrosation and mixed disulfide formation represents a target for nitrosative stress due to NO or to peroxinitrite, or for subsequent post-translational modification with NAD+, culminating in nonenzymatic ADP-ribosylation (53, 74). Oxidative inactivation of GAPDH has also been reported during regulation of glucose balance toward glycolysis or PPP. Indeed, hydrogen peroxide exposure inactivates GAPDH with a consequent block in glycolysis within seconds, redirecting glucose flux into the oxidant-resistant PPP (55, 81, 112). Behind this initial and rapid response, glycolysis could be re-established by re-reduction due to the NADPH increase, but the balance between glycolysis and PPP is maintained in favor of the latter by additional mechanisms mainly due to PKM2 inhibition, sensitive to oxidants, but also to tyrosine phosphorylation and acetylation to maintain its inhibition (3, 92, 138).
Adenylate monophosphate kinase
AMPK is not only an energy-responsive enzyme, sensitive to increased AMP/ATP ratio, but it also senses redox signals. Hence, beside its complex regulation by the adenylate pool, AMPK has been reported to respond to pro-oxidative or nitrosative conditions, thus enhancing cell survival via autophagy in response to nutrient starvation (109). Hence, AMPK behaves as a versatile molecular effector in integrating metabolic and oxidative stimuli.
Several conditions sharing wide oxidative stress, as reduced nutrient availability, block of mitochondrial electron transport chain, or hypoxia, enhance the AMPK activity (64). Mild hypoxia, for example, leads to a ROS-dependent activation of AMPK, still without appreciable changes in the adenylate pool (32). Proof of a direct redox regulation of AMPK has been given by Zmijewski et al., who suggest that exposure to hydrogen peroxide results in the oxidation of Cys299 and Cys304 of the α- and β-subunits of AMPK, leading to enhancement of its kinase activity by inducing an allosteric rearrangement of the AMPK αβγ heterotrimer (144). Similar to ROS, NO and peroxynitrite have been implicated in AMPK activation, although the proofs for direct nitrosylative modifications of the kinase have been never described. It is conceivable that an increase of nitrosylating equivalents can promote S-nitrosylation of reactive Cys299 and Cys304 of the AMPK α-subunit (12).
In line with the strong correlation between tumor progression and metabolic oxidative changes, germline mutations in the liver kinase B1 tumor suppressor gene, the main upstream activator of AMPK, have been correlated with inherited predisposition to cancer (67). It is also known that tumors with reduced levels of AMPK phosphorylation have a poor prognostic outcome, supporting the hypothesis that the loss of AMPK activity could help and sustain cancerogenesis increasing malignancy (61). Recent evidence show that autophagy activation by AMPK, a process for which its redox sensitivity may play a key role, enables long-term survival for cancer cells when hypoxia and/or ischemia lead to nutrient deprivation. Of note, AMPK-mediated autophagy also enhances the resistance of cancer cells to chemotherapeutic agents or kaempferol, a redox-active compound causing metabolic oxidative stress (40, 65).
Ataxia telangiectasia mutated
ATM is a serine/threonine protein kinase activated under genotoxic or oxidative stress conditions. It phosphorylates several proteins involved in cell proliferation, cell survival, and DNA repair (70, 122). However, the molecular mechanisms of the activation of ATM by DNA damage and oxidative stress are molecularly different. Upon double-strand DNA breaks, induction cells recruit the Mre11–Rad50–Nbs1 (MRN) complex to damaged sites together with ATM, triggering autophosphorylation of monomeric ATM and activating ATM protein kinase activity, finally leading to phosphorylation of downstream signaling proteins, such as p53, checkpoint kinase 2 or H2AX. Conversely, ATM responds to ROS through oxidation at specific cysteines leading to ATM dimerization and activation (105, 122). Cys2991 is critical to the formation of these active ATM dimers through the disulfide bond formation, as mutation to Leu of Cys2991 results in insensitivity of ATM to oxidative insults (59). It is remarkable that the C2991A ATM mutant was fully activable by the DNA breaks pathways lead by MRN, but not by oxidative stress, confirming that ATM activation through the DNA damage pathway does not involve the ATM dimer formation via Cys-2991 oxidation and intermolecular disulfide bridge formation (59).
Cosentino recently revealed that ATM, behaving as a functional sensor of cellular redox homeostasis and undergoing reversible oxidation-dependent dimerization, stimulates PPP providing NADPH for both detoxification or re-reduction of oxidized molecules, as well as ribose for nucleotide synthesis required for DNA repair (26). ATM oxidation/activation promotes phosphorylation of the small heat shock protein Hsp27. This leads to increased association between Hsp27 and the rate-limiting PPP enzyme glucose-6-phosphate dehydrogenase (G6PD), showing increased enzymatic activity, thereby enhancing engagement of uptaken glucose in PPP and NADPH formation (54, 75). P53 acts by opposing this function of ATM behaving as a post-transcriptional inhibitor of G6PD, blocking its dimerization/activation (75). In keeping, Jiang presents quantitative evidence that G6PD inhibition and block of PPP is a mandatory function of wild-type p53, lost in common cancer-causing p53 mutations (75).
The metabolic shift toward the Warburg metabolism, linked to PPP and to the ability of ATM, via its redox sensing, to re-route central carbon metabolism toward PPP upon oxidative stress, plays also an important role in tumor immune escape, progression, and resistance to immune-, radiation-, and chemotherapy (55). Indeed, nevertheless oxidants activate ATM through a different biochemical mechanism than that of DNA damage (59), we should consider that chemotherapeutic drugs often show both the ability to cause oxidative stress and DNA double-strand breaks. For example, an increased activity of G6PD is mandatory to some multidrug-resistant colon cancer cells to increase the NADPH and GSH content, necessary to safely handle oxidative stress and to extrude anticancer drugs from the cell (110). Recent studies also accredit the nonoxidative branch of PPP for cancer progression and metastases dissemination, mainly acting through transketolase-like-1 enzyme to guarantee the connection between glycolysis and PPP (83, 142).
Taken together, the above considerations underline the strict link between the Warburg metabolism, PPP, and tumor malignancy, support the idea that glycolytic switch may primarily represent a cancer cell strategy to resist oxidative stress and validate many of the key players of this metabolic reprogramming, several of which are redox sensors or themselves redox molecular machines, as potential targets for anticancer therapy. To this end, an attractive speculation has been given recently by Ralser, who proposes to induce ROS overload in cancer cells, while inhibiting the protective effects of the Warburg effect. This could be achieved by reactivating glycolysis circumventing PKM2 inhibition or by inhibition of G6PD, while maintaining cancer cells exposed and vulnerable to oxidative damage (55). The effect of such a strategy, leading to unbalancing of redox homeostasis, likely embraces both proliferation and motility of cancer cells. Indeed, the block of the Warburg phenotype leads to a decrease of anabolic intermediates and limitations for enhancing cellular biomass, but also to a NADPH decrease and consequently to a deregulation of redox-based cycles orchestrating cytoskeleton dynamics driving cell motility. The metabolic reprogramming of cancer cells toward a Warburg phenotype, and, in particular, its dependence on redox homeostasis, is an attractive and central target for future anticancer therapies, likely leading to pleiotropic effects. To grow this approach, a much complete and comprehensive insight in redox homeostasis within cancer cells is highly warranted.
Footnotes
Acknowledgments
This work was supported by the Italian Association for Cancer Research, the Tuscany Tumor Institute, MIUR PRIN grant, and the Tuscany Project TUMAR.