
Abstract
Introduction
F
Among the molecular mechanisms compromised in most neurodegenerative diseases (ND), those related to oxidative folding of proteins in the endoplasmic reticulum (ER) and to the degradation of misfolded proteins are of critical importance. Using Caenorhabditis elegans models that recapitulate pathological aspects of Alzheimer, Parkinson, and Huntington diseases, we have demonstrated that DNJ-27/ERdj5 plays a protective role in these models by modulating the aggregation levels of beta amyloid peptide, α-syn, and polyglutamine proteins. Moreover, our data suggest that DNJ-27/ERdj5 exerts this protective function by impacting on several subcellular compartments, such as ER, cytoplasm, and mitochondria. Thus, DNJ-27/ERdj5 arises as a novel therapeutic target to investigate the molecular basis of these ND.
The progression of many ND share common cellular and molecular mechanisms, including mitochondrial dysfunction, oxidative stress, protein aggregation, and inclusion body formation (56, 74). Indeed, aberrant protein deposition has been consistently linked to etiologically diverse ND. Hence, it is widely accepted that beta amyloid peptide (Aβ) aggregation plays a central role in the pathogenesis of Alzheimer Disease (AD) (27), the most prevalent ND worldwide. Increasing evidence points to a causative role of the presynaptic protein alpha-synuclein (α-syn), a polypeptide with a propensity towards intracellular aggregation, in Parkinson Disease (PD) pathogenesis (66, 67). Likewise, CAG repeat-disorders, including Huntington Disease (HD), spinocerebellar ataxias, spinal, and bulbar muscular atrophy (50), are typically associated with polyglutamine expansions (polyQ) of greater than 40 residues that cause the specific disease-associated proteins to misfold and aggregate. However, whether aggregates are causal in ND progression remains controversial and a matter of intense debate as they have been linked to both toxicity (by being themselves the toxic species) and protection (by sequestering soluble oligomeric products regarded as the toxic species) (2, 47, 56, 77).
Caenorhabditis elegans has emerged as a very useful model organism for the study of ND due to its advantageous characteristics, including a well-defined cell lineage and anatomy, its transparency through its entire life cycle and its simple, well-established genetics. Several C. elegans transgenic models have been developed to study many pathological aspects of ND (7, 41, 58). In these models, the specific expression of aggregation-prone proteins, such as human Aβ, human α-syn, and polyQ fusion proteins in muscle and neuronal cells allows the study of the cellular and molecular processes disrupted by, and behind, the aggregation and the toxicity of these proteins (18, 29, 49).
Within cells, quality control mechanisms maintain protein homeostasis by ensuring both accurate protein folding and degradation of proteins that fail to adopt their native conformation (5). Disturbances of these cytoprotective processes can favor abnormal protein conformations with toxic effects. In this sense, among the different pathways that lead to, or protect from the pathological effects associated to the aggregation-prone proteins in ND, those involved in protein folding and degradation are among the most relevant (85). The endoplasmic reticulum (ER) is the subcellular organelle where proper protein folding is monitored and achieved. Proteins that pass ER quality control criteria continue to their final destinations through the secretory pathway, whereas non-native and unassembled subunits of multimeric proteins are degraded by the ER-associated degradation (ERAD) pathway (36). Alterations in ERAD or in the oxidative folding of proteins containing disulfide bonds have been unequivocally implicated in ND (13, 33, 78).
Thioredoxins (Trxs) are the major cellular protein disulfide reductases and, among other functions, they act as potent antioxidant defenses, have redox regulatory roles and display chaperone properties. An RNA interference (RNAi) screen designed to identify modulators of protein aggregation and toxicity among the members of the Trx family of redox proteins in C. elegans models of AD and PD (12, 25), identified dnj-27 as the only gene that provided a protective role in the two models assayed. C. elegans dnj-27 is the ortholog of mammalian ERdj5, an ER-resident protein involved in ERAD (11, 80). ERdj5 works as an ERAD enhancer in concert with ER-degradation enhancing mannosidase-like protein (EDEM), which selectively recognizes misfolded glycoproteins, and BiP, an ER-resident Hsp70 family chaperone (80). ERAD substrates frequently contain disulfide bonds that must be cleaved before their retrotranslocation and ERdj5 has been proposed to be involved in the reduction of the disulfide bonds of these misfolded proteins through its Trx domains, before their retrotranslocation (80).
We have found that dnj-27 protects against the aggregation of Aβ, α-syn and polyQ containing proteins in C. elegans, as well as their associated pathological phenotypes. Importantly, this protective effect is also achieved to some extent when ERdj5, the human dnj-27 ortholog, is expressed in worm ND models. Our findings point to dnj-27/ERdj5 as a new potential target for protection from proteotoxicity. These results also emphasize how small disturbances in ER protein homeostasis can affect the aggregation and toxicity of pathological proteins leading to worsening of the phenotypes associated with ND.
Results
C. elegans dnj-27 is the ortholog of mammalian ERdj5
We previously identified the C. elegans dnj-27 gene as the ortholog of human ERdj5 by an in silico approach (11). C. elegans DNJ-27 and human ERdj5 proteins display a high amino acid sequence homology consisting of an N-terminal DnaJ/Hsp-40 domain followed by four Trx-like domains, with different CXXC redox active site motifs (Fig. 1A) (11). The three-dimensional structure of mouse ERdj5 has been recently solved (24) and revealed that the interface domain flanked by the first and second Trx domains (Trx-1 and Trx-2) also folds as two additional, but more divergent, Trx-like domains, that lack a redox-active CXXC motif (Trxb-1 and Trxb-2). These two newly identified Trx-like domains are also present in the DNJ-27 protein sequence (Fig. 1A).

We first aimed to determine the cells and tissues expressing dnj-27 within the nematode. For this purpose, we generated transgenic animals harboring the transcriptional reporter Pdnj-27::GFP, which expresses green fluoresecent protein (GFP) under the control of the dnj-27 promoter (2 kb upstream the ATG codon) (All strains used in this work are described in Supplementary Table S1; Supplementary Data are available online at
Both human ERdj5 and C. elegans DNJ-27 proteins are flanked by an N-terminal signal peptide and by a C-terminal ER-retention signal tetrapeptide (KDEL for ERdj5 and HDEL for DNJ-27) that are required to target and retain the proteins in the ER lumen, respectively (Fig. 1A). To confirm this subcellular localization [and taking advantage of an available DNA construct encoding yellow fluorescent protein (YFP) engineered to harbor the KDEL tetrapeptide at its C-terminus (37)], we generated the reporter Pdnj-27::dnj-27::YFP::KDEL, that expresses DNJ-27 fused to YFP::KDEL. The KDEL tetrapeptide has been shown to be functionally similar to HDEL in yeast (40) and this seems to be the case for C. elegans as well, where several worm ER resident proteins containing either C-terminal tetrapeptide are found in the C. elegans proteome. As shown in Figure 1H, transgenic nematodes simultaneously expressing DNJ-27::YFP::KDEL (under the control of its own promoter) and the C. elegans ER-marker mCherry::TRAM-1 (under the control of the myo-3 promoter) (75) demonstrated a marked colocalization of both proteins in the muscle cells of these animals, confirming that DNJ-27 is also an ER luminal protein in C. elegans.
