
Abstract
Introduction
S
The prevention of a majority of the acute radiation-induced liver injury in SIRT3−/− mice after exposure to Cs-137 γ-rays by the small-molecular-weight superoxide dismutase (SOD) mimic GC4401 indicates that ionizing radiation-induced damage in this model system can be caused by O2 •−. Previous studies have shown that similar SOD mimics can delay lethality from higher doses of radiation; however, prevention of sublethal injury by GC4401 represents a substantial innovation that is potentially relevant to several different organ systems during whole-body radiation exposure. This may have broad applications in therapeutic settings requiring radioprotection or in cases of risk associated with accidental exposures.
Sirtuin 3 (SIRT3) is the primary mitochondrial protein deacetylase and an emerging regulator of mitochondrial responses to IR as well as other oxidative stresses (11, 39). Since SIRT3 loss is associated with increased levels of O2 •−, SIRT3 is thought to play a critical role regulating manganese superoxide dismutase (MnSOD) expression and activity both transcriptionally through FOXO3a and post-translationally via deacetylation of lysine residues on MnSOD (20, 40). SIRT3 also plays a critical role in regulating metabolic output from the mitochondria and has been associated with carcinogenesis (16, 23, 35). SIRT3−/− mouse embryonic fibroblasts (MEFs) have been reported to produce excess O2 •− and demonstrate evidence of chronic oxidative stress without application of exogenous stresses (1, 23). Here, we are using SIRT3−/− mice to determine whether O2 •− generation associated with mitochondrial dysfunction contributes to liver injury after exposure to whole-body irradiation doses similar to those given during the course of bone marrow transplantation.
The relatively high level of oxidative metabolism in the liver is thought to necessitate high levels of O2 •− metabolizing enzymes, including MnSOD and copper-zinc SOD (CuZnSOD) found in the liver. MnSOD is localized to the inner mitochondrial membrane and matrix, while CuZnSOD resides in the intermembrane space of the mitochondria, the cytoplasm, and the nucleus (13). MnSOD is critical for oxygen-breathing organisms, as indicated by the <3 week survival times, depending on the genetic background of MnSOD homozygous knockout mice (2, 19). In addition, enforced overexpression of MnSOD has also been shown to prevent increased sensitivity to radiation-induced cell killing as well as to prevent chronically elevated levels of mitochondrial reactive oxygen species in cultured cells with defects in succinate dehydrogenase (complex II) (7, 37).
SIRT3 also participates in several pro-survival pathways, preventing cell death as a result of nutrient deprivation or application of other exogenous stress (24, 39). Increased SIRT3 activity is directly implicated in increasing cell survival through p53 degradation in vitro and in vivo as well as through negative regulation of the mitochondrial permeability transition pore in heart mitochondria (15). Further, reports showing p53 translocation to mitochondria preceding nuclear activity of p53 seem to suggest that this process may be related to mitochondrial O2 •− levels and SIRT3 activity (24, 28, 43). SIRT3-mediated prevention of oxidative damage by increased O2 •− removal coupled with the blunting of p53 activation may, therefore, play a critical role in the radiation responses of mitochondria-rich, metabolically active organs such as the liver.
The current study builds on previous work identifying SIRT3 as regulating MnSOD in the liver (40) to clearly delineate the importance of O2 •− in the enhancement of acute liver injury induced by IR in SIRT3−/− mice. We hypothesized that excess O2 •− sensitizes liver tissue to acute IR-induced injury in SIRT3−/− mice and that this sensitization could be prevented with the addition of an O2 •− specific SOD mimetic, GC4401 (3).
GC4401, a small molecule, highly specific SOD mimic with a rate constant similar to the native enzyme (109 mol−1·s−1), is the S,S-dimethyl derivative of the biscyclohexylpyridine Mn-based SOD-mimetic GC4403 (M40403) that has been shown to be stable in vivo with a plasma clearance half life in rats of 2 h and no known toxicity at the doses and times utilized in this study (30, 41). Homozygous wild-type (WT) and SIRT3−/− mice were exposed to two fractions of 2 Gy doses of whole-body Cs-137 irradiation separated by 24 h. SIRT3−/− mice demonstrated substantial acute liver injury 20 h after 2×2 Gy as indicated by evidence of hydropic degeneration, increased 3-nitrotyrosine modified proteins detected by immunohistochemistry, and loss of proliferation as assayed by Ki67 staining. This injury was prevented by pretreatment with GC4401. These results support the hypothesis that mitochondrial O2 •− generation after exposure to IR plays a critical role in acute radiation-induced liver injury in SIRT3−/− animals.
