Significance: Control over reversible changes to molecular structure forms the basis for artificial molecular machines that could eventually lead to the development of molecule-based nanotechnology. Recent Advances: Particular applications in information storage and processing could emerge where the structural rearrangements give rise to bistability and molecular hysteresis effects. Critical Issues: Oxidation-state-dependent coordination and bonding preferences in transition metal complexes and organometallic compounds provide a versatile approach to the control of molecular motions by redox input, but so far, few structural motifs have been applied in redox-actuated molecular machines. Future Directions: Further progress toward molecule-based nanoscale devices might be accomplished with molecular components derived from a wider range of structural themes and forms of molecular motion. Examples of redox-stimulated rearrangements in metal complexes and organometallic compounds are described that have been employed in molecular machines or could be considered for the design of new functional molecules. Antioxid. Redox Signal. 19, 1803–1814.
Introduction
Redox processes that involve major, yet reversible, changes to molecular structure have attracted a great deal of attention due to their potential applications in redox-actuated nanoscale devices (5, 6, 9, 19, 39, 71). Depending on the potential applications in mind, the molecular motion itself may be the desired effect, and supramolecular assemblies that feature large-amplitude translocations can be employed as engines of molecular machines (59). In other cases, the structural change as such is not the desired effect, yet a requirement for a bistable, that is, hysteresis-like, electrochemical response. Such hysteresis effects at the purely molecular scale (7, 37, 40, 61), that is, for noninteracting molecules, are essential to potential applications of molecular switches (27) in molecule-based devices for information storage and processing (21, 30, 48, 65).
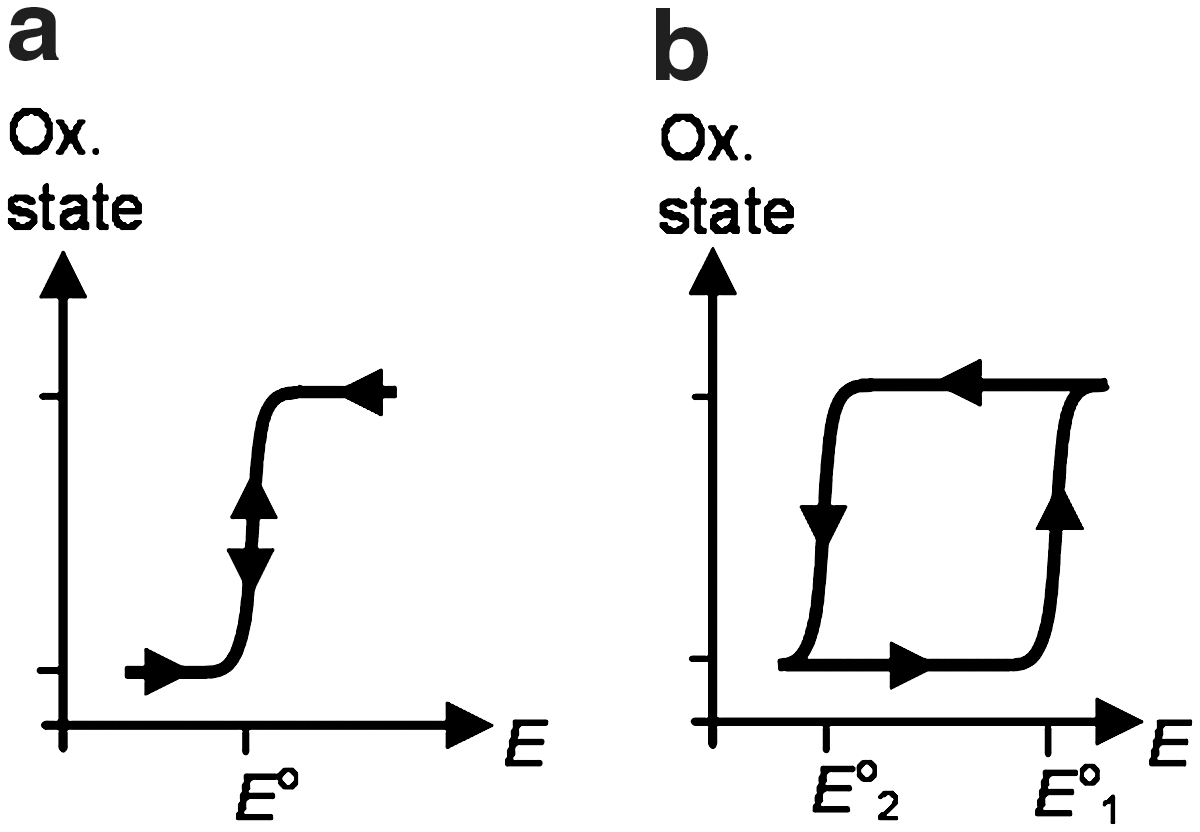
Bistability generally describes a system that can switch between two different states depending on an external trigger in such a way that for a certain interval of this parameter, the system can exist in either state, depending on its history. For a redox-driven system, bistability requires two redox states of a molecule that exist alternatively in a range of electrochemical potential, but can be reversibly interconverted at potentials outside the bistability interval (Fig. 1b). Pronounced bistability effects can be expected to occur where reversible, redox-triggered structural rearrangements exert major feedback on the redox properties of the compound. This review is focused on systems where bistability arises from redox-stimulated structural rearrangements in metal complexes and organometallic compounds. It will be illustrated how molecular entities can be set in motion by oxidation-state-dependent preferences of the metal center for certain coordination geometries or ligand sets. The examples discussed range from linkage isomerizations and geometric rearrangements in simple metal complexes to ligand translocation or metal translocations in larger frameworks or supramolecular assemblies.
Oxidation state as a function of electrode potential. Common redox response (a) and response of a bistable system (b).
Redox-Stimulated Structural Changes in Electrochemistry
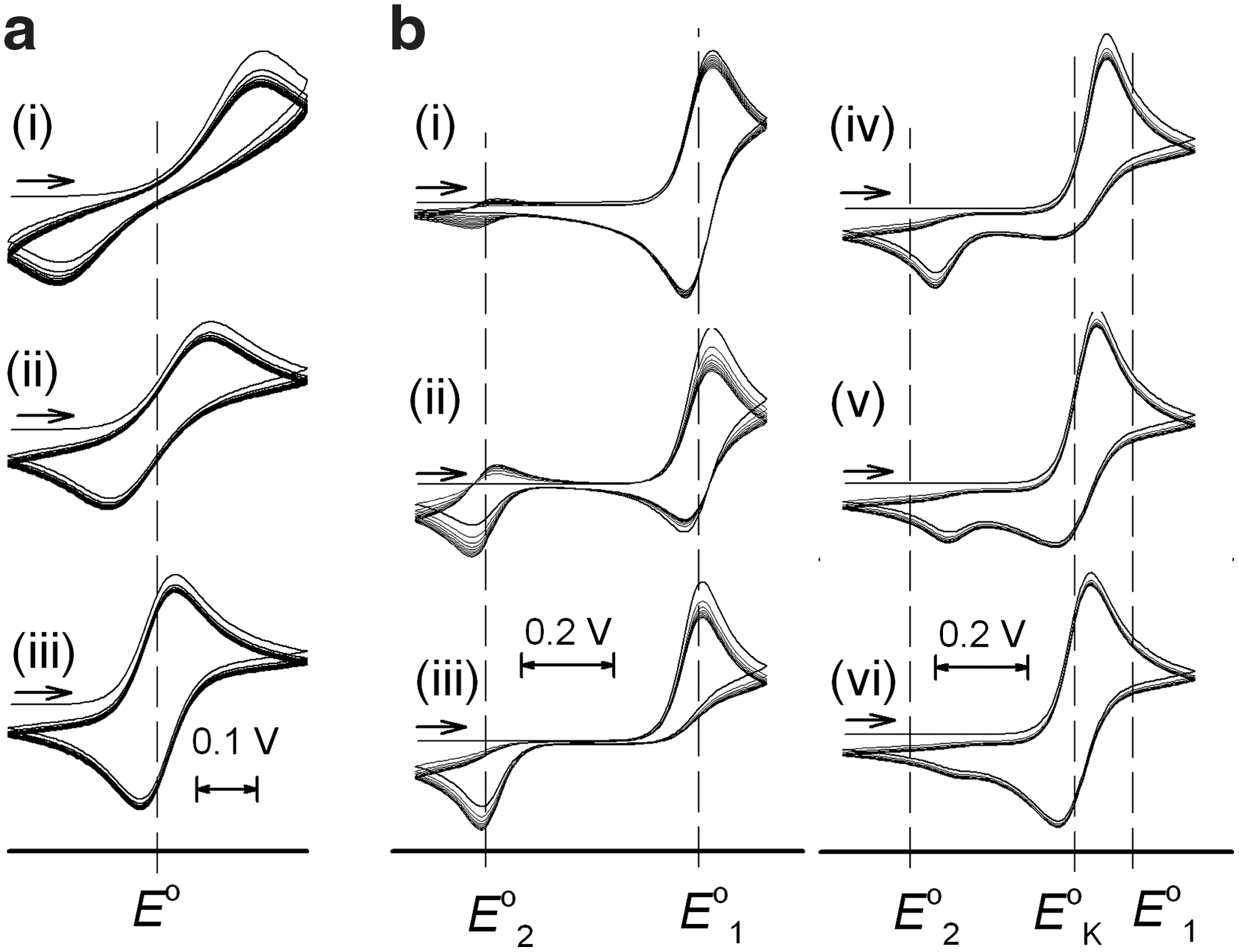
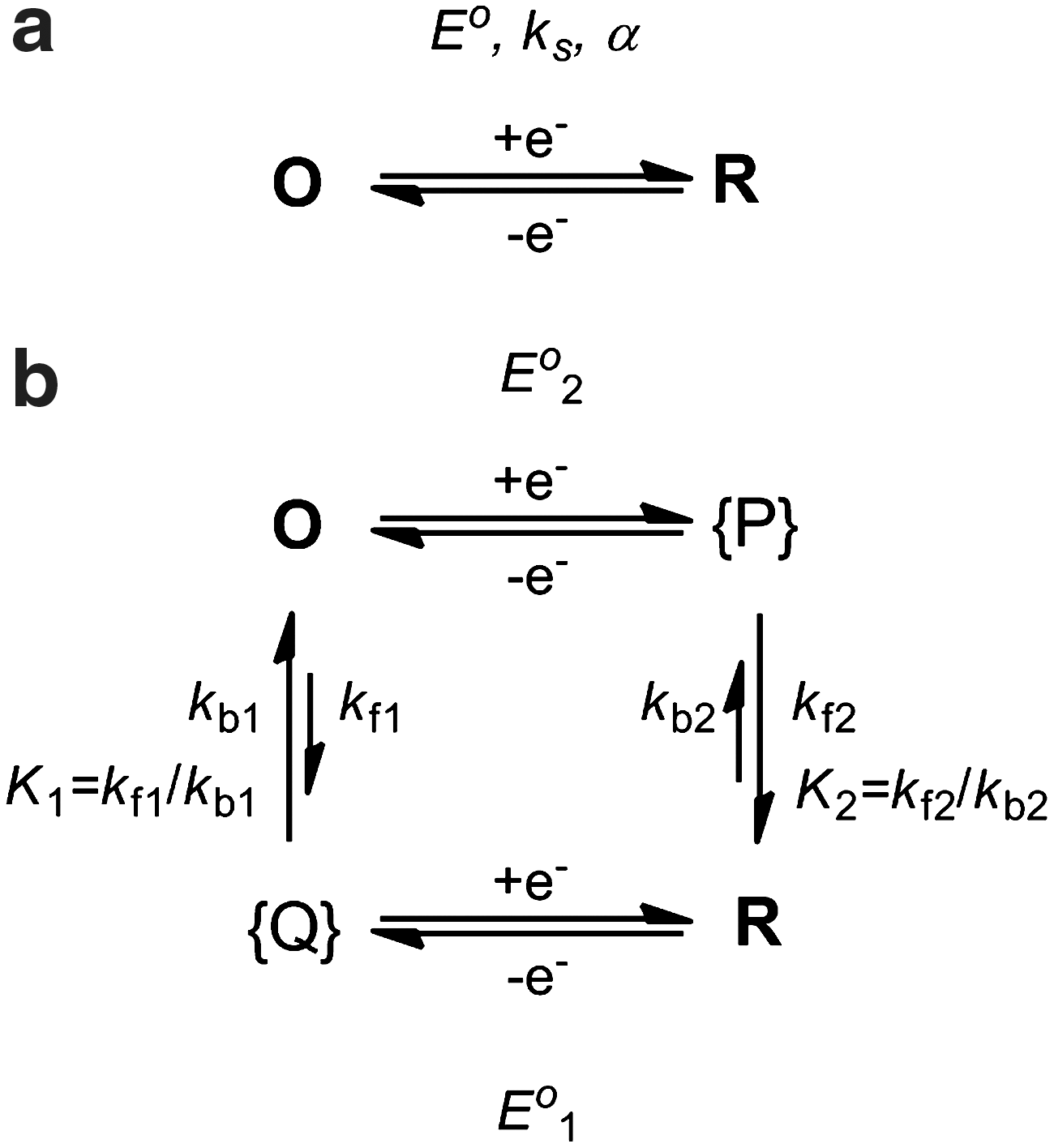