The unfolded protein response (UPR), a set of conserved intracellular signaling pathways that sense protein misfolding within the ER and work to regulate ER stress, is intimately coordinated with ERAD, which eliminates terminally misfolded non-native proteins via retrograde translocation and degradation in the cytosol (76). In mammals, the UPR is activated through three different pathways involving the proteins IRE1/XBP1, PERK, and ATF6 (44) (termed IRE-1, XBP-1, PEK-1, and ATF-6 in C. elegans). ERdj5 has been previously found to be upregulated by ER stressors that induce UPR (11). To determine whether dnj-27 is also upregulated upon ER stress, we treated transgenic animals expressing the Pdnj-27::GFP reporter with tunicamycin, a typical ER stress inducer (61). Treatment with 10, 20, and 30 μg/ml tunicamycin resulted in a strong increase of GFP fluorescence (Fig. 1I and data not shown). Using RNAi, we also determined that tunicamycin-induced expression of dnj-27 requires IRE-1/XBP-1 but is ATF-6 and PEK-1 independent, as only RNAi downregulation of ire-1 and xbp-1, but not atf-6 or pek-1, caused a marked decrease in the expression of the Pdnj-27::GFP transcriptional reporter. Indeed, pek-1 downregulation produced a significant increase in dnj-27 reporter expression (Fig. 1I). Inhibition of pek-1, but not atf-6 or ire-1/xbp-1, activates the UPR in C. elegans (61). Therefore, a possible explanation for this effect could be that pek-1 RNAi would lead to a further activation of the UPR, thus resulting in a higher induction of dnj-27. The induction of dnj-27 through the IRE-1/XBP-1 pathway is consistent with a role of DNJ-27 in ERAD as this pathway has been specifically linked to the transcriptional induction of genes that function in ERAD, such as the ERdj5 partner EDEM (89).
Using the ER stress reporter Phsp-4::GFP (79), we next examined whether efficient knockdown of dnj-27 by RNAi [demonstrated by the decreased expression of the Pdnj-27::dnj-27::GFP translational reporter (Supplementary Fig. S1A)] induces UPR activation. Similar to mammalian ERdj5 (80), ER stress was not induced in worms where dnj-27 was downregulated (data not shown). Moreover, wild type animals treated with dnj-27 RNAi showed no obvious phenotype, in consonance with the healthy and viable phenotype reported for the ERdj5 knockout mice (31). Importantly, dnj-27 RNAi downregulation did not cause any motility defects in wild type worms as demonstrated by bending and thrashing assays (Supplementary Figs. S1B and S6).
As a whole, the high homology between ERdj5 and DNJ-27 together with the DNJ-27 ER-localization and induction upon ER-stress via IRE-1/XBP-1 pathway, strongly suggests that the function of dnj-27/ERdj5 in ERAD is conserved.
dnj-27 protects against intracellular human Aβ toxicity and aggregation in a transgenic C. elegans model of AD
To evaluate whether members of the Trx family of redox proteins modulate human Aβ and α-syn aggregation, we performed an RNAi screen of all the seven C. elegans Trxs with the conserved active site sequence WCGPC plus the two thioredoxin reductases in transgenic worm models that express human Aβ or human α-syn::GFP in worm body wall muscle cells (Supplementary Table S2). Aβ aggregation causes an easily scored aging-dependent paralysis phenotype (43), while α-syn::GFP aggregation results in the formation of fluorescent aggregates that can be readily identified in vivo under a fluorescence microscope thanks to the transparency of the animal (25). As a result of this RNAi screen we identified the worm gene dnj-27 as the only member of the Trx family playing a protective role in both models (Supplementary Table S2).
Initially, for the Aβ RNAi screen, we used the transgenic strain CL647 (Supplementary Table S1) that overproduces human Aβ in body wall muscle cells in an inducible manner after temperature upshift (from 16°C to 25°C) at the L3 larval stage, since it allows a rapid scoring of paralysis (12). This strain also carries the rrf-3(pk1426) mutation that hypersensitizes worms against feeding RNAi (65). However, to rule out the effect of a higher temperature on the ER-stress machinery, we decided to perform the subsequent analyses with the CL2006 strain that overexpresses the human Aβ peptide constitutively, also in body wall muscle cells (hereafter Aβ worms) (41). As shown in Figure 2A, dnj-27 downregulation caused a significant acceleration of the onset of paralysis in Aβ worms.
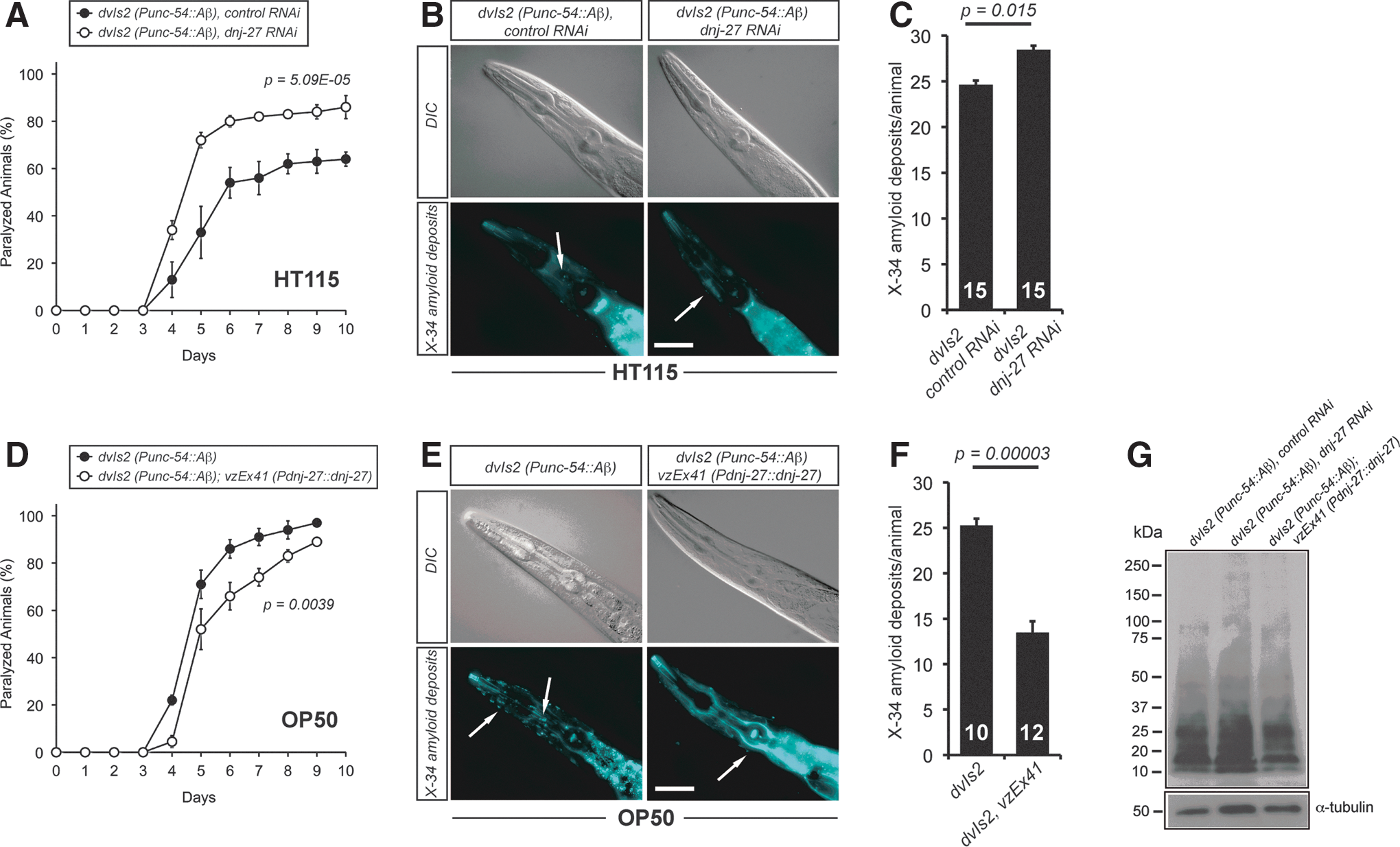
To gain deeper insight into the mechanisms by which dnj-27 exerts this protective function, we wondered whether this enhancement on the paralysis phenotype was associated with differences in the formation of Aβ aggregates. Immunohistochemistry with the fluorescent amyloid vital dye X-34 (42) on Aβ worms demonstrated that dnj-27 RNAi induces a mild, although statistically significant, increase of amyloid deposit formation (Fig. 2B, C). To determine whether this increase of amyloid deposits was due to changes in total Aβ content or changes in the Aβ oligomeric state, we performed immunoblotting on Aβ worm extracts using the Aβ-peptide specific antibody 6E10, which detects all forms of Aβ. We found no major differences in either total Aβ levels or Aβ oligomeric species in dnj-27 downregulated Aβ worms as compared to control animals (Fig. 2G; Supplementary Fig. S2A).