Results
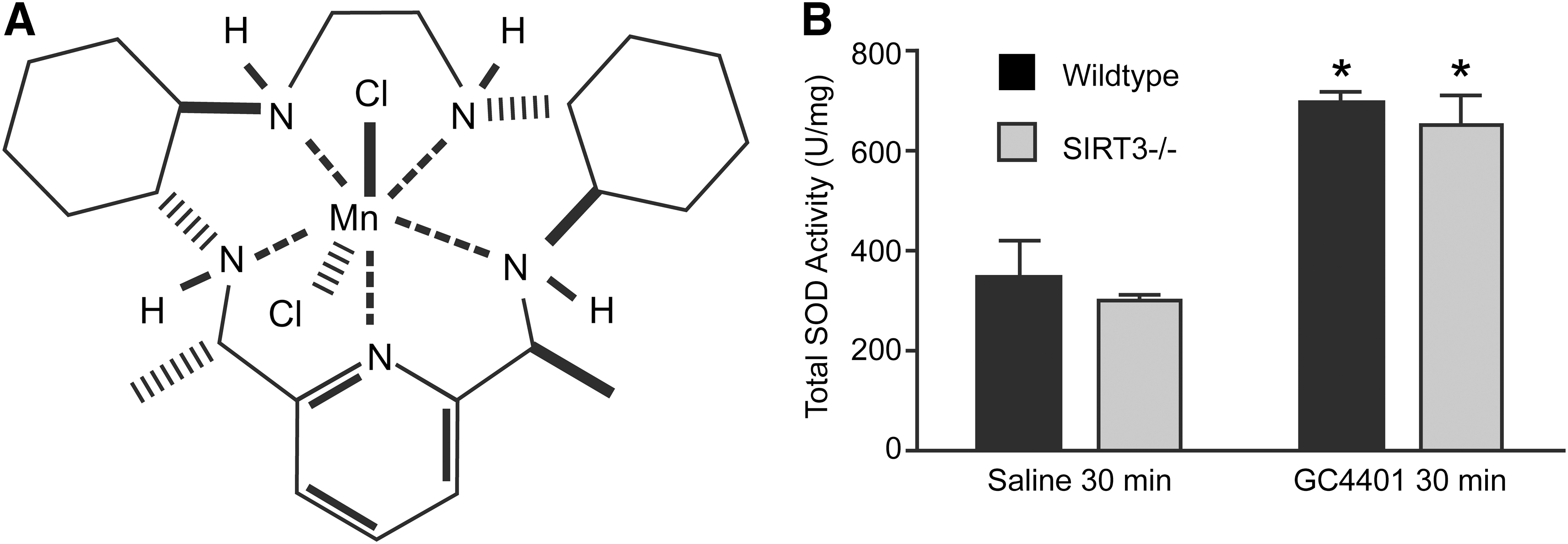
GC4401 delivery and liver SOD activity
GC4401 has a catalytic rate constant similar to the SOD enzymes at pH 7.4 (1.2×109 mol−1·s−1), freely diffuses across cellular membranes, and members of this class of SOD mimics have been shown to be stable in vivo with a half life of ∼2 h (3, 30, 41). When 2 mg/kg GC4401 (Fig. 1A) was injected intraperitoneally (IP) into mice and livers were harvested 30 min later, the total SOD activity detected in both WT and SIRT3−/− liver homogenates demonstrated a two-fold increase (Fig. 1B). These results show that GC4401 significantly increases liver tissue levels of SOD activity in vivo and can be used to determine whether O2 •− contributed to the histological liver injury previously reported in the SIRT3−/− model system (40).