Chemical reactions coupled to redox processes can be conveniently studied by electrochemical methods (63). Generally, redox-stimulated reactions can occur concerted with the electron-transfer step or in a subsequent reaction that follows electron transfer. In the first case, the barrier to electron transfer increases, and electron transfer is slowed down as a consequence of the concomitant structural change. The reaction involves two states O and R that differ in the oxidation state and molecular structure (Scheme 1a). The electrochemical behavior of such systems is determined by the rate constant of electron transfer ks relative to the time scale of the experiment. In, for example, cyclic voltammetry, the relation is described by the parameter Λ=ks(RT/FvD)1/2, where v=dE/dt is the scan rate, and D is the diffusion coefficient of O and R. With decreasing Λ, voltammetric peaks shift to more extreme potentials (Epa and Epc), and ks can be determined from the variation of peak potentials, the increasing separation between the potentials of the forward and reverse peaks ΔEp=Epa – Epc with scan rate (Fig. 2a).
Principal effects of reactions coupled to redox processes on voltammograms (scans 1–10) at different scan rates. (a) Concerted reaction (Scheme 1a) with Λ=5×10−7 (i), 3×10−6 (ii), and 1.5×10−4 (iii) (ks=0.1 cm s−1, α=0.5, v=10,000, 300, 0.1 Vs−1, D=10−5 cm2 s−1). (b) Consecutive reactions (Scheme 1b) with K2=106 and K1=0.01 (Eo1 – Eo2=0.473V) for λ1=λ2=0.003 (i), 0.1 (ii), 2.5 (iii), 250 (iv), 2500 (v), and 25,000 (vi) (kb1=kf2=1000 s−1, v=10,000, 300, 10, 0.1, 0.01, 0.001 Vs−1.)
Redox-stimulated structural changes concerted with the electron-transfer step (a) or as subsequent reactions in a square scheme with Eo1>Eo2 (K2>K1) (b).
Bistability arises if the structural change is following after the electron transfer reaction, and the redox cycle involves four distinct species, that is, the two structural isomers in their two oxidation states. For this ECEC mechanism, the electron transfer reactions (E) are without kinetic constraints from the structural rearrangements (C), that is, electrochemically reversible. The two metastable species P and Q generated by electron transfer convert with characteristic rate and equilibrium constants to the isomer more stable in the oxidized state (O) and reduced state (R), respectively. The redox couples of each isomeric form are characterized by their standard potentials that are related to the driving forces for the rearrangements by a thermodynamic cycle (K2/K1=exp[(F/RT)(Eo1 – Eo2)]). For K2>>1 and K1<<1, kb2 and kf1 can be neglected, and the voltammetric response depends on the kinetic parameters λ1=(kb1/v)(RT/(nF)) and λ2=(kf2/v)(RT/(nF)). The rate constants can be obtained from the variation of peak potentials Ep with scan rate if λ>5 [Ep=Eo – 0.780RT/nF+(RT/2nF) ln λ] or from the ratio ipa/ipc for values of λ between 0.1 and 1. For λ<0.1, reversible waves are observed that reveal the standard potential Eo1 or Eo2 when starting from R or O, respectively. At very low scan rates, when kb2 and kf1 cannot be neglected; reverse peaks emerge again; and equilibrium constants K2 and K1 can be readily obtained if the scan rate can be lowered to the point where the irreversible waves coalesce in one reversible wave with EoK=Eo1 – (RT/F)ln(1+K1−1)=Eo2+(RT/F)ln(1+K2). In practice, the extremes of very high and very low scan rates required for straightforward determination of all thermodynamic and kinetic parameters cannot always be covered, and digital simulations are a powerful complementary tool for data analysis.
In the following sections, examples of redox-driven molecular motions and bistability according to Scheme 2b are described in terms of molecular structures (indices O and R indicating the stable isomer in the oxidized and reduced state, respectively) and key parameters such as bistability intervals Eo1 – Eo2 and conversion rate constants kb1 and kf2. Starting out from simple structures that illustrate common motifs for redox-induced structural changes, subsequent sections exemplify how the basic motifs can be incorporated in increasingly complex molecular assemblies to generate the driving forces for more complex forms of molecular motion.