Since downregulation of dnj-27 enhances the Aβ-dependent paralysis phenotype, we next asked whether higher levels of DNJ-27 would improve the paralysis onset. To this aim, we generated transgenic strains expressing the construct Pdnj-27::dnj-27, which produces high levels of DNJ-27 under the control of its endogenous dnj-27 promoter (Supplementary Fig. S3A). Transgenic Aβ worms overexpressing dnj-27 showed a significant reduction of the paralysis onset compared to their corresponding nontransgenic control siblings (Fig. 2D). dnj-27 overexpressing transgenic Aβ worms also showed a strong decrease of amyloid deposits formation (Fig. 2E, F), while, similarly to dnj-27 RNAi downregulation, total Aβ and Aβ oligomers were not significantly affected (Fig. 2G; Supplementary Fig. S2A).
Another important issue arising from our results was the evident effect of the type of food source on Aβ-induced toxicity. In RNAi experiments, animals were grown on Escherichia coli HT115 (an RNAi E. coli feeding strain), while E. coli OP50 (the standard laboratory food source) was used for overexpression experiments. Thus, when comparing Aβ worms grown on OP50 and HT115, we detected a much faster paralysis onset in the animals grown on OP50 (Fig. 2A vs. D, black circles), while formation of amyloid deposits remained practically the same (Fig. 2B, C vs. E, F). We then repeated the paralysis assays growing transgenic Aβ worms overexpressing dnj-27 on HT115 bacteria. As expected, the paralysis phenotype was also improved in these transgenic animals. However, the paralysis onset of the dnj-27 overexpressing worms grown on HT115 bacteria was dramatically reduced in comparison to that reached by the same animals grown on OP50 bacteria (Supplementary Fig. S4A vs. Fig. 2D, white circles).
Overall, these data indicate that dnj-27 plays an in vivo protective role on Aβ toxicity and amyloid deposit formation in C. elegans.
dnj-27 protects against intracellular human α-syn toxicity and aggregation in different transgenic C. elegans models of PD
Next, we set to quantify the effect of dnj-27 on α-syn toxicity and aggregation. Intracytoplasmic α-syn inclusions, called Lewy bodies, are the characteristic hallmark of PD and other synucleinopathies (67). C. elegans models of PD that recapitulate distinct aspects of this ND upon human α-syn expression in body wall muscle cells and dopaminergic (DA) neurons have been developed (25, 82).
Initially, for the α-syn RNAi screen, we used the transgenic strain UA50 (Supplementary Table S1) that overproduces the fusion protein α-syn::GFP along with TOR-2 in worm body wall muscle cells. TOR-2 ameliorates the formation of α-syn::GFP aggregates, thus facilitating the identification of aggregation enhancers in RNAi screens (25). However, for the subsequent analyses we used the C. elegans strain NL5901 that overexpresses α-syn::YFP within worm body wall muscle cells (hereafter called α-syn worms). This strain exhibits age-dependent mobility defects associated with α-syn::YFP aggregation, which can be easily monitored (81,82). As shown in Figure 3A, an increase in the percentage of animals with impaired mobility was observed in α-syn worms upon dnj-27 RNAi downregulation, although the differences were not statistically significant. In contrast, decreased levels of dnj-27 clearly worsen α-syn::YFP aggregation (Fig. 3B, C) without affecting the total α-syn::YFP levels (Fig. 3G; Supplementary Fig. S2B).

Similarly to Aβ worms, we next aimed to determine whether dnj-27 overexpression would protect from α-syn::YFP toxicity and aggregation in C. elegans. To this end, α-syn worms also expressing the Pdnj-27::dnj-27 construct were generated. However, the results obtained with this strain were not reproducible and we decided to force the expression of DNJ-27 in body wall muscle cells by using the myo-3 promoter (Supplementary Fig. S3B). Transgenic α-syn worms overexpressing dnj-27 in muscle cells showed a clear improvement in mobility compared to nontransgenic control animals (Fig. 3D). Moreover, the reduction in α-syn::YFP toxicity was accompanied by a strong decrease in α-syn::YFP aggregates (Fig. 3E, F), while no changes in total α-syn::YFP levels were observed (Fig. 3G; Supplementary Fig. S2B).
Differences in toxicity between α-syn worms grown on HT115 or OP50 were not as obvious as for Aβ worms (compare Fig. 3A, D, black circles for α-syn worms and Supplementary Figs. S4B and Fig. 3D, white circles, for α-syn worms overexpressing dnj-27). Nonetheless, α-syn::YFP aggregation was clearly higher in α-syn animals grown on OP50 than on HT115 (62 aggregates/worm in OP50 vs. 43 in HT115) (Fig. 3C, F).
DA neurons are the main neuronal type affected in PD (67). We next set to determine whether dnj-27 protection could also be extrapolated to a C. elegans model of DA neurodegeneration induced by α-syn (7). For this purpose, we used the strain UA44 that expresses human α-syn in DA under the control of the dopamine transporter dat-1 promoter and that causes an age-dependent DA neurodegeneration (7, 10). DA neurons are easily identified by the simultaneous expression of GFP under the control of the dat-1 promoter. Thus, animals overexpressing dnj-27 under the control of its own promoter were analysed for evidence of degenerative changes in DA neurons. Higher levels of DNJ-27 significantly improved both the percentage of animals with the normal number of neurons (Fig. 3H) and the percentage of total nondegenerated neurons (Fig. 3I). Together, our data indicate that dnj-27 is protective against α-syn dependent deleterious phenotypes.
dnj-27 protection is also extended to the toxicity associated to polyQ aggregation in C. elegans
To investigate the protective role of dnj-27 in other aggregation-prone proteins, we resorted to a previously developed C. elegans model of CAG-repeat disorders (47). The expression of either GFP or YFP fused to different numbers of glutamine residues in C. elegans body wall muscle cells leads to polyQ-dependent aggregation and toxicity when the number of glutamine expansions is greater than 35–40 residues (47, 49). In this work, we used the AM141 strain which shows mobility impairment upon expression of Q40::YFP in body wall muscle cells (referred to as Q40 worms hereafter) (47).
First, we studied the effect of knocking down dnj-27 by RNAi on the mobility of the AM141 strain. As shown in Figure 4A, decreased levels of DNJ-27 resulted in a higher percentage of animals with impaired mobility, accompanied by a striking fourfold increase in the number of Q40::YFP aggregates (Fig. 4B, C). Similarly to Aβ and α-syn worms, we performed inmunoblotting on Q40 worms using anti-GFP antibodies (which efficiently cross-react with YFP) to rule out that dnj-27 inhibition could be affecting the total Q40::YFP levels. As shown in Figure 4G and Supplementary Figure S2C, total Q40::YFP levels were not affected by dnj-27 downregulation.