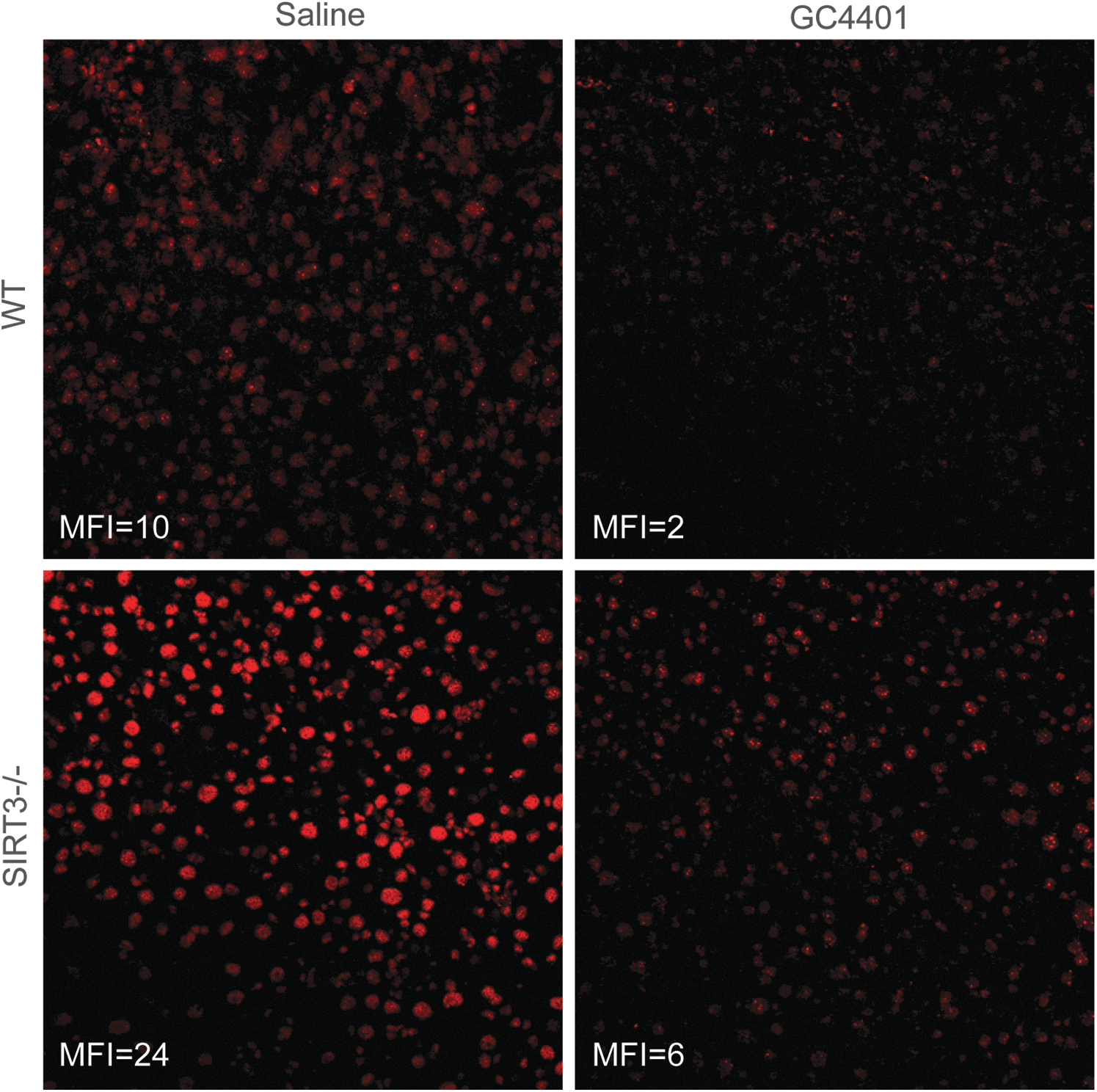
Dihydroethidium (DHE) is oxidized to red fluorescent products by O2
•− as well as other oxidants and is used as a marker for superoxide levels in cells and tissues (36). Data in Figure 2 show that fresh frozen liver sections from SIRT3−/− animals demonstrated a two-fold increase in the ability to oxidize DHE to its red fluorescent products, relative to WT livers. In addition, 4 μM GC4401 added directly to tissue sections before DHE labeling suppressed 70%–85% of DHE oxidation in all slides examined, indicating that in this experimental model, DHE oxidation is largely mediated by O2
•− (Fig. 2). The SIRT3−/− phenotype was then confirmed with immuno-blotting (Supplementary Fig. S1A; Supplementary Data are available online at
To confirm that mitochondrial O2 •− contributed to changes in dye oxidation, fresh frozen liver sections were labeled with MitoSOX Red (a mitochondrial-targeted triphenylphosphonium derivative of DHE) as well as MitoTracker Green FM (MTG) (Supplementary Fig. S1B). Quantitation of MitoSOX Red staining which co-localized with MTG fluorescence revealed that mitochondrial MitoSOX Red oxidation was increased 30%–40% in the SIRT3−/− liver sections, relative to WT (Supplementary Fig. S1B).