Linkage Isomerism in Complexes with Monodentate Ligands
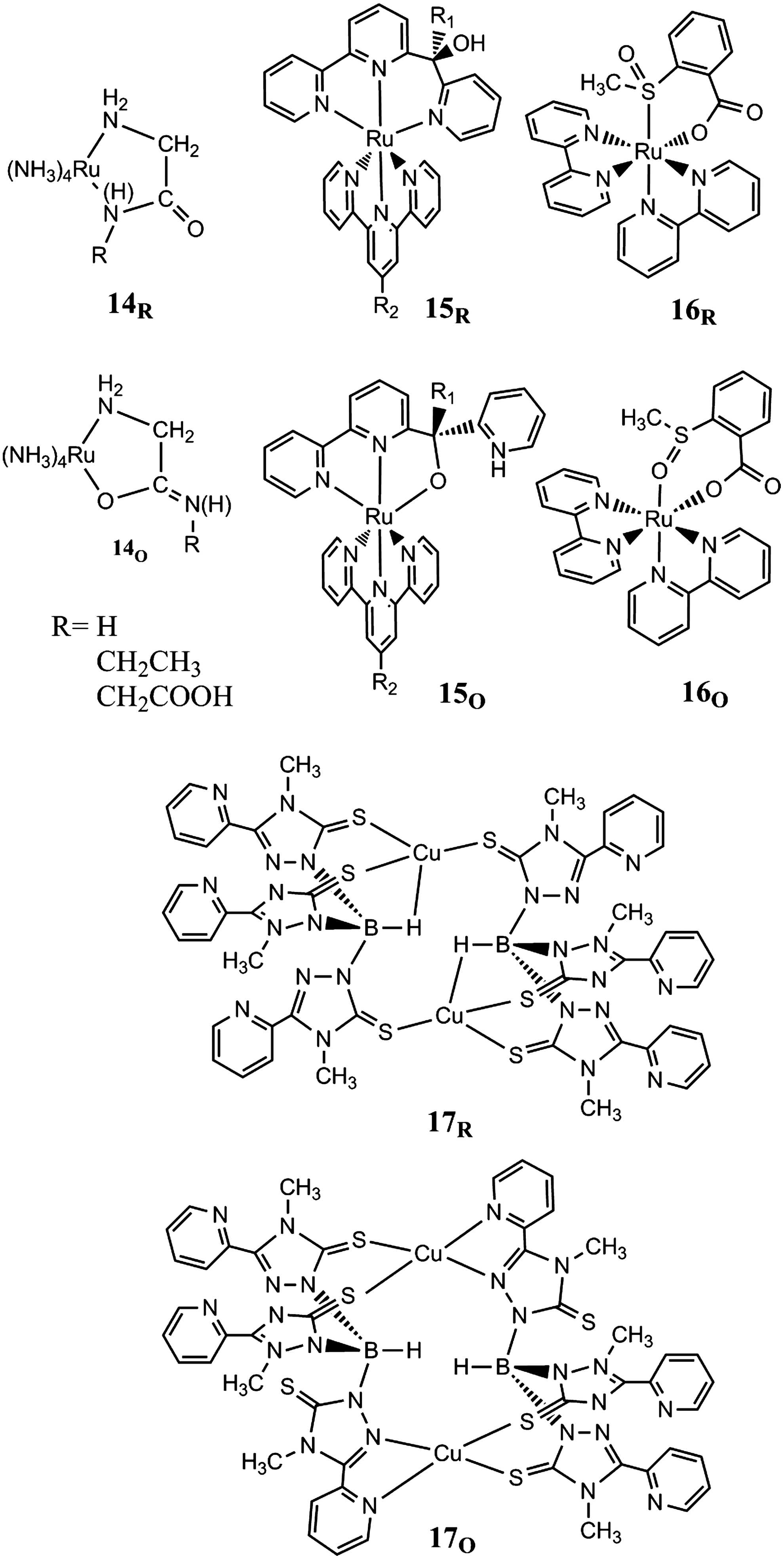
Linkage isomerization reactions can be used to drive molecular motions in a wide range of coordination compounds. In the simplest case, a single ligand can bind via one of two different binding sites that provide different degrees of stabilization to alternative oxidation states of the metal center. Figure 3 compiles examples of the widely studied M(L)5(LA) motif, where M is a transition metal ion, Fe2+, Fe3+, Co2+, Co 3+, Ru2+, or Ru3+, in an octahedral complex with one ambidentate ligand LA. Ambidentate ligands with alternative N/O-binding groups are common and will typically coordinate to the higher oxidation state via the O-donor atom, while N-coordination is favored by the M(II) state. This behavior is featured by complexes of, for example, amino acids such as glycine (22) (1), or amides such as the carboxamide (28) in complex 2. In these complexes, the linkage isomerizations are frequently coupled to protic equilibria (e.g.,2O⇌2O′+H+) or tautomerizations, and hence under a combination of redox and pH control. In addition, the investigation of the isomerization processes can be complicated by solvolysis, as the nonchelating ambidentate ligands may be readily replaced by coordinating solvents such as water. Isomerization rates faster than solvolysis however provide evidence for the isomerization proceeding via an intramolecular mechanism that holds for all examples discussed here.
Redox-driven linkage isomerism in complexes with monodentate ligands.
Taube, Sano, and coworkers have studied extensively the S/O linkage isomerism of sulfoxide ligands (67, 74). Initially, dimethylsulfoxide (dmso) has been combined with the Ru-pentamine motif (3). The site preference of Ru(III) for O-coordination is drastic in the sulfoxide complexes and results in a 0.9-V difference in the reduction potentials. O→S isomerization in the Ru(II) state is rapid (70 s−1), but not matched by the slow S→O isomerization of the Ru(III) complex (0.04 s−1). Detailed investigations of substituents on the sulfoxide (3a–3h, 3k, 3l) and of coligands (3i, 3j) have shown that the S→O isomerization on Ru(III) is accelerated to values on the order of 103 s−1 by bulky substituents on the sulfoxide, while the O→S isomerization rate on Ru(II) was largely invariant to the substituent effects. Similarly, large acceleration of isomerization on Ru(III) was found for the SS→SO isomerization of 3i with a cis-dmso coligand. The isomerization of the second sulfoxide ligand (SO→OO) is however slow (0.01 s−1). Dinuclear Ru complexes with sulfoxide ligands have been shown to exhibit bistability based on isomeric mixed-valence states that are discussed in a separate paragraph.
The N/N linkage isomerism is found in complexes with N-heterocycle ligands and can involve only endocyclic N atoms such as in pyrazole complexes 4 (66) or exo-/endocyclic isomers as in complexes of aminopyrimidines 5 and isocytosine (41) or isonicotinamide 6 (14). The pyrazole ligands in the Ru-pentamine complexes favor N2 coordination to Ru(II) (K=83) and partly isomerizes to the N1 isomer upon oxidation to Ru(III). The isomerization equilibrium is however still on the N2 side (K=1.4), and also in the Ru(III) state, as the site preference is primarily arising from comparatively small differences in back-bonding effects. More pronounced differences in site preferences of the Ru(II) and Ru(III) states are observed for the endo-/exocyclic ambidentate ligands such as aminopyrimidine where the Ru(III/II) reduction potential is lowered by ∼0.4 V upon isomerization to the exocyclic amine N that is favored by Ru(III). For the nicotinamide ligand, the rapid intramolecular isomerization occurs between binding sites separated by six bonds, and it has been suggested that the Ru-pentamine moiety walks the aromatic ring via an intermediate that is η2-bound to the C=C bond of the ring (14).
Linkage isomerizations involving stable isomers with η2-bound ligands are also known for the Ru-pentamine motif when complemented with, for example, acrylamide 7 (38) or acetone 8 (51) ligands. In the first example, the ambidentate ligand coordinates to Ru(II) via a η2-bond to the olefin site as a strong η-acceptor, while oxidation to the Ru(III) state triggers isomerization to the N-bound amide complex. The rate constants for the isomerizations in both oxidation states are 8 s−1. Parallel solvolysis in the Ru(III) state by acetonitrile is observed however. For the keto group of the acetone ligand, linkage isomers of different hapticity are also found to have strong oxidation-state preferences with the η1 (O-bound) isomer preferably formed with Ru(III) and the η2 (CO-bound) isomer with Ru(II).
Linkage Isomerism in Complexes with Chelating Ancillary Ligands
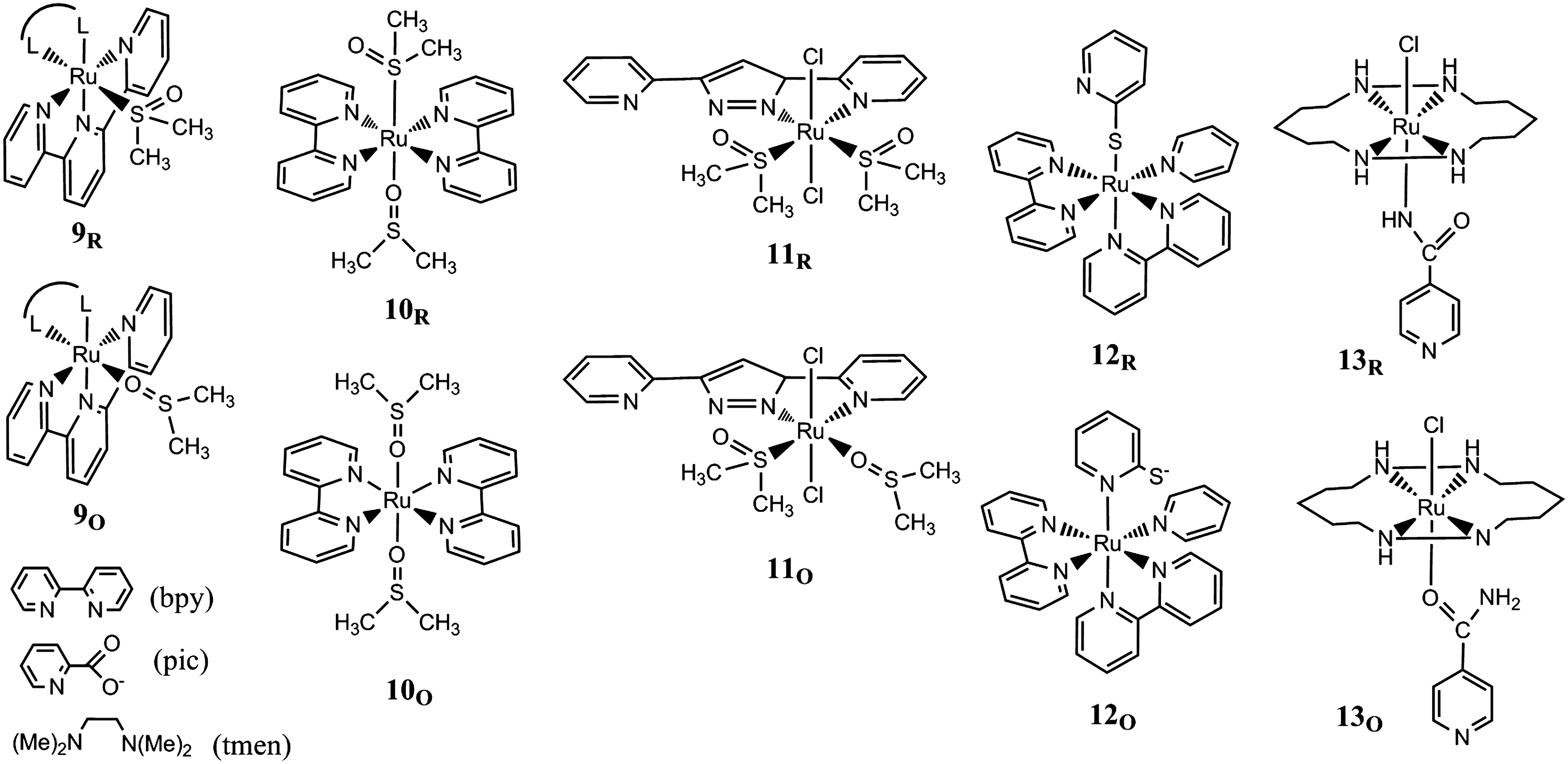
Ambidentate ligands have also been combined with chelating ancillary ligands of, for example, polypyridyl or macrocycle type, and selected examples are shown in Figure 4. Ru-polypyridyl complexes with sulfoxide ambidentate ligands (57, 58) have been extensively studied by Rack and coworkers (53), primarily with regard to their photochemical isomerization reactions and the resulting photochromism, but redox-induced isomerizations have also been documented for several of these complexes. Compounds of the type [Ru(tpy)(L2)(dmso)]z+ (9) (tpy is 2,2′:6′,2′′-terpyridine; L2 is 2,2′-bipyridine [bpy], N,N,N′,N′-tetramethylethylenediamine [tmen], and 2-pyridine carboxylate [pic]) feature redox-induced isomerizations with the S→O rate constants kIIIS→O>50 s−1 and the kIIO→S on the order of 10−3 s−1 (52, 55). Interestingly, the isomerization is impeded when the O donor bidentate ancillary ligands (L2=acetylacetonate, malonate, or oxalate) are incorporated in the complex. These ligands apparently render Ru(III) too soft as to promote formation of the O-bonded isomer. Ru-polypyridyl complexes with two ambidentate dmso ligands have also been studied in form of cis- and trans- [Ru(bpy)2(dmso)2]z+ (10) (54). Oxidation of the cis-[S,S] isomer at 2.2 V (vs. Ag/AgCl in acetonitrile) triggers formation of the cis-[O,O] isomer that is reduced at 0.79 V. For the trans-configuration, the [S,O] isomer was obtained that is oxidized at 1.26 V and isomerizes to the trans-[O,O] complex (kIIIS→O=0.5 s−1) and back to trans-[S,O] upon reduction at 0.71 V (kIIO→S=3×10−3 s−1).