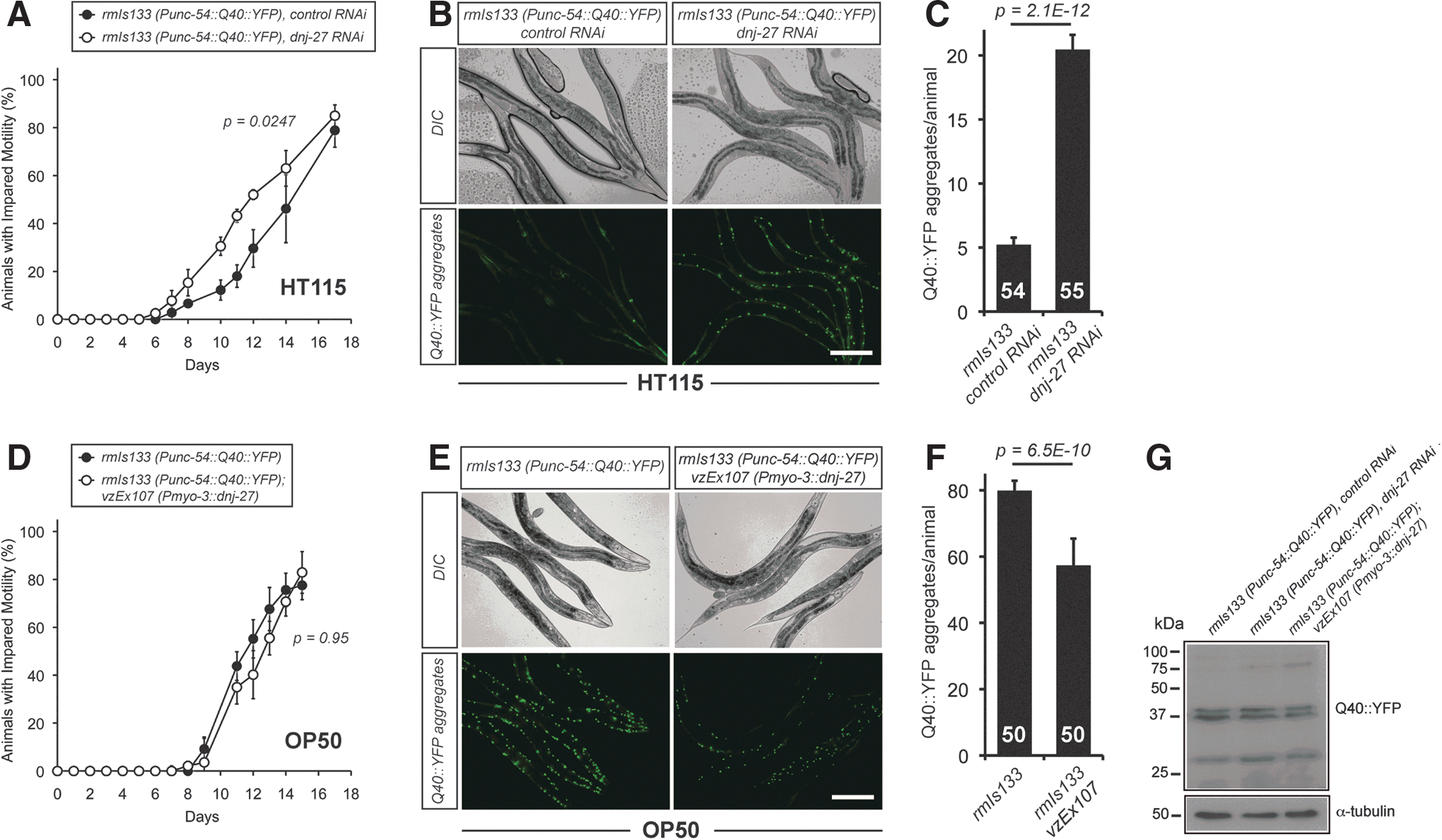
On the contrary, overexpression of dnj-27 in body wall muscle cells did not improve the impaired mobility associated with Q40 animals when DNJ-27 transgenic worms were compared to their corresponding nontransgenic control siblings (Fig. 4D; Supplementary Fig. S4C), although a statistically significant decrease in the number of aggregates was observed (Fig. 4E, F).
Our results highlight that Q40-dependent toxicity and aggregation are also influenced by the type of food source, as demonstrated in the Aβ model (Fig. 2). Thus, compared to animals grown on HT115, Q40 worms grown on OP50 show a strong increase in both the percentage of animals with impaired mobility (Fig. 4A; Supplementary Fig. S4C vs. Fig. 4D black circles) and in the numbers of aggregates (79 aggregates/worm on OP50 vs. 5 on HT115) (Fig. 4B, C vs. E, F).
As a whole, these data demonstrate a general in vivo protective role of dnj-27 on Aβ, α-syn and Q40-dependent toxicity and aggregation in C. elegans.
ERdj5, the mammalian ortholog of dnj-27, is also protective in some of the C. elegans ND models
To determine whether the protective effects of dnj-27 described above are conserved across evolution, we overexpressed human ERdj5, the mammalian ortholog of dnj-27, in the body wall muscle cells of the three various worm models of ND. For Aβ worms we evaluated the paralysis phenotype and, in contrast to dnj-27 overexpressing Aβ worms, we found no improvement of the paralysis onset in worms expressing human ERdj5 (Supplementary Fig. S5). However, it should be noted that overexpression of human ERdj5 in Aβ animals caused a severe embryonic and larval arrest phenotype. Given that the paralysis assay is performed in adult worms, it is plausible that the animals selected for the assay were those segregants expressing the lowest levels of ERdj5, thus explaining the lack of paralysis rescue. In contrast, for the other two models, we found a significant decrease in the number of α-syn::YFP and Q40::YFP aggregates when ERdj5 was overexpressed (Fig. 5B, C, E, F). Regarding toxicity, and similarly to DNJ-27 overexpression, human ERdj5 was protective against α-syn toxicity (Fig. 5A), while it had no such effect in the Q40 animals (Fig. 5D). In summary, our results show that the protective role of dnj-27 in alleviating cytoplasmic proteostasis is recapitulated (at least in the α-syn and Q40 models) by human ERdj5, further supporting dnj-27 as the ortholog of ERdj5 and suggesting a potential protective role of ERdj5 in human ND pathologies.

Other C. elegans ERAD genes are also protective in models of ND
ER misfolded proteins to be degraded by the ERAD pathway transit through different steps that entail substrate recognition, targeting, retrotranslocation, ubiquitination, and proteasomal degradation (84). Among these steps, the supramolecular functional ERAD complex formed by EDEM and ERdj5 is implicated in the substrate recognition and targeting steps (84).
Previously described C. elegans ERAD genes include cup-2 and R151.6, which encode Derlin proteins and are candidates for the retrotranslocation channel (59, 60, 88); sel-1, ortholog of human sel1 (79), a member of the HRD complex that degrades misfolded ER-resident proteins (26); sel-11/hrd-1 and hrdl-1, two E3 ubiquitin ligases (57) and cdc-48.1 and cdc-48.2, orthologues of human p97 that functions as a ubiquitin-selective chaperone (48). Thus, cup-2 and R151.6 are part of the retrotranslocation step, sel-1, sel-11/hrd-1 and hrdl-1 would be involved in ubiquitination, while cdc-48.1 and cdc-48.2 would be implicated in proteasomal targeting and degradation.
To determine whether these other ERAD genes, similarly to dnj-27, would also affect the aggregation and toxicity of aggregation-prone proteins in the cytoplasm, we studied the effect of downregulating these genes by RNAi on the paralysis of Aβ worms as well as on polyQ aggregation. Control RNAi downregulation of these genes in wild type worms does not cause any motility phenotype as demonstrated by a thrashing assay (Supplementary Fig. S6). As shown in Table 1, we found an enhancement of the paralysis phenotype of Aβ worms as well as an increase in polyQ aggregation in four out of the five ERAD genes tested. It is worth noting that a RNAi screen performed by Nollen et al. in polyQ worms found no effect in polyQ aggregation when downregulating dnj-27 or any of the genes studied in Table 1 (49). This apparent discrepancy can be explained by two facts: (i) the study by Nollen et al. was performed in animals grown for one generation on the respective RNAi, while our work used animals grown for two generations on the respective RNAi to ensure effective gene downregulation and (ii) the screen by Nollen et al. used Q35::YFP animals in which the polyQ stretches cause an intermediate aggregation phenotype, while our study uses Q40::YFP worms where the polyQ stretches produce a more severe aggregation phenotype (49).