Evaluation of liver injury, oxidative damage, and oxidant production
To produce acute liver injury in SIRT3−/− mice, a previously characterized (40) fractionated whole-body Cs137 γ-irradiation protocol was utilized where 2×2 Gy doses were delivered 24 h apart and tissues were collected 20 h after the second dose. Consistent with the previous study (40), 20 h after the second 2 Gy dose, livers from SIRT3−/− animals demonstrated significantly increased hepatocellular hydropic degeneration in the periportal and midzonal hepatocytes that were characterized by poorly defined cytoplasmic space and clear space within hepatocytes (Fig. 3A). SIRT3−/− mice exposed to radiation also demonstrated increased immunohistochemical liver staining for the oxidative damage endpoint, 3-nitrotyrosine (Fig. 3B). No significant increases in immuno-reactive inducible nitric oxide synthase were observed in liver homogenates from SIRT3−/− animals (Supplementary Fig. S2). Consistent with a causal role for O2 •− in these radiation-induced injury processes, evidence of hydropic degeneration and oxidative damage were significantly attenuated by an IP injection of 2 mg/kg of GC4401, 30 min before each radiation exposure (Fig. 3C, D).

It was anticipated that SIRT3−/− livers would demonstrate significantly increased levels of DHE oxidation both before and after radiation exposure. Not surprisingly, DHE oxidation showed a significant 40% increase in the SIRT3−/− livers from un-irradiated animals, relative to WT (representative images in Fig. 4A, quantified in C). Exposure of WT animals to 2×2 Gy caused no change in DHE oxidation 20 h after the second exposure; however, SIRT3−/− livers appeared to oxidize 30% less DHE, relative to un-irradiated SIRT3−/− livers 20 h after exposure (Fig. 4A, C). This reduction in apparent DHE oxidation in irradiated SIRT3−/− livers was accompanied by a 20%–25% decrease in staining for MTG when compared with WT as well as the SIRT3−/− sham (Fig. 4B, D). In contrast, quantitation of MitoSOX Red co-localization with MTG revealed that mitochondrial MitoSOX Red oxidation was still increased 30% in the SIRT3−/− liver sections from irradiated animals, relative to WT (Supplementary Fig. S1B). These results combined with the DHE and MTG results in Figure 4 suggest that SIRT3−/− animals sustain greater radiation-induced injury to liver mitochondria (relative to WT) which leads to apparent reductions in DHE oxidation and MTG uptake and retention; but in mitochondria that maintain the membrane potential necessary to take up and retain both MitoSOX Red and MTG, O2 •− levels remain unchanged, relative to WT.