Redox-driven linkage isomerism in complexes with chelating ancillary ligands.
Ru(II) complexes 11 containing the 3,5-bis(2-pyridyl)pyrazole (Hbpp) ligand together with Cl− and dmso ligands have been studied by Llobet and coworkers for catalytic applications (64). Also in these compounds, the Ru–S-to-Ru–O linkage isomerization of the dmso ligands is triggered by oxidation to the Ru(III) state, and the kinetic and thermodynamic parameters of the isomerization equilibria in both oxidation states have been obtained (KIIIO→S=0.25±0.025, kIIIO→S=0.017 s−1, and kIIIS→O=0.065 s−1; KIIO→S=6.45×109, kIIO→S=0.132 s−1, kIIS→O=2.1×10−11 s−1).
Electrochemically induced S/N linkage isomerism was also reported for the 2-mercaptopyridine ligand in [Ru(bpy)2(py)(2-S−-pyH+)]2+ (12) and [Ru(tpy)(2-S−-pyH+)]2+ (31, 32). The complexes obtained in the Ru(II) state are coordinated by thiolate and are protonated on the pyridine, which prevents isomerization to the κ-N complex. After deprotonation, the 2-S−-py ligands, unlike their 4-S−-py analogs, undergo isomerization in both complexes. The (bpy)(tpy) ancillary ligands was found to have favorable effects on the rate constants kIIIS→N=50 s−1 (0.40) and kIIN→s=1×103 s−1 [50] and the equilibrium constant KIIIN→S=0.068 [0.64] as compared to the (bpy)2(py) ancillary ligand set (values in brackets).
Amide-N-to-amide-O linkage isomerization was, for example, observed for 4-pyridinecarboxamide on trans-chloro(cyclam)ruthenium complexes 13 (20) (cyclam=1,4,8,11-tetraazacyclotetradecane) with the N and O preferences of the Ru(II) and Ru(III) states, respectively, which are known from amide complexes of the Ru-pentaamine type. The cyclam complex is more inert toward hydrolysis at low pH, but also linkage isomerization is slower (2×10−2 s−1) than in the pentaamine analog.
Linkage Isomerism in Complexes with Chelating Ambidentate Ligands
Complexes with ambidentate ligands that bind in an alternative, but monodentate, fashion, as in the previously discussed examples, do not benefit from stabilization by chelation effects. Incorporating the ambidentate function in a chelating ligand has therefore intrinsic advantages with respect to stability of the complex. Furthermore, when the ambidentate unit is a part of a larger chelating ligand together with a nonambidentate donor unit, linkage isomerizations will also drive molecular motion within the ligand that needs to adopt different conformations as the ambidentate unit changes its binding mode. Examples of complexes with chelating ambidentate ligands are shown in Figure 5.
Redox-driven linkage isomerism in complexes with chelating ambidentate ligands.
Coordination of chelating amide ligands such as glycylglycine, glycinamide, and ethylglycinamide to, for example, ruthenium tetraammine (14) or its cobalt analog yields five-membered chelate rings that close on the nitrogen of the amide group [the (N,N′) form] or the oxygen or the amide group [the (N,O) form] (33). The equilibria between the (N,N′) and (N,O) isomers in these compounds are under redox and pH control. For, for example, complex 14 with the glycinamide ligand, the (N,O) isomer is largely favored in the Ru(II) state (KIIN,O/N,N′=6×103) when both isomers are protonated, whereas the deprotonated ligand [pKa=4.3 for Ru(II)(N,N′) and pKa∼13 for Ru(II)(N,O)] prefers (NN′)-coordination to Ru(II) (KIIN,N′/N,O=105). Similarly, the equilibrium constants in the Ru(III) state change from KIIIN,O/N,N′=1.6×10−4 to KIIIN,O/N,N′=1.6×102 upon ligand protonation.
Tridentate ligands with an ambidentate pyridyl-N/alkoxy-O− moiety tethered to a bipyridine moiety were employed in Ru complexes 15 with both functional and ancillary ligands of the polypyridyl type (35). The Ru(II) state of these complexes strongly prefers the N6 donor set jointly provided by the tpy ancillary ligand, the bpy moiety of the functional ligand, and the pyridine of its ambidentate unit. Oxidation to the Ru(III) state favors linkage isomerization to an N5O ligand set, where the alkoxy-O replaces the pyridine N. The isomerizations involve a rotational motion of the ambidentate unit about the connecting bond to the static bpy unit and are coupled to tautomerization of the alkoxylproton to the pyridine. The deprotonated complex binds to Ru(II) and Ru(III) in the N5O form. Kinetics and thermodynamics of the isomerizations can be tuned by steric and electronic effects of substituents on the ambidentate and ancillary ligand, respectively (34). For, for example, R1=methyl and R2=Cl, the isomerizations are rapid in both directions (kIIIN→O=6.5×102 s−1, kIIO→N=4×102 s−1) and generate pronounced bistability (ΔEo=0.53, KIIN/O=2.3×106, KIIIN/O=2.5×10−3). Complexes of this type were immobilized on a nanoparticulate metal oxide (70) and undergo rapid isomerization on gold microelectrodes (69). Dinuclear complexes with one bistable unit based on 15 feature isomeric mixed-valence states and are described in a separate paragraph (36).
Recently, chelating sulfoxide ligands has also been introduced in the Ru-polypyridyl complexes studied by Rack and coworkers. The [Ru(bpy)2(OSO)]+ complexes 16 employ 2-methylsulfinylbenzoate (OSO) as a chelating ligand that binds via its benzoate group and the ambidentate sulfoxide moiety with its usual S/O preference to the Ru(II)/Ru(III) oxidation states (45). The chelating sulfoxide features however drastically slow the isomerization rates on Ru(III) (kIIIS→O=0.090 s−1 in propylene carbonate and kIIIS→O=0.11 s−1 in acetonitrile) as compared to kIIIS→O≈50–100 s−1 for complexes 9 of the type [Ru(tpy)(L2)(dmso)]n+ with the simple dmso ligand. Also, the bistability interval (EOS=0.86 V, EOO=0.49 V vs. Ag/Ag+) is markedly smaller than for 9 with the 2-pyridine carboxylate ligand (EOS=1.38; EOO=0.63V vs. Ag/Ag+).
Copper complexes 17 with S/N homoscorpionate and heteroscorpionate ligands are examples of more complex ditopic ligand structures with ambidentate moieties (11). The hydrotris(thioxotriazolyl)borate or hydro[bis(thioxotriazolyl)-3(2-pyridyl)pyrazolyl] borate ligands are capable of S3H/S2N2 and S2N2/S2NH linkage isomerization triggered by Cu(I)/Cu(II) redox processes due to stabilization of Cu(II) by involvement of N atoms for the metal coordination.