+, moderate enhancement of the phenotype.
++, strong enhancement of the phenotype.
ER, endoplasmic reticulum; ERAD, ER-associated degradation; GFP, green fluorescent protein; RNAi, RNA interference; YFP, yellow fluorescent protein.
We also mined data from two published RNAi screens in C. elegans whereby modulation of α-syn aggregation was assessed (25, 82). There were some differences in identified modifiers between these distinct studies, likely because they employed different genetic backgrounds. Specifically, Hamamichi et al. (25) utilized a transgenic background wherein α-syn misfolding was attenuated with a chaperone concurrent to RNAi treatment, while van Ham et al. (82) screened for enhanced α-syn aggregation in nonattenuated backgrounds. Importantly, when these two screens are combined, four out of the five ERAD candidates were examined in these studies but only two of the candidates enhanced the α-syn aggregation phenotype (hrdl-1 and R151.6). We therefore conclude from these studies that α-syn aggregation does not result from the knockdown of all ERAD components, since RNAi of sel-11 and cup-2 does not cause any enhancement of the aggregation phenotype (Table 1).
The proteasomal targeting and degradation genes, cdc-48.1 and cdc-48.2, could not be targeted for RNAi as their downregulation causes embryonic and larval lethality. However, it has been reported that overexpression of either cdc-48.1 or cdc-48.2 suppresses the formation of polyQ aggregates in C. elegans (87), which also suggests a protective role of these ERAD genes in this model.
dnj-27 inhibition induces proteosomal and autophagic dependent cytoplasmic protein degradation and mitochondrial fragmentation
To get insight into the mechanisms behind the increased cytoplasmic aggregation of Aβ, α-syn::YFP, and Q40::YFP upon dnj-27 downregulation, we aimed to determine whether dnj-27 RNAi affects the homeostasis of the cytoplasm. Previous studies have demonstrated that ERdj5 accelerates ERAD by reducing disulfide bonds in misfolded glycoproteins (24, 80). Therefore, it is reasonable to propose that inhibiting dnj-27 function will lead to an increase in the ER load of misfolded proteins. To alleviate this stress, an increased retrotranslocation of these misfolded proteins from the ER to the cytoplasm would happen. This, in turn, would raise the load of misfolded proteins in the cytoplasm meddling the cytoplasmic folding machinery and proteostasis (22).
To test this hypothesis, we used worms expressing a Punc-54::LacZ transgene which can be used as a reporter of protein degradation and proteostasis in the cytoplasm of body wall muscle cells (90). Muscle cells have a well-defined network of signals which modulate protein degradation (38), thus allowing direct testing of the relevance of these pathways to pathology. Because LacZ is synthesized only until adulthood and remains stable for at least the first 72 h of adulthood (90), it can be used to determine if acute experimental manipulations in adult muscle, for example RNAi treatment (62), trigger increased degradation. Thus, LacZ transgenic animals were grown on dnj-27 RNAi and the LacZ reporter protein was assessed. As shown in Figure 6A, dnj-27 downregulation promotes cytosolic protein degradation, indicating that dnj-27 modulates cytoplasmic protein homeostasis in otherwise wild type healthy animals. Moreover, we were able to determine that the increase in degradation is related to autophagy since it was blocked by reduction-of-function mutations in genes such as unc-51 (a serine/threonine protein kinase orthologous to Saccharomyces cerevisiae autophagy protein Atg1p and the vertebrate ULK proteins), mpk-1 (a mitogen-activated protein kinase, ortholog of ERK), and daf-18 (a lipid phosphatase, orthologous to the human PTEN tumor suppressor) (Fig. 6A). These three genes are key components of diverse signaling pathways that have been shown to modulate autophagy in C. elegans muscle (71, 72). Similarly, the proteasome inhibitor MG132, which has previously been shown to block proteasome mediated degradation in C. elegans muscle (14), was also effective in blocking the increased cytosolic protein degradation upon dnj-27 downregulation. However, this increase was not dependent on the apoptotic pathway as it was not blocked by a loss-of-function mutation in the caspase ced-3 (Fig. 6A).
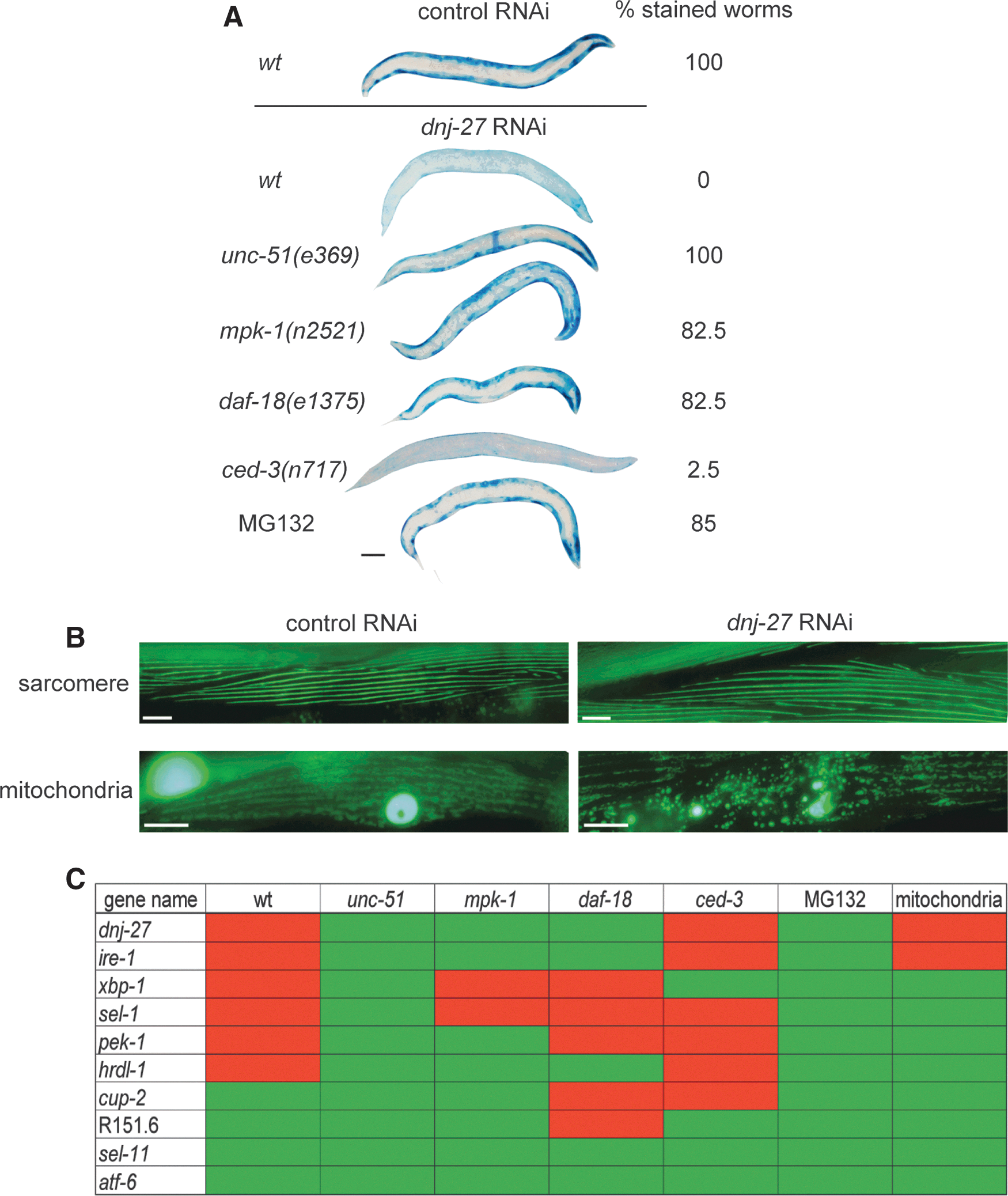
Since toxicity was scored by means of paralysis and impaired mobility phenotypes in most cases, this raised the possibility that dnj-27 was affecting muscle development and maintenance. However, we ruled out this possibility as we checked myofibrillar structure upon dnj-27 RNAi downregulation using a Pmyo-3::GFP transgene and no myofibrillar structural changes were observed (Fig. 6B). The absence of muscle function alteration upon dnj-27 downregulation is further supported by the lack of phenotype in bending and thrashing assays (Supplementary Figs. S1B and S6).