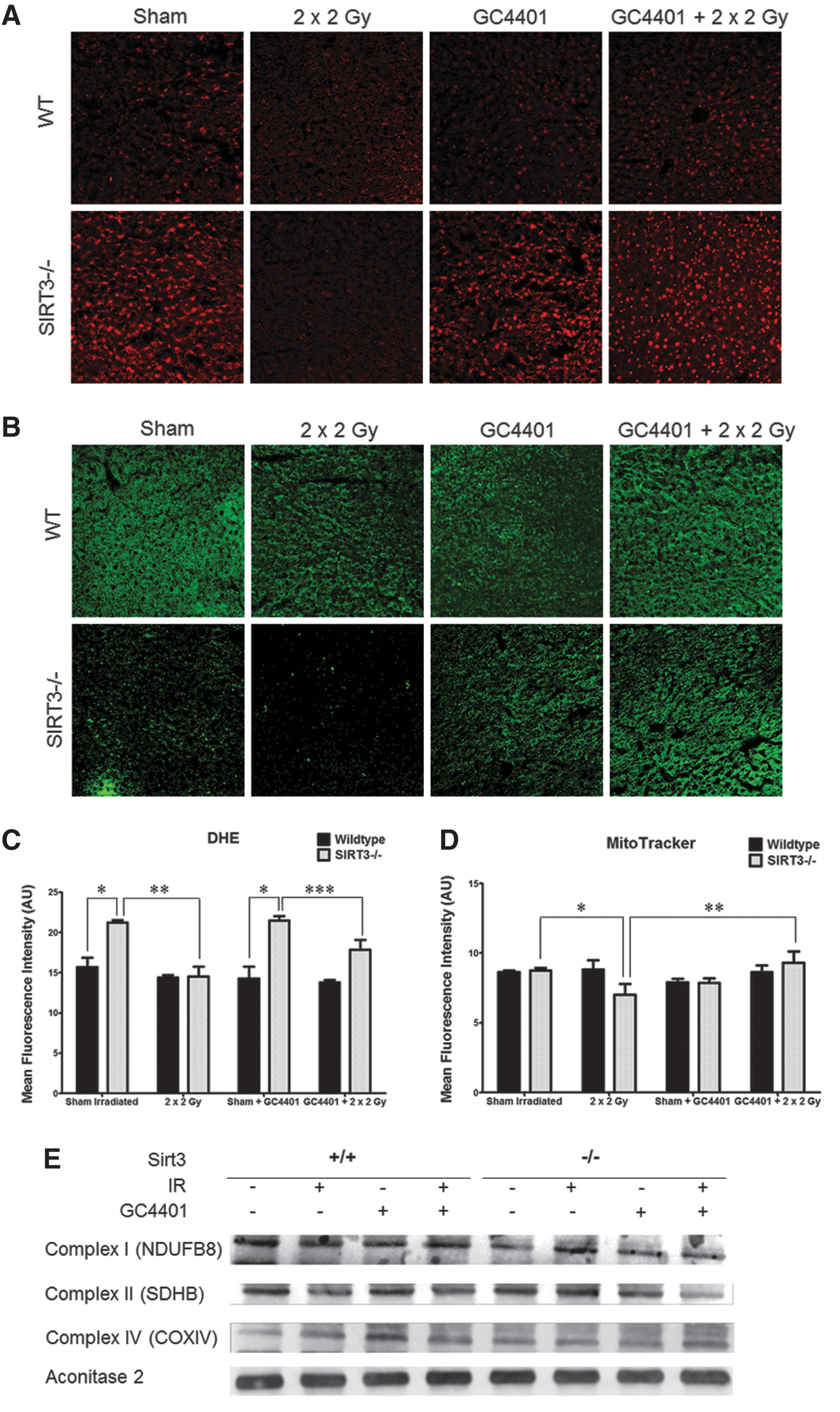
The specific mechanisms responsible for the decrease in DHE and MTG staining in SIRT3−/− livers 20 h after radiation exposure are unclear, as no significant loss of immuno-reactive mitochondrial proteins (NDUFB8, SDHB, COXIV, and Aconitase 2) was noted in any of these groups (Fig. 4E; Supplementary Fig. S3). However, consistent with increased radiation-induced injury to SIRT3−/− liver hepatocytes contributing to decreased dye uptake and retention, significant increases in serum aspartate aminotransferase and alanine aminotransferase were detected in serum samples from irradiated SIRT3−/− mice, relative to WT (Supplementary Fig. S4). In addition, since MTG dye retention is thought to rely on free thiol groups in the mitochondria, increased glutathione oxidation (Supplementary Fig. S5) in irradiated SIRT3−/− liver homogenates (that was inhibited by GC4401) may also partially explain the loss of MTG staining which was observed in Figure 4.
Consistent with decreased DHE and MTG staining being associated with O2 •—mediated damage to mitochondria, GC4401 pretreatment of SIRT3−/− mice 30 min before radiation exposure prevented radiation-induced decreases in DHE oxidation and MTG staining in SIRT3−/− livers (Fig. 4A–D). In addition, no statistically significant alterations in catalase or total glutathione peroxidase activity were detected in isolated mitochondria from these livers (Supplementary Fig. S6A, B). Interestingly, a significant 20%–30% increase in total glutathione content was detected in irradiated SIRT3−/− samples, which was unaffected by GC4401 treatment (Supplementary Fig. S6C).