Linkage Isomerism in Dinuclear Complexes with Isomeric Mixed-Valence States
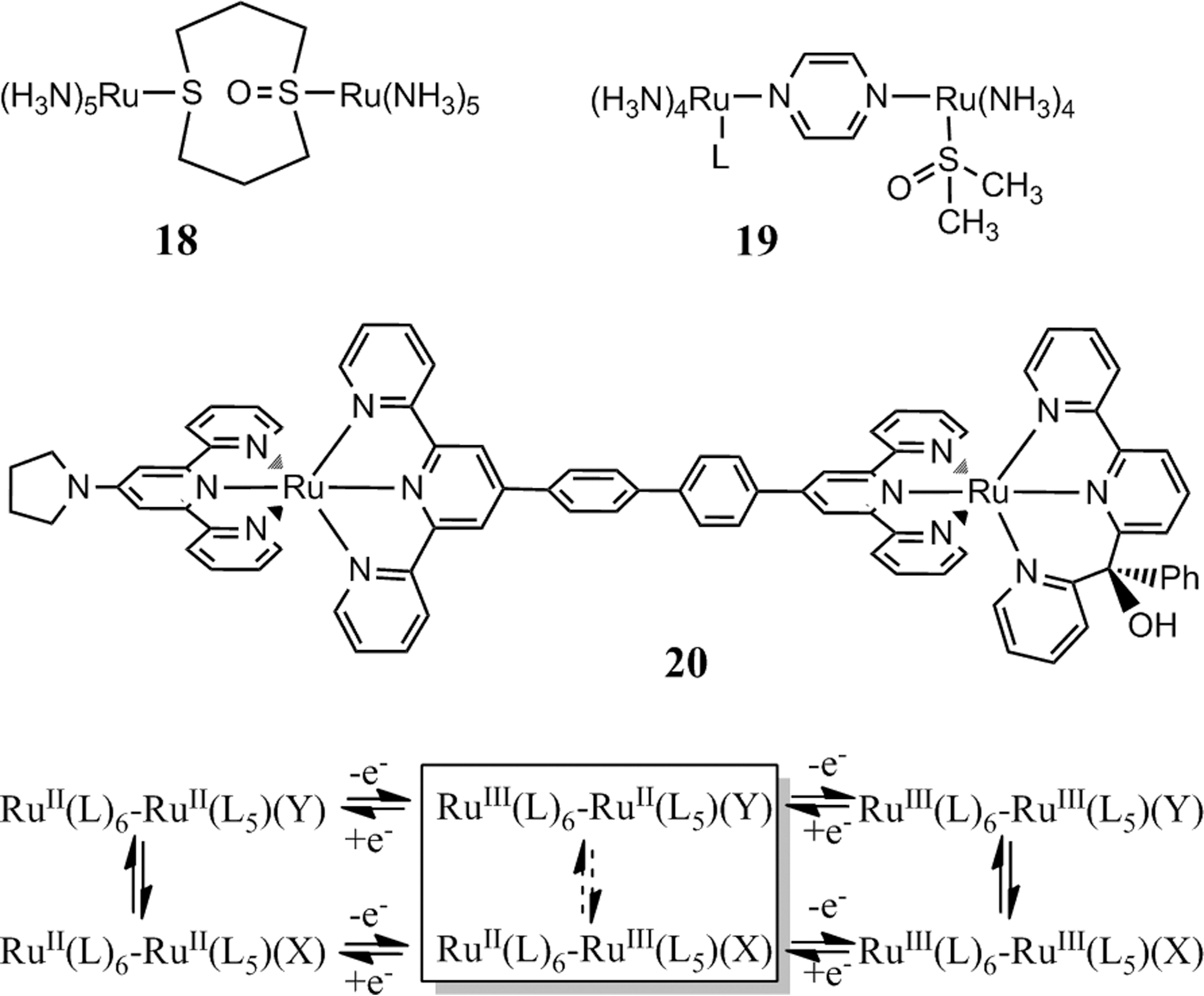
A particular form of bistability or molecular hysteresis can be accomplished in dinuclear metal complexes where one of the metal centers features a bistable redox behavior, while the second metal center undergoes a simple reversible redox process. If the standard potential of the latter couple is placed in between the two potentials of the two-bistable unit, the dinuclear complex will form alternative mixed-valence states depending on the isovalent state previously adopted. For example, an Ma(II)Mb(III) or Ma(III)Mb(II) state can be obtained from the same complex by either oxidation of the Ma(II)Mb(II) state or reduction of the Ma(III)Mb(III) state. The redox behavior is hence described by a double-square scheme if the undesirable direct interconversion (dashed arrows) of the isomeric states is included (Fig. 6). This concept was initially implemented in form of the dinuclear Ru-sulfoxide complexes studied by Sano and coworkers. The ambidentate coordination to one of the Ru centers was provided by either the bridging ligand (1,5-dithiacyclooctane 1-oxide) itself (61, 62) or a terminal dmso ligand on the pyrazine-bridged complexes (60, 68). In contrast to the original complex 18, strong electronic communication via the pyrazine bridge in complexes 19 (L=NH3, pyridine, and benzonitrile) promotes intervalence charge transfer with distinct intervalence bands of the two isomeric mixed-valence states.
Binuclear Ru complexes with one nonbistable center Ru(L)6 and one bistable center Ru(L)5(X/Y) and resulting double-square scheme with isomeric mixed-valence states.
Isomeric mixed-valence states were also obtained with the dinuclear complex 20 that contains the above-mentioned chelating ambidentate ligand motif consisting of a pyridyl-N/alkoxy-O− function tethered to a bipyridine moiety (36). The pyrolidine substituent on the bis-terpyridine unit tunes the potential of the reversible Ru(III/II) couple to a value close to the center between the Ru(III/II) couples of the N6 and N5O isomers of the bistable unit to create the desired redox behavior. Direct interconversion of the isomeric states was insignificant in this system.
Geometric Rearrangements and Assembly/Disassembly Reactions
Redox-driven molecular motion due to linkage isomerization always involves transitions between complexes with alternative ligand sets, that is, structural isomers, according to the electronic preferences of the metal center in its alternative oxidation states. Molecular motion in coordination compounds can be stimulated alternatively by geometric rearrangements of the same ligand set that adopts alternative configurations or coordination geometries depending on the electronic configuration of the metal center, the electronic structure of the ligands, and their steric bulk. Examples of redox-driven geometric rearrangements are shown in Figure 7.
Redox-driven geometric rearrangements and assembly/disassembly reactions.
Carbonyl complexes 21 of chromium, molybdenum, or tungsten [M(CO)2(P–P)2]+/0 with a chelating bisphosphine ligand (P–P) are examples of redox-driven geometric rearrangements in form of cis–trans isomerizations (73). In the M(0) oxidation states, these complexes adopt the cis-configuration, and oxidation to the M(I) state favors isomerization to the trans-isomer. The isomerization results in marked bistability with differences in the standard reduction potentials of the M(I/0) couples of Eocis – Eotrans=0.43, 0.33, and 0.30 V for the Cr, Mo, and W complexes, respectively (in dichloromethane, P–P=1,2-bis(diphenylphosphino)ethane [dpe]). The easier oxidation of the trans-isomers is a consequence of the larger splitting of the filled t2g orbitals, and hence the easier removal of electrons from the dxy orbital. The isomerization rate constants follow the order Cr<Mo<W with up to 45 s−1 for the M(I) cis→trans process in [W(CO)2(dpe)2]+, but much slower M(0) trans→cis reactions with 0.1 s−1 in [W(CO)2(dpe)2] or 1.2 s−1 in [W(CO)2(dpm)2] [P–P=dpm=1,2-bis(diphenylphosphino)methane] (8).
The Cu(II/I) redox couple with the preference of Cu(I) for tetrahedral and Cu(II) for square-planar coordination is also readily employed for redox-induced structural changes in coordination compounds. In, for example, complexes 22, the tetradentate bis-thia-bis-quinoline ligands adopt a tetrahedral configuration with Cu(I) and rearrange to the square-planar configuration with Cu(II) (4). Also, this isomerization reactions result in pronounced bistability, as the Cu(II/I) couple is about 0.4 to 0.6 V lower in the square-planar isomer.
Tetradentate ligands that cannot adopt a tetrahedral configuration preferred by Cu(I) feature instead a redox-driven reversible assembly of copper-containing double-stranded helicates (23). This behavior was found for Cu complexes of quaterpyridine and quinquepyridine ligands (23a) (50) and bisimino bisheterocyclic ligands (23b) (2,3) that can be converted between monomeric (1:1), planar Cu(II) complexes, and tetrahedral, dimeric (2:2) Cu(I) helicates.
Also, the redox-induced motion of the noncoordinating subunits in the ferrocenyl-appended cyclam copper complex 24 operates through the geometric preferences of the Cu(II/I) couple (10). In the Cu(I) state, the cyclam ligand (1,8-dimethylferrocene-4,11-dimethyl-1,4,8,11-tetraazacyclotetradecane) adopts a type V geometry (R,R,R,R-configuration of the N atoms) with ferrocenyl and methyl groups on opposite sides of the N4 plane, according to the tetrahedral geometry preferred by Cu(I). The five-coordinate square pyramidal geometry around the Cu(II) center involves one CH3CN nitrogen together with the cyclam ligand in a type I arrangement (R,S,R,S) with both ferrocene and methyl substituents on the same side of the N4 plane. The two isomers differ by 0.32 V in the standard potentials of the Cu(II/I) couples and transform with kI=2 s−1 and kII=0.1 s−1.