Besides affecting their aggregation, dnj-27 could be influencing the toxicity of aggregation-prone proteins by other ways. For instance, mitochondrial deficits are key events in most ND (54, 68) and have also been related to pathological phenotypes in C. elegans models of ND (6, 35, 86). Indeed, increased mitochondrial fragmentation has been reported in Aβ and α-syn worm models (17, 23, 32). In polyQ worm models, although no direct evidence of mitochondrial fragmentation has been reported, downregulation of the mitochondria fission drp-1 gene improves the motility of polyQ worms (86). Furthermore, ER and mitochondria are known to be in direct contact. Biochemical studies have revealed that they both are physically connected through a specialized subcompartment called the mitochondria-associated membrane (MAM) (83), which supports bidirectional communication regulating fundamental physiological processes between these two organelles (30, 64, 70). Therefore, we wondered whether dnj-27 RNAi would have any impact on mitochondrial structure and integrity. As shown in Figure 6B, a Pmyo-3::MitGFP transgene (that expresses a fusion protein of the mitochondrial targeting sequence of chicken aspartate aminotransferase and GFP in worm muscle cells under the control of the myo-3 gene promoter) allowed us to determine that, indeed, dnj-27 downregulation induces mitochondrial fragmentation in these cells.
Next, to determine whether this effect in cytosolic degradation and mitochondrial network is specific for dnj-27 downregulation or, instead, is a more general consequence of compromising ER function, RNAi of other genes involved in ERAD and UPR was performed in the LacZ and mitochondrial reporter strains. Interestingly, the downregulation of some, but not all, of UPR/ERAD genes resulted in an increase of protein degradation as shown in Figure 6C. Increased protein degradation resulted from the knockdown of ire-1, xbp-1, pek-1, sel-,1 and hrdl-1. However, knockdown of atf-6, cup-2, R151.6 and sel-11/hrd-1 did not increase protein degradation, maybe due to functional redundancy with other genes. In addition to dnj-27, only ire-1 downregulation was shown to affect mitochondrial integrity.
Collectively, our data demonstrate a requirement of DNJ-27 for the maintenance of protein homeostasis in the cytosol, most likely as a consequence of an increased load of misfolded proteins retrotranslocated from the ER when dnj-27 function is compromised, which also appears to affect mitochondrial network integrity.
dnj-27 modulates the mitochondrial fragmentation phenotypes of C. elegans models of ND
To further evaluate the impact of dnj-27 in the mitochondrial network of ND worms, we generated transgenic strains expressing a fusion protein of C. elegans mitochondrial import receptor subunit TOMM-20 and mRFP in body wall muscle cells (a kind gift of Dr. Amir Sapir) (6) in the background of the three ND worm models. This way, we avoid overlapping with the green fluorescence derived of α-syn::YFP and Q40::YFP fusion proteins by labeling the mitochondrial network with red fluorescence.
As shown in Figure 7A, we confirmed a severe disruption of the mitochondrial network in Aβ worms, while α-syn worms displayed a milder mitochondrial fragmentation phenotype, as previously reported (17, 23, 32). Interestingly, we demonstrate for the first time that the mitochondrial network of Q40 animals is also dramatically disrupted (Fig. 7A). Moreover, dnj-27 RNAi downregulation increased the mitochondrial fragmentation of wild type animals (Fig. 7A, B) as well as α-syn worms (Fig. 7A, C). However, the severe disruption of the mitochondrial network in Aβ and Q40 worms precluded any quantification of the effect of dnj-27 downregulation in these two models.

Next, we set to determine whether increased levels of DNJ-27 would result in any improvement of the mitochondrial fragmentation phenotypes of the ND worms. For this purpose, we generated derivatives of Aβ and α-syn worms carrying the mitochondrial mRFP reporter and overexpressing DNJ-27 in body wall muscle cells (we were unsuccessful to generate the corresponding Q40 derivative strains; this precluded the assessment of DNJ-27 in rescuing the mitochondrial fragmentation phenotype of Q40 worms) (Supplementary Table S1). High levels of DNJ-27 strongly reduced the mitochondrial fragmentation in both Aβ and α-syn worms (Fig. 7D, E). We conclude that DNJ-27 levels modulate the mitochondrial network integrity at least in the Aβ and α-syn models, providing a possible mechanism for the protective function of dnj-27/ERDj5 in AD and PD.
Discussion
Cells have developed elaborate protein quality-control systems to assure the maintenance of proteostasis. Within the crowded cellular environment, proteins are always at risk of misfolding. Furthermore, cellular protein homeostasis is continuously challenged by changing genetic and environmental factors, as during pathology. For instance, aberrant protein deposition, that has been shown to influence the initiation and progression of many aging-associated ND (56), is known to challenge the proteostasis network. A key component of this complex protective network is the ER, wherein quality-control mechanisms assure proper folding of all secretory and integral proteins, in such a way, that unrefoldable non-native proteins are degraded by the ERAD pathway (36).
In this study we have identified dnj-27, the C. elegans ortholog of the ERAD mammalian gene ERdj5 (80), as a protective gene in three different nematode models of ND caused by cytoplasmic proteotoxicity. We first demonstrated that C. elegans dnj-27 gene encodes a protein that is targeted to ER luminal space in vivo, as suggested by its ER-retention signal (HDEL) at the C-terminus. Moreover, dnj-27 was induced upon ER-stress caused by tunicamicyn and this induction occurs via IRE-1/XBP-1 pathway, one of the three branches of the UPR. Interestingly, the transcriptional induction of EDEM, and other genes involved in ERAD, is specifically mediated by IRE-1/XBP-1 (89), strongly suggesting an evolutionarily conserved role of ERdj5/dnj-27 in ERAD.
We have shown that DNJ-27 plays a protective role in the cytoplasmic aggregation of human Aβ, α-syn, and polyQ proteins in C. elegans body wall muscle cells, results that were corroborated, to some extent, when human ERdj5 was expressed in these models. These data raise the question of how DNJ-27, an ER-resident protein, is able to influence the aggregation state of aggregation-prone proteins in a different subcellular compartment, such as the cytoplasm. Indeed, dnj-27 knockdown results in an alteration of protein homeostasis in the cytoplasm, inferred by the increased cytoplasmic protein degradation via autophagy and proteasome pathways. A plausible explanation is that dnj-27 inhibition compromises ERAD [as it has been previously shown for ERdj5 inhibition in mammals (80)] leading to an alteration of ER homeostasis that, in turn, would result in an increase of the load of misfolded proteins retrotranslocated into the cytosol for degradation. Ultimately, the cells of a healthy animal would try to reduce this extra cargo of misfolded proteins in the cytoplasm by increasing their degradation. However, in the context of the ND models used in this work, it has been demonstrated that the accumulation of Aβ, α-syn, and polyQ causes an impairment of the proteasome and autophagy function (4, 16, 34, 51). Thus, the altered cytoplasmic proteostasis caused by dnj-27 downregulation in these ND models cannot be relieved by these clearing mechanisms, hence resulting in a more sustained alteration of protein homeostasis and a net increase of misfolded proteins in the cytoplasm. As Gidalevitz et al. have elegantly shown (22), this disturbance of cytoplasmic proteostasis would favor the rapid aggregation of any aggregation-prone proteins, therefore explaining the effect on Aβ, α-syn and Q40 worms found upon dnj-27 downregulation.