Evaluation of SIRT3−/− ETC complex activities in response to IR and GC4401
To further probe the mechanisms involved in decreased DHE and MTG staining in irradiated SIRT3−/− livers, the activity of ETC complexes I, II, III, and IV, as well as aconitase activity were measured in mitochondria isolated from sham and irradiated SIRT3−/− and WT livers, with and without treatment with GC4401 (Fig. 5). Complexes I, II, and III as well as mitochondrial aconitase have been shown to be inactivated in the presence of increased mitochondrial O2 •− levels (presumably by disruption of Fe-S clusters by O2 •−), and so, we expected that some of these complexes would display decreased activity. Relative to WT, liver mitochondria from sham-treated SIRT3−/− animals displayed decreases in Complex II and III activity as well as decreased aconitase activity (relative to WT; Fig. 5B, C, E), in the absence of detectable decreases in mitochondrial immunoreactive proteins (Fig. 4E). These results support the hypothesis that some ETC complex activities in SIRT3−/− liver cells are being inhibited by O2 •−. Consistent with this interpretation, treatment with GC4401 significantly increased the activity of complexes II and III in un-irradiated Sirt3−/− animals, but not in un-irradiated WT animals (Fig. 5B, C); supporting the hypothesis that increased O2 •− dismutation by GC4401 was preventing inactivation of these complexes in Sirt3−/− liver cells.
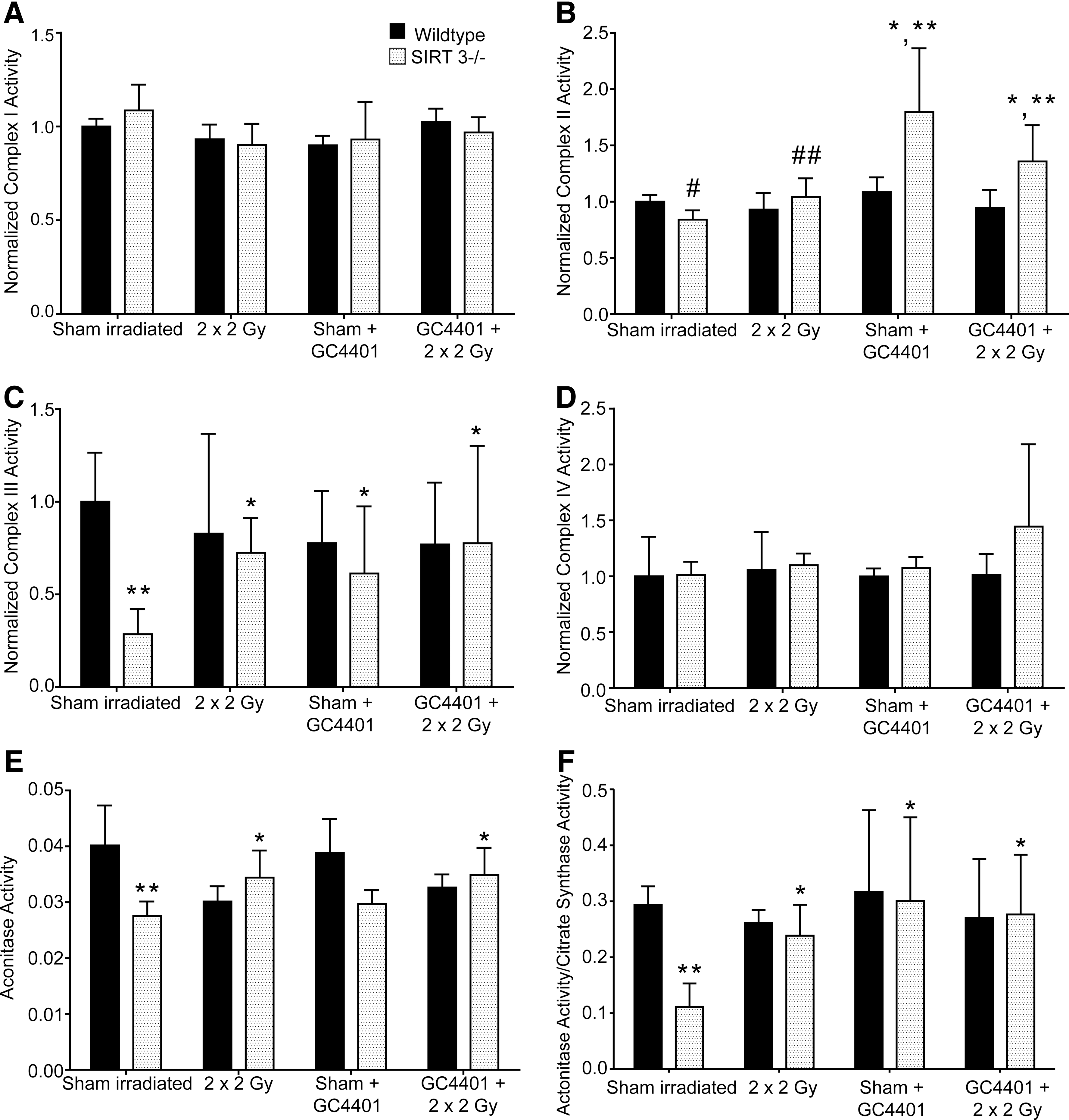
Unexpectedly, after irradiation, SIRT3−/− liver mitochondria demonstrated no decreases in any ETC activities, but demonstrated increases in complexes II and III as well as aconitase activity, relative to sham-exposed SIRT3−/− (Fig. 5B, C, E). This unexpected increase in ETC complex activity in irradiated liver mitochondria from Sirt3−/− animals could be related to the unexpected decrease in DHE oxidation seen in irradiated Sirt3−/− livers (Fig. 4) if an increase in basal levels of O2 •− in sham-exposed SIRT3−/− liver cells was, indeed, suppressing ETC complex activity. Consistent with this interpretation, GC4401 treatment of SIRT3−/− animals before radiation also caused increases in ETC complexes II and III as well as aconitase activity in liver