Metal Translocations and Insertions
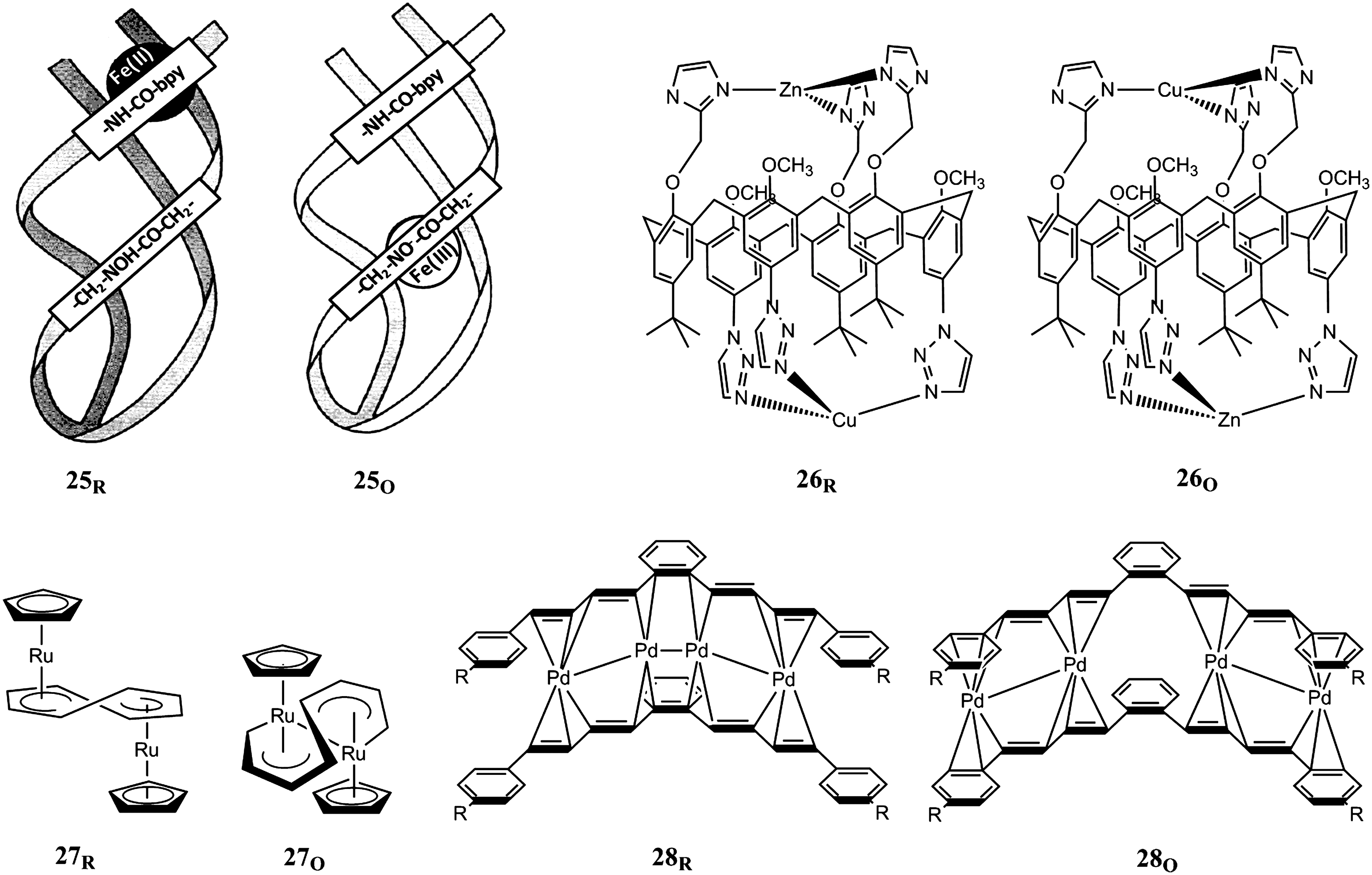
Redox-controlled site preferences of metal ions can also drive the translocation of metal ions within a larger ligand framework (Fig. 8). Triple-stranded helical peptides 25, where an Fe ion can be shuttled between an internal hydroxamate-binding site preferred by Fe(III) and an external bipyiridine-binding site for Fe(II), have been put forward as molecular redox switches (75). A remarkable double translocation of two metal ions can be triggered in ditopic ligands with two sets of ligating groups on the rims of callix[6]arene rings (26) (15). Three imidazole groups at the small rim form the preferred binding site for Cu(II), while Zn(II) is bound by the three pyrazole groups at the large rim. Reduction of the copper ion to Cu(I) leads to the reverse situation by translocation of the metal ions. Interestingly, the redox-inactive Zn(II) is required for the translocation of Cu(II) to occur. In the mononuclear complex, the Cu ion resides in the imidazole site in either oxidation state. The translocations in the dinuclear complex are about equally fast (on the order of 10 s−1) in both directions and result in pronounced electrochemical bistability, as the reduction potential of the Cu(II/I) couple is 0.65 V higher for the Cu ion at the triazole site as compared to the imidazole-bound Cu ion.
Metal translocations and insertions.
The redox-driven insertion or translocation of metal atoms or metal ions in organometallic compounds is yet another form of molecular motion and bistability. The reactivity of compound 27 exemplifies a reversible metal insertion reaction where the Ru atoms of the pseudo-triple-decker complex upon oxidation are inserted in a C–C bond of the cyclooctatetraene ligand in the resulting flyover complex (29). The rearrangement occurs already at the one-electron oxidation level, but the flyover structure is more easily oxidized than the pseudo-triple-decker complex, which results in an overall two-electron oxidation. In the metal sandwich framework 28 (47), translocation of metal atoms with formation/cleavage of metal–metal bonds is giving rise to redox bistability. Upon two-electron oxidation, the Pd atoms confined between π-conjugated sp2-carbon planes slide along the π-system under cleavage of the central Pd–Pd bond.
Ligand Translocations in Interlocked Supramolecular Assemblies
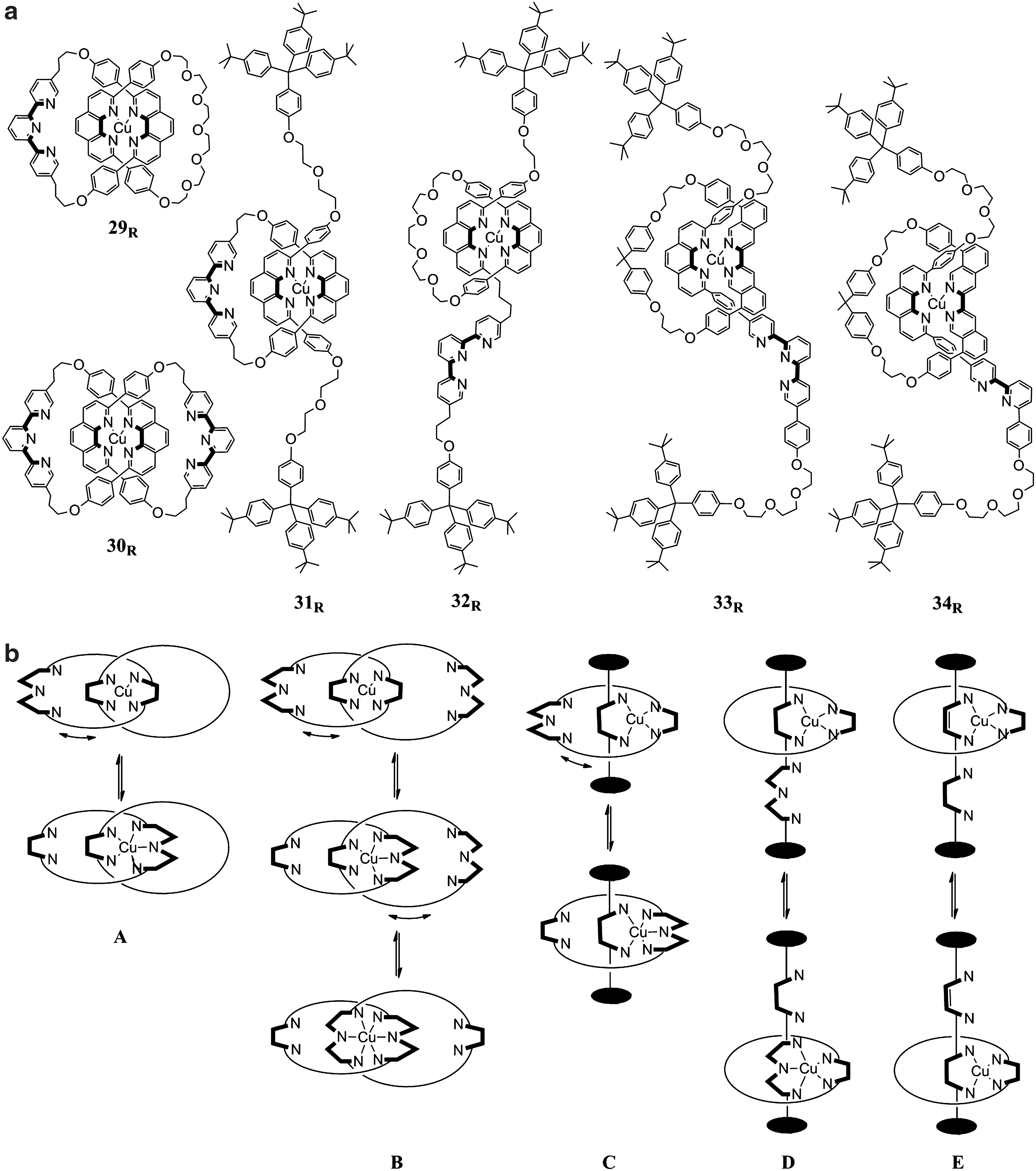
Highly sophisticated molecular structures that can be set in motion by redox processes in coordination compounds are metallorotaxanes and metallocatenanes extensively studied by Sauvage and coworkers (26). These structures (Fig. 9a) consist of either interlocked rings (catenanes) or rings threaded onto an axle with stoppers on both ends (rotaxanes). Due to metal-binding sites with distinct preferences for alternative metal oxidation states, diverse forms of molecular motion as schematically depicted in Figure 9b can be initiated by redox processes of the metal center. In Cu(I) [2]pseudorotaxane (46), reduction of a ligand [3,6-bis(5-methyl-2-pyridine)-1,2,4,5-tetrazine] was instead used to trigger the formation of a [3]pseudorotaxane as one of the fewer examples of reversible structural changes in metal complexes driven by redox-active ligands (1). Generally, the motions driven by metal-redox processes can be described as ligand translocation reactions that are driven by the different stereoelectronic requirements of the Cu ion in its Cu(I) and Cu(II) state, respectively. In the original copper catenates 29, one of the rings coordinates via a bidentate phenantroline unit, while the second ring represents an ambidentate ligand with bidentate (phenantroline) and terdentate chelates (2,2′,6′,2′′-terpyridine). The coordination number preferences (4 vs. 5 or 6) between Cu(I) and Cu(II) result in the Cu(I)N4/Cu(II)N5 linkage isomerism that drives a ring-gliding motion (A) (43, 44). The stabilization of Cu(II) by the N5 donor set lowers the Cu(II/I) potential by 0.70 V, but Cu(II)N4→Cu(II)N5 is very slow, taking minutes or longer depending on solvent conditions. Also, the Cu(I)N5→Cu(I)N4 isomerization (1 s−1) is still much slower than substitution rates in Cu(I) complexes, and topological effects are likely to cause the sluggishness of the process. Further examples of copper catenanes also include compounds 30 where both rings are ambidentate with bidentate and terdentate chelates, which results in three stage systems (B) that involve isomers with coordination numbers of 3, 4, and 5 that differ in Cu(II/I) potentials by 0.4 V (N6vs. N5) and 0.6 V (N5vs. N4), respectively (12).