Supporting our hypothesis, similar results were obtained when other genes involved in ERAD and UPR were assayed. RNAi downregulation of ire-1, xbp-1, pek-1, sel-1, and hrdl-1 resulted in increased cytosolic protein degradation. Nevertheless, knockdown of atf-6, cup-2, R151.,6 and sel-11/hrd-1 did not affect protein degradation. This could be explained by partially redundant gene function, as has been previously described for cup-2 and R151.6 in C. elegans (59). It is important to point out that, with the exception of R151.6, the protection observed with these genes was not extended to all the three ND models. This implies putative different mechanisms (from that proposed for dnj-27) for the impact of cup-2, R151.6 and sel-11/hrd-1 on polyQ and/or α-syn aggregation and on Aβ-dependent paralysis. Previous studies have related several aspects of the ERAD pathway to Aβ, α-syn or polyQ toxicity (10, 13, 33). For instance, α-syn has been found to inhibit ER-Golgi trafficking in yeast, resulting in the accumulation of specific misfolded proteins to be degraded by ERAD (10). Moreover, it has been demonstrated that entrapment of some essential ERAD proteins by polyQ-expanded huntingtin is an early event of toxicity in yeast and mammalian cells (13). Therefore, it is conceivable that different mechanisms may contribute to the cellular consequences observed in these C. elegans ND models and that other ERAD genes might then affect toxicity by distinct mechanisms.
Cytoplasmic aggregation of Aβ, α-syn, or polyQ in C. elegans has been linked to the pathological phenotypes associated to these peptides (25, 41, 58). The protective role of the dnj-27 gene in paralysis and mobility tests certainly appears to correlate well with the level of aggregation of the specific toxic peptide in most cases, although a 100% correlation was not found. This is the case for α-syn worms treated with dnj-27 RNAi and Q40 animals overexpressing dnj-27, where a mild (although statistically significant) increase or decrease in aggregation, respectively, did not result in any changes in toxicity. The most plausible explanation for the lack of correlation in these two specific cases is that an upper/lower aggregation threshold must be reached to induce the cellular and behavioral symptoms we measure in our studies.
In this context, whether aggregates are causal in ND progression remains controversial, as aggregates and inclusions have been linked both to toxicity and protection (2, 47, 56, 77). A recent study suggests that each aggregation-suppressor gene will have differential effects on toxicity depending on the affected cellular function and on its own network of interacting partners (63). For dnj-27, this appears to be the case since there is a strong correlation between ND proteins aggregation and toxicity, but other mechanisms underlying dnj-27 protection cannot be ruled out. Key events in most ND include oxidative stress and mitochondrial dysfunction, among others (12, 21, 54, 68). In this study, we have confirmed the previously reported mitochondrial network disruption in Aβ and α-syn worms (17, 23, 32) and demonstrated for the first time that this is also the case for Q40 animals. We have found mitochondrial fragmentation upon dnj-27 downregulation, thus suggesting that mitochondrial dynamics represents another potential factor implicated in dnj-27 protection. Given the fact that the ER and the mitochondria are physically connected through MAMs (83), it is conceivable that disruption of ER function caused by dnj-27 downregulation could negatively affect mitochondrial function. Indeed, dnj-27 downregulation worsens the mitochondrial fragmentation of α-syn worms, while overexpression of DNJ-27 alleviates the mitochondrial fragmentation of Aβ and α-syn animals, supporting the notion that dnj-27 might exert its protective function, at least in part, by regulating mitochondrial function.
Another important issue that has emerged from this study is the influence of the type of food source on the toxicity and aggregation phenotypes of the ND worm models. In this context, it has been shown that differences in food sources can have a profound effect on C. elegans lifespan and other parameters (45, 55). These differences can be explained by differing nutritional values of both HT115 and OP50 E. coli strains, since metabolic profiles and fat storage levels are not the same in worms grown on each bacteria (45, 55). Central to these issues is the insulin pathway, since it is known to mediate a large part of the sensory inputs on lifespan (1, 45) and to modulate pathogen resistance to bacteria (20). Importantly, toxicity in C. elegans ND models is influenced by the insulin pathway [(8, 9, 16, 47) and our own unpublished observations] providing a link between food source and proteotoxicity. Although the differences between the nematode and human diet are obvious, our observation is consistent with an increasing number of studies that support a role of environmental factors, such as diet and environmental toxins, in ND (52, 53, 73).
In conclusion, we have identified DNJ-27/ERdj5 as a novel regulator of age-related proteotoxicity in transgenic C. elegans models of ND. Different subcellular compartments appears to mediate the DNJ-27 protection since mitochondrial dynamics and cytoplasmic proteostasis disturbances, mechanisms previously identified in ND progression, are involved.
Materials and Methods
C. elegans strains and culture conditions
The standard methods for culturing and maintenance of C. elegans were used as described previously (69). The strains used in this work are described in Supplementary Table S1. All experiments were performed at 20°C unless otherwise noted.
To induce ER stress, L4 worms were placed onto seeded nematode growth medium (NGM) plates containing 10, 20 and 30 μg/ml of tunicamycin (Sigma). One day adult animals were analyzed after 16 h of tunicamycin treatment.
RNA interference
HT115 E. coli strain transformed with either pL4440 empty vector or the respective RNAi test clones were grown in liquid Luria-Bertani medium containing 100 μg/ml ampicillin for 15 h at 37°C prior seeding the RNAi plates containing 1 mM isopropyl β-
Plasmid constructs and transgenesis
All plasmid constructs used to generate the transgenic strains described in this work (see Supplementary Table S1) were made using the vector pPD95.77 as backbone (Fire Vector Kit), with the following exceptions: pVZ378 used a modified version of pPD95.85 containing an ER signal peptide and a KDEL retention signal as described (37); pVZ451 was constructed using a modified pDEST vector backbone (from the Gateway Cloning System; Invitrogen). pVZ448 was generated using the Pges-1::mCherry::TRAM-1 construct as described (75). The Punc-122::GFP and Pmyo-2::mCherry reporters were kind gifts from Piali Sengupta and Peter Askjaer, respectively. The Ptrx-3::mCherry reporter was generated in our lab (unpublished data). Information on baits, primers, cloning sites, sizes, and sequences of the different inserts as well as the amount of DNA injected for the constructs used to generate the corresponding transgenic strains will be provided upon request. Germline transformation was performed as described (46).
Microscopy
Animals were mounted in a 5 μl drop of 10 mM levamisole (Sigma) on a 3% agarose pad covered with a 24×24 mm coverslip. Differential interference contrast and fluorescence imaging was performed on a Zeiss AxioImager M2 ApoTome fluorescence microscope equipped with an AxioCam MRn. Images were captured with the AxioVision 4.8 Software (Zeiss). Confocal images were obtained with Leica AOBS SP2 and equal adjustment of brightness and contrast on control and matched experimental images was done using Adobe Photoshop 10 Software (Adobe Systems) and ImageJ (NIH).
Paralysis phenotype and β-amyloid deposit quantification
Synchronous populations of Aβ worms were generated by time-limited (2–3 h) egg lay at 16°C. Parents were then removed, and the progeny were grown continuously at 20°C. Paralysis scoring was initiated at the first day of adulthood and determined daily, whereby paralyzed worms were removed from plates. A worm was scored as paralyzed if it did not respond to a gentle touch stimulus with a platinum wire. In experiments measuring the paralysis of transgenic strains carrying extrachromosomal arrays, worms containing the transgene were identified by the fluorescence of the reporter included in the extrachromosomal array.
For amyloid deposit staining, worms were stained with the amyloid-specific dye X-34 (a kind gift from Prof. William Klunk) as previously described (42). Briefly, worms were propagated at 20°C and first-day adults were incubated for 2 h in 20 μl drops of 1 mM X-34 in 10 mM TRIS pH 7.5. Worms were then destained by rinsing once in a drop of phosphate buffered saline and then transferring them to NGM plates seeded with E. coli strain OP50 for overnight recovery at 20°C. Stained worms were anaesthetized with sodium azide and imaged with a Zeiss Axiophot epifluorescence microscope.