mitochondria (Fig. 5E), relative to saline-treated SIRT3−/− animals. These results support the hypothesis that the SOD mimic was decreasing O2 •− mediated inactivation of these ETC complexes in SIRT3−/− livers, which also appears consistent with the SOD mimic inhibiting the formation of 3-nitrotyrosine and the histological injury seen in Figure 3.
To further address the mechanisms of sensitivity of SIRT3−/− cells to oxidative stress using an in vitro model, mouse embryo fibroblasts were isolated from SIRT3−/− embryos as previously described (40) and grown in different O2 tensions. These studies showed that SIRT3−/− fibroblasts demonstrated an O2 concentration-dependent inhibition of cell growth at both 4% and 20% oxygen, relative to WT cells (Supplementary Fig. S7). Furthermore, the growth inhibition seen in 4% O2 with SIRT3−/− MEFs could be completely returned to WT levels, with the addition of GC4401, supporting the hypothesis that dysfunctional mitochondria and increased O2 •− production were also significantly contributing to slower growth rates in SIRT3−/− MEFs in vitro (Supplementary Fig. S7B).
Investigation of GC4401 effects on p53 mitochondrial translocation and liver cell proliferation rates after IR
Since SIRT3 has been reported to have functions related to p53 activation and liver injury is often followed by liver regeneration, p53 and Ki67 were evaluated in this model system. We anticipated that the injury seen in irradiated SIRT3−/− livers would be accompanied by increased p53 stabilization and inhibition of growth, as indicated by loss of Ki67 staining. Interestingly, livers from SIRT3−/− animals showed elevated p53 levels in isolated mitochondria in both the presence and absence of radiation (Fig. 6A). This elevated level of mitochondrially localized p53 was abrogated by the addition of GC4401 (Fig. 6A). A corresponding, significant decrease in immunohistochemical staining for Ki67, indicative of decreased hepatocellular proliferation, was detected in SIRT3−/− livers relative to WT livers after irradiation (Fig. 6B, C). This IR-induced difference in liver cell proliferative index was partially ameliorated in both WT and SIRT3−/− mice exposed to IR on addition of GC4401 (Fig. 6C).