Metallocatenanes and metallorotaxanes.(a) Structures; (b) Schematic representation of their redox-driven molecular motions: ring gliding (A), ring gliding, three stage (B), pirouetting (C), ring shuttling (D, E).
In copper rotaxanes, the ambidentate N2/N3 rings perform redox-driven pirouetting motions (C) when threaded onto an axis with a single bidentate-binding site as initially observed with rotaxane 31 (56) or analogous [3]rotaxanes with two N2/N3 rings on an axis composed of two identical subunits linked by a disulfide bridge (72). The relatively slow pirouetting in 31 (kIIN4→N5=7×10−3 s−1, kIN5→N4=17 s−1) was accelerated in rotaxanes with a less-bulky bipyridine (42, 49) or 8,8′-diphenyl-3,3′-biisoquinoline (dpbiiq) (17) chelate in the axis, and maximal rates were observed with bipyridine in combination with a longer axis (kIIN4→N5=12 s−1, kIN5→N4>1.2×103 s−1) (42).
Alternatively, a shuttling motion (D) of the ring along the axis can be initiated if the latter contains bidentate (phenantroline) and terdentate (2,2′,6′,2′′-terpyridine) stations as in the first example 32 (18). While the translocations of the ring in 32 were slow (kIIN4→N5=1.5×10−4 s−1; kIN5→N4≈10−4–10−2 s−1), the rates were largely increased when rings with reduced steric hindrance based on the dpbiiq ligand were introduced (33) (kIIN4→N5=2 s−1; kIN5→N4=35 s−1) (23–25).
The above-mentioned examples of metallocatenanes and metallorotaxanes are set in motion by ligands that satisfy the different coordination number preferences of Cu(I) and Cu(II) by alternative N2-/N3-binding sites. However, ring-shuttling motions in rotaxane with two bidentate chelate ligands in both isomers have also been described (E). Compound 34 combines a ring with the nonhindering dpbiiq ligand and a rod with shielding 2,9-diphenyl-1,10-phenatroline (dpp) and an alternative nonshielding bpy (16). Coordination to the dpp site is preferred by Cu(I), while the stable isomer in the Cu(II) state involves the nonshielding bpy site. The latter allows for additional coordination of solvent that is prevented by the shielding dpp. The isomerizations are reasonably fast (kIIdpp→bpy=0.8 s−1; kIbpy→dpp>50 s−1), but result in a smaller difference in standard potentials between the Cu(II/I) couples (250 mV) compared to the systems based on the isomerism between bidentate-/tridentate-binding sites.
Conclusions and Perspectives
The chemistry of coordination and organometallic compounds offers a wealth of oxidation-state-dependent structural preferences. These provide a versatile platform for the design of compounds that features the redox-driven molecular motion of various kinds. In many cases, simpler structural rearrangements based on linkage isomerism or geometric rearrangements are employed to drive larger-scale motions in more complex molecular assemblies. Examples range from the rotation of ligand moieties or translocation of metals within larger ligand frameworks, via assembly/disassembly phenomena, to long-range shuttling motion in supramolecular assemblies and demonstrate the potential that coordination compounds hold for the design of redox-driven engines in molecular machines. To this end, coordination compounds with redox-driven unidirectional rotation (13, 71) remain a formidable challenge. As a consequence of the structural changes triggered by an electron transfer according to Scheme 1b, all structures presented in this review feature redox bistability (Fig. 1b), a response to applied potential that directly emulates the performance of logic gates known as sequential SET/RESET latches (48). By analogy to these fundamental elements of digital electronics, the redox state of bistable molecular switches responds to SET and RESET inputs, for example, ESET>E02 and ERESET<E01, depending on the previous input. This behavior relies on the prevalence of alternative oxidation state in the bistability interval {E02; E01}, and the frequency of SET/RESET operations is accordingly limited by the time required for the structural rearrangements. With the fastest redox-actuated systems operating in the millisecond time regime, faster reactions in both oxidation states remain a central design objective. Regarding further progress toward the device level, immobilization and electrochemical addressing of the molecular components without deleterious effects on the structural rearrangements will be equally important.
Footnotes
Abbreviations Used
References
1.
AllgeierAM, MirkinCA. Ligand design for electrochemically controlling stoichiometric and catalytic reactivity of transition metals. Angew Chem Int Ed, 37:894–908. 1998.
2.
AmendolaV, FabbrizziL, GianelliL, MaggiC, ManganoC, PallaviciniP, ZemaM. Electrochemical assembling/disassembling of helicates with hysteresis. Inorg Chem, 40:3579–3587. 2001.
3.
AmendolaV, FabbrizziL, LinatiL, ManganoC, PallaviciniP, PedrazziniV, ZemaM. Electrochemically controlled assembling/disassembling processes with a bis-imine bis-quinoline ligand and the Cu-II/Cu-I couple. Chemistry, 5:3679–3688. 1999.
BallardiniR, BalzaniV, CrediA, GandolfiMT, VenturiM. Artificial molecular-level machines: which energy to make them work?Acc Chem Res, 34:445–455. 2001.
6.
BalzaniV, CrediA, SilviS, VenturiM. Artificial nanomachines based on interlocked molecular species: recent advances. Chem Soc Rev, 35:1135–1149. 2006.
7.
BolvinH, KahnO, VekhterB. Hysteresis for noninteracting molecules. N J Chem, 15:889–895. 1991.
8.
BondAM, GrabaricBS, JackowskiJJ. Investigation of isomerism rates and mechanism of some 17- and 18-electron substituted carbonyl complexes of chromium, molybdenum, and tungsten using double potential step chronoamperometry. Inorg Chem, 17:2153–2157. 1978.
9.
BrowneWR, FeringaBL. Making molecular machines work. Nat Nanotechnol, 1:25–35. 2006.
10.
BucherC, MoutetJ-C, PécautJ, RoyalG, Saint-AmanE, ThomasF. Redox-triggered molecular movement in a multicomponent metal complex in solution and in the solid state. Inorg Chem, 43:3777–3779. 2004.
11.
CammiR, GennariM, GiannettoM, LanfranchiM, MarchiòL, MoriG, PaiolaC, PellinghelliMA. Synthesis, structure, and electrochemical properties of copper(I) complexes with S/N homoscorpionate and heteroscorpionate ligands. Inorg Chem, 44:4333–4345. 2005.
12.
CardenasDJ, LivoreilA, SauvageJP. Redox control of the ring-gliding motion in a Cu-complexed catenane: a process involving three distinct geometries. J Am Chem Soc, 118:11980–11981. 1996.
ChouMH, BrunschwigBS, CreutzC, SutinN, YehA, ChangRC, LinCT. Facile amino to pyridyl isomerization: pentaammineruthenium(II) walks the nicotinamide and isonicotinamide rings. Inorg Chem, 31:5347–5348. 1992.
15.
ColassonB, PoulNL, MestYL, ReinaudO. Electrochemically triggered double translocation of two different metal ions with a ditopic calix[6]arene ligand. J Am Chem Soc, 132:4393–4398. 2010.
16.
CollinJ-P, DurolaF, LuxJ, SauvageJ-P. A copper-based shuttling [2]rotaxane with two bidentate chelates in the axis: steric control of the motion. N J Chem, 34:34–43. 2010.
17.
CollinJ-P, DurolaF, MobianP, SauvageJ-P. Pirouetting copper(i)-assembled pseudo-rotaxanes: strong influence of the axle structure on the motion rate. Eur J Inorg Chem, 2007:2420–2425. 2007.
18.
CollinJ-P, GaviñaP, SauvageJ-P. Electrochemically induced molecular motions in copper-complexed threaded systems: From the unstoppered compound to the semi-rotaxane and the fully blocked rotaxane. N J Chem, 21:525–528. 1997.
19.
CrediA, SemeraroM, SilviS, VenturiM. Redox control of molecular motion in switchable artificial nanoscale devices. Antioxid Redox Signal, 14:1119–1165. 2011.
20.
da RochaZN, FerreiraKQ, SilvaM, de OliveiraÉC, ChiericatoG, TfouniE. 4-Cyanopyridine and amide-N and amide-O linkage isomers of 4-pyridinecarboxamide on trans-chloro(1,4,8,11-tetraazacyclotetradecane)ruthenium(II/III)Inorg Chem, 40:5385–5392. 2001.
21.
de SilvaAP, UchiyamaS. Molecular logic and computing. Nat Nanotechnol, 2:399–410. 2007.
22.
DiamondSE, TaubeH. Nitrogen to oxygen isomerization of the pentaammineruthenium(III) glycine ion. J Am Chem Soc, 97:5921–5923. 1975.
23.
DurolaF, LuxJ, SauvageJ-P. A fast-moving copper-based molecular shuttle: synthesis and dynamic properties. Chemistry, 15:4124–4134. 2009.
24.
DurolaF, SauvageJ-P. Fast electrochemically induced translation of the ring in a copper-complexed [2]rotaxane: the biisoquinoline effect. Angew Chem Int Ed, 46:3537–3540. 2007.
25.
DurolaF, SauvageJ-P, WengerOS. The magic effect of endocyclic but non-sterically hindering biisoquinoline chelates: from fast-moving molecular shuttles to [3]rotaxanes. Coord Chem Rev, 254:1748–1759. 2010.
26.
DurotS, ReviriegoF, SauvageJ-P. Copper-complexed catenanes and rotaxanes in motion: 15 years of molecular machines. Dalton Trans, 39:10557–10570. 2010.
FairlieDP, IlanY, TaubeH. Oxygen versus nitrogen bonding of carboxamides to pentaammineruthenium(II/III)Inorg Chem, 36:1029–1037. 1997.
29.