Impaired mobility phenotype quantification
Synchronous populations of α-syn or Q40 worms were generated by time-limited (2–3 h) egg lay at 20°C. Parents were removed, and progeny were grown continuously at 20°C. Impaired mobility scoring was initiated at the first day of adulthood and determined daily, whereby worms with compromised mobility were removed from plates. A worm was scored as mobility impaired if it did not move or was barely able to move a few millimeters after a gentle touch stimulus with a platinum wire. In experiments measuring the mobility of transgenic strains carrying extrachromosomal arrays, worms containing the transgene were identified by the fluorescence of the reporter included in the extrachromosomal array.
Quantification of aggregates
The number of α-syn aggregates was determined from the tip of the nose to the end of the second pharyngeal bulb using a Zeiss AxioImager M2 ApoTome fluorescence microscope equipped with an AxioCam MRn and ImageJ software for image analysis. Ten animals at day 1 of adulthood were included for each condition and each independent experiment.
The number of polyQ aggregates present in whole animals were counted at 40× magnification using a Zeiss AxioImager M2 ApoTome fluorescence microscope equipped with an AxioCam MRn. Twenty animals at day 1 of adulthood were included for each condition and each independent experiment.
Analysis of DA neurodegeneration
Worms were synchronized by time-limited (2–3 h) egg lay on NGM plates seeded with OP50 E. coli at 20°C. Progeny were transferred to a fresh plate containing 40 μg/ml 5-Fluoro-2′-deoxyuridine, to prevent eggs from hatching, when they reached late L4 stage. For each independent experiment, thirty 7-days old worms of each strain were examined under a Nikon Eclipse E800 epifluorescence microscope equipped with an Endow GFP HYQ filter cube (Chroma). The six anterior DA neurons (four CEP and two ADE DA neurons) were scored for neurodegeneration according to previously described criteria (3, 7).
Preparation of worm protein extracts and western blotting
For protein extraction, 100 worms from each strain (grown for two generations on the corresponding RNAi bacteria or grown on OP50 for one generation) were manually collected at their first day of adulthood in 15 μl of Laemmli buffer. After freezing in liquid nitrogen, the mix was heated at 95°C for 10 min. For all immunoblots, proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred to Immobilon-P polyvinylidene fluoride membranes (Millipore). For Aβ, a 4–20% gradient polyacrylamide gel (BioRad) was used and blots were probed with anti-Aβ monoclonal 6E10 (Covance) at a 1:1000 dilution and mouse anti-immunoglobulin G (IgG; Sigma) at 1:10,000 as secondary antibody. For DNJ-27 detection (Supplementary Fig. S3A, B), a 1:500 dilution of rabbit anti-DNJ-27 primary antibody and a 1:10,000 dilution of horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (Sigma) were used. For α-syn::YFP and Q40::YFP detection, blots were probed with rabbit anti-GFP antibody (Santa Cruz) at a 1:500 dilution and a 1:10,000 dilution of the anti-rabbit as secondary antibody. The ECL kit (GE) was used for signal detection, following manufacturer's instructions. Monoclonal anti-α-tubulin (Sigma) 1:10,000 was used as loading control.
Analysis of muscle protein degradation, sarcomere structure, and mitochondrial network structure
Worms containing different transgenic reporters were used to assess potential subcellular defects in muscle (Supplementary Table S1). We used the same protocol as previously utilized for an analysis of known C. elegans muscle mutants (62); [for detailed consideration of the RNAi approach utilized see ref. (39)]. We employed two modifications to this past approach: (i) The effect of acute RNAi against a gene in a fully developed adult was assessed even if chronic growth on RNAi produced no subcellular defects; (ii) ced-3(n717) was used to assess if observed protein degradation required the protease CED-3. Briefly, animals were grown on RNAi for two generations. Subcellular phenotypes were recorded for 20–30 animals at the first day of adulthood and 24 h later in both the F1 and F2 generations (data not shown). Next, animals were age synchronized and grown to adulthood at which point the presence of normal cytosolic reporter protein, sarcomere structure, or mitochondrial network structure was confirmed [as previously described (62)], and then 100 adults, per condition, were placed on fresh plates containing RNAi against the gene of interest or control RNAi. Twenty-four hours later the presence of cytosolic reporter protein was assessed in 20–30 animals (not shown). At 48 (not shown) and 72 h post introduction to RNAi, the presence of cytosolic reporter protein, normal sarcomere structure, or normal mitochondrial network structure was assessed. For each individual time point, an RNAi treatment was scored as giving a subcellular defect in an individual animal if: (i) For cytosolic protein degradation: there was a visual loss of at least 30% of LacZ stain (e.g., the worms looked light blue or “washed out” rather than dark blue); (ii) For sarcomere structure: there were at least two disorganized arrays of sarcomeres or at least two breaks in the normal array of sarcomeres in at least two muscle cells; (iii) For mitochondrial networks: at least a 25% lack of networked mitochondria in at least two muscle cells. For each individual time point, at least 20% of animals (above any baseline defects in the control RNAi animals) had to have been scored as having a defect for an overall defect to have been scored for the population.
Quantification of extent of fragmentation of mitochondrial networks within muscle
Pmyo-3::tomm-20::mRFP animals were grown on control RNAi (HT115) or dnj-27 RNAi for two generations at 16°C before age synchronizing as previously described (90) then transferring L1's to 20°C for 72h (early adulthood, t=0 h of experiment). Animals which expressed all the necessary transgenes were picked and imaged using a Zeiss AX10 microscope with an Axiocam MRC Digital camera and Axiovision LE software (Nottingham). Mitochondrial fragmentation was quantified in a three tier scoring system: “Baseline” which represented typical wt mitochondria expressing TOMM-20::mRFP (NB this baseline would be considered minor fragmentation in strain CB5600 which was used for the initial experiments in Figure 6); “Moderate fragmentation” which represented multiple obvious gaps in the networks of mitochondria; and “Severe fragmentation” which represented networks which were difficult to reconcile as mitochondria. Scoring was completed for n=20 animals every 24 h from t=0 h to t=72 h inclusive and each experiment was completed in triplicate. In the dnj-27 overexpression experiments roughly 20 adult animals which over expressed DNJ-27 and which were grown at 16°C, were picked to a fresh plate at 20°C, they were allowed to lay eggs for 24 h, and were then removed. At young adulthood, (roughly 96 h later) n=20 animals which expressed all relevant transgenes were picked and scored. All DNJ-27 overexpression experiments were completed in triplicate. Image analysis and figure preparation was done with the GNU Image Manipulation Program. Scale bars represent 20 μm.
Statistical analysis
Microsoft Excel two-tail Student's t-test was used to calculate p-values in neurodegeneration assays and in Aβ deposits and aggregates quantification assays. A two-way analysis of variance was used to determine significance in the paralysis and mobility assays.
Footnotes
Acknowledgments
We thank the Caenorhabditis Genetics Center for providing worm strains and Drs. Piali Sengupta, Ellen Nollen, Xiaochen Wang, Peter Aksjaer, Richard Morimoto, Alexander Van der Bliek, Andrew Fire, Paul Sternberg, Amir Sapir and William Klunk for strains, constructs, and reagents. Continuous support from Peter Askjaer's and Manuel Muñoz's groups is deeply acknowledged. Drs. Laura Berkowitz, Adam J. Harrington and Alexander J. Burdette are gratefully acknowledged for excellent technical support and fruitful discussions. A.M.-V. was supported by the Instituto de Salud Carlos III (Projects PI080557 and PI1100072, cofinanced by the Fondo Social Europeo [FEDER]), Junta de Andalucía (Projects P07-CVI-02697 and P08-CVI-03629) and CSIC (Project PIE 200920I118). F.S. and N.J.S. were supported by NIH (AR-05342).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.