GeigerWE, SalzerA, EdwinJ, VonphilipsbornW, PiantiniU, RheingoldAL. Structural consequences of electron-transfer reactions. 22. Electrochemically induced insertion of ruthenium atoms into a C-C bond—reversible slicing of a cyclooctatetraene ligand. J Am Chem Soc, 112:7113–7121. 1990.
30.
GreenJE, ChoiJW, BoukaiA, BunimovichY, Johnston-HalperinE, DeIonnoE, LuoY, SheriffBA, XuK, ShinYS, TsengH-R, StoddartJF, HeathJR. A 160-kilobit molecular electronic memory patterned at 1011 bits per square centimeter. Nature (London, United Kingdom), 445:414–417. 2007.
31.
HamaguchiT, InoueY, UjimotoK, KawataS, AndoI. Control of isomerization of pyridinethiol ruthenium complexes via external stimuli and factors affecting isomerization behavior. Bull Chem Soc Jpn, 85:61–68. 2012.
IlanY, TaubeH. Isomeric forms of the complexes of tetraammineruthenium(III) and (II) with glycinamide and derivatives. Inorg Chem, 22:1655–1664. 1983.
34.
JohanssonO, JohannissenLO, LomothR. Bistable molecular switches based on linkage isomerization in ruthenium polypyridyl complexes with a ligand-bound ambidentate motif. Chemistry, 15:1195–1204. 2009.
35.
JohanssonO, LomothR. Rapid electrochemically induced linkage isomerism in a ruthenium(II) polypyridyl complex. Chem Commun, 1578–1580. 2005.
36.
JohanssonO, LomothR. Molecular hysteresis in a rigid dinuclear ruthenium polypyridyl complex incorporating a ligand-bound ambidentate motif. Inorg Chem, 47:5531–5533. 2008.
37.
KahnO, KröberJ, JayC. Spin transition molecular materials for displays and data recording. Adv Mater, 4:718–728. 1992.
38.
KatzNE, FagaldeF. Redox-induced linkage isomerizations of acrylamide complexes of pentaammineruthenium(II) and pentaammineruthenium(III)Inorg Chem, 32:5391–5393. 1993.
39.
KayER, LeighDA, ZerbettoF. Synthetic molecular motors and mechanical machines. Angew Chem Int Ed, 46:72–191. 2007.
40.
KolleU. Energetic and constitutional hysteresis in bistable molecules. Angew Chem Int Ed Engl, 30:956–958. 1991.
41.
LaChance-GalangKJ, MaldonadoI, GallagherML, JianW, ProckA, ChacklosJ, GalangRD, ClarkeMJ. Terpsichorean movements of pentaammineruthenium on pyrimidine and isocytosine ligands. Inorg Chem, 40:485–492. 2000.
42.
Létinois-HalbesU, HanssD, BeierleJM, CollinJ-P, SauvageJ-P. A fast-moving [2]rotaxane whose stoppers are remote from the copper complex core. Org Lett, 7:5753–5756. 2005.
43.
LivoreilA, Dietrich-BucheckerCO, SauvageJ-P. Electrochemically triggered swinging of a [2]-catenate. J Am Chem Soc, 116:9399–9400. 1994.
44.
LivoreilA, SauvageJ-P, ArmaroliN, BalzaniV, FlamigniL, VenturaB. Electrochemically and photochemically driven ring motions in a disymmetrical copper [2]-catenate. J Am Chem Soc, 119:12114–12124. 1997.
45.
McClureBA, MockusNV, ButcherDP, LuttermanDA, TurroC, PetersenJL, RackJJ. Photochromic ruthenium sulfoxide complexes: evidence for isomerization through a conical intersection. Inorg Chem, 48:8084–8091. 2009.
46.
McNittKA, ParimalK, ShareAI, FahrenbachAC, WitlickiEH, PinkM, BediakoDK, PlaisierCL, LeN, HeeringaLP, GriendDAV, FloodAH. Reduction of a redox-active ligand drives switching in a Cu(I) pseudorotaxane by a bimolecular mechanism. J Am Chem Soc, 131:1305–1313. 2009.
47.
MurahashiT, ShiratoK, FukushimaA, TakaseK, SuenobuT, FukuzumiS, OgoshiS, KurosawaH. Redox-induced reversible metal assembly through translocation and reversible ligand coupling in tetranuclear metal sandwich frameworks. Nat Chem, 4:52–58. 2012.
48.
PeriyasamyG, CollinJP, SauvageJP, LevineRD, RemacleF. Electrochemically driven sequential machines: an implementation of copper rotaxanes. Chemistry, 15:1310–1313. 2009.
49.
PoleschakI, KernJ-M, SauvageJ-P. A copper-complexed rotaxane in motion: pirouetting of the ring on the millisecond timescale. Chem Commun, 474–476. 2004.
50.
PottsKT, Keshavarz-KM, ThamFS, AbrunaHD, AranaCR. Metal ion-induced self-assembly of functionalized 2,6-oligopyridines. 2. Copper-containing double-stranded helicates derived from functionalized quaterpyridine and quinquepyridine: redox state-induced transformations and electron communication in mixed-valence systems. Inorg Chem, 32:4422–4435. 1993.
51.
PowellDW, LayPA. Linkage isomerization reactions of (acetone)pentaammineruthenium(III) and -ruthenium(II) complexes. Inorg Chem, 31:3542–3550. 1992.
RackJJ. Electron transfer triggered sulfoxide isomerization in ruthenium and osmium complexes. Coord Chem Rev, 253:78–85. 2009.
54.
RackJJ, MockusNV. Room-Temperature Photochromism in cis- and trans-[Ru(bpy)2(dmso)2]2+Inorg Chem, 42:5792–5794. 2003.
55.
RackJJ, RachfordAA, ShelkerAM. Turning off phototriggered linkage isomerizations in ruthenium dimethyl sulfoxide complexes. Inorg Chem, 42:7357–7359. 2003.
56.
RaehmL, KernJ-M, SauvageJ-P. A Transition metal containing rotaxane in motion: electrochemically induced pirouetting of the ring on the threaded dumbbell. Chemistry, 5:3310–3317. 1999.
57.
RoeckerL, DobsonJC, ViningWJ, MeyerTJ. Oxygen atom transfer in the oxidations of dimethyl sulfide and dimethyl sulfoxide by bis(bipyridine)oxo(pyridine)ruthenium(2+)Inorg Chem, 26:779–781. 1987.
58.
RootMJ, DeutschE. Synthesis and characterization of (bipyridine)(terpyridine)(chalcogenoether)ruthenium(II) complexes. Kinetics and mechanism of the hydrogen peroxide oxidation of [(bpy)(tpy)RuS(CH3)2]2+ to [(bpy)(tpy)RuS(O)(CH3)2]2+. Kinetics of the aquation of [(bpy)(tpy)RuS(O)(CH3)2]2+Inorg Chem, 24:1464–1471. 1985.
SanoM. Molecular hysteresis by linkage isomerizations induced by electrochemical processes. Struct Bond (Berlin, Germany), 99:117–139. 2001.
61.
SanoM, TaubeH. Molecular hysteresis. J Am Chem Soc, 113:2327–2328. 1991.
62.
SanoM, TaubeH. “Molecular hysteresis” in an electrochemical system revisited. Inorg Chem, 33:705–709. 1994.
63.
SaveantJ-M. Elements of Molecular and Biomolecular Electrochemistry. Hoboken: Wiley-Interscience, 2006; 398.
64.
SensC, RodriguezM, RomeroI, LlobetA, ParellaT, SullivanBP, Benet-BuchholzJ. Synthesis, structure, and spectroscopic, photochemical, redox, and catalytic properties of ruthenium(II) isomeric complexes containing dimethyl sulfoxide, chloro, and the dinucleating bis(2-pyridyl)pyrazole ligands. Inorg Chem, 42:2040–2048. 2003.
65.
SzacilowskiK. Digital information processing in molecular systems. Chem Rev, 108:3481–3548. 2008.
66.
TomaHE, GiesbrechtE, RojasRLE. Linkage isomerism in penta-ammineruthenium(II),(III) complexes of benzotriazole. J Chem Soc-Dalton Trans, 2469–2472. 1985.
67.
TomitaA, SanoM. Linkage isomerizations of (sulfoxide)ammineruthenium complexes induced by electrochemical processes. Inorg Chem, 33:5825–5830. 1994.
68.
TomitaA, SanoM. Preparations and electrochemical properties of pyrazine-bridged ruthenium-binuclear complexes exhibiting molecular hysteresis. Inorg Chem, 39:200–205. 1999.
69.
TsekourasG, JohanssonO, LomothR. A surface-attached Ru complex operating as a rapid bistable molecular switch. Chem Commun, 3425–3427. 2009.
70.
TsekourasG, MinderN, FiggemeierE, JohanssonO, LomothR. A bistable electrochromic material based on a hysteretic molecular switch immobilised on nanoparticulate metal oxide. J Mater Chem, 18:5824–5829. 2008.
71.
VivesG, de RouvilleH-PJ, CarellaA, LaunayJ-P, RapenneG. Prototypes of molecular motors based on star-shaped organometallic ruthenium complexes. Chem Soc Rev, 38:1551–1561. 2009.
72.
WeberN, HamannC, KernJ-M, SauvageJ-P. Synthesis of a copper [3]rotaxane able to function as an electrochemically driven oscillatory machine in solution, and to form SAMs on a metal surface. Inorg Chem, 42:6780–6792. 2003.
73.
WimmerFL, SnowMR, BondAM. Electrochemical investigations of isomerism in manganese and Group (VI) dicarbonyl-bis [1,2-bis(diphenylphosphino)ethane] complexes. Inorg Chem, 13:1617–1623. 1974.
74.
YehA, ScottN, TaubeH. S to O and O to S linkage isomerization in sulfoxide complexes of pentaammineruthenium. Inorg Chem, 21:2542–5. 1982.
75.
ZelikovichL, LibmanJ, ShanzerA. Molecular redox switches based on chemical triggering of iron translocation in triple-stranded helical complexes. Nature, 374:790–792. 1995.