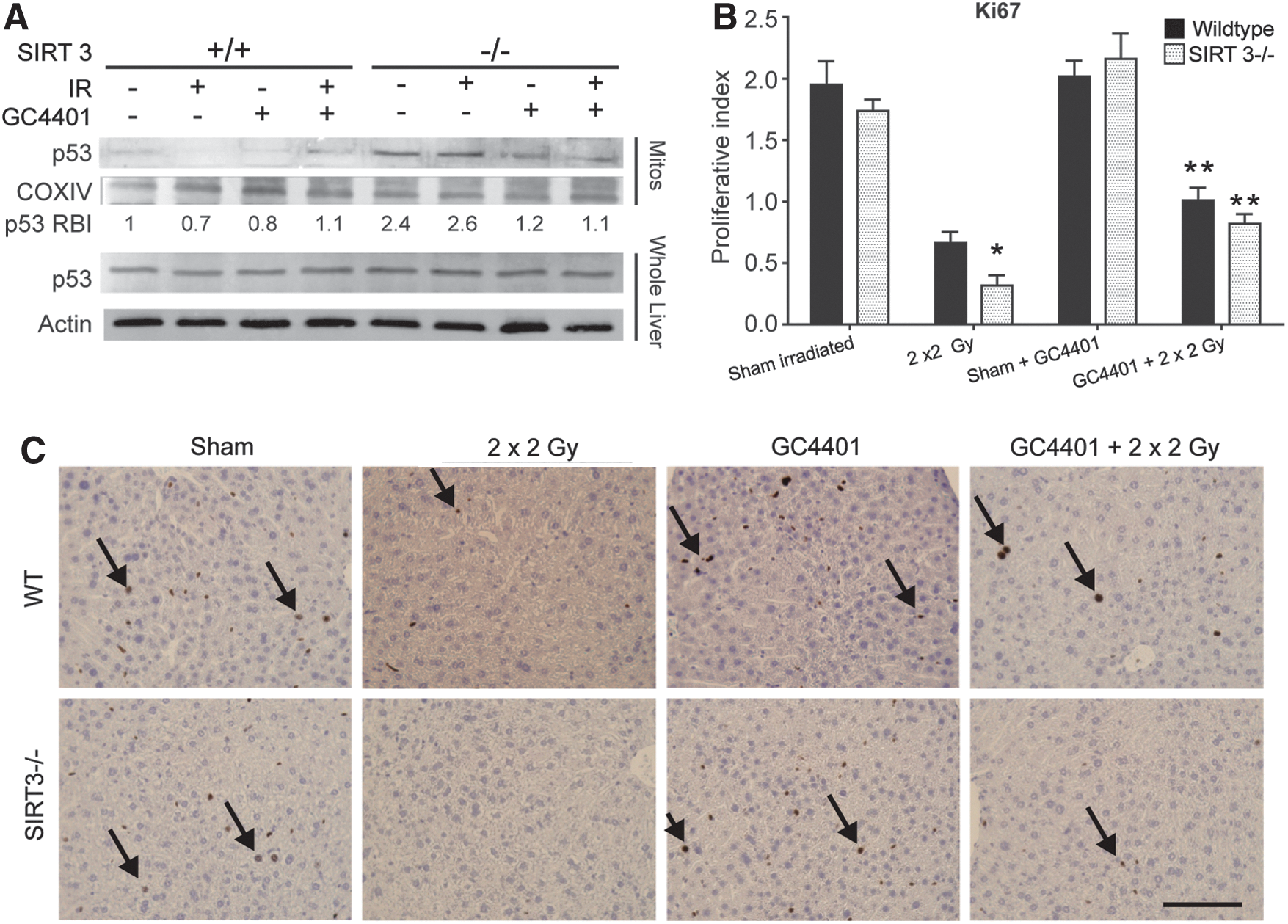
Discussion
The current study provides evidence for a critical role of O2 •− in murine liver acute radiation response by preventing O2 •− mediated damage using a highly specific SOD mimic, GC4401. Previous studies have shown that SIRT3 deacetylated MnSOD in WT mouse liver mitochondria, resulting in increased MnSOD activity after exposure to radiation which correlated with less radiation-induced tissue injury (40). However, no direct involvement of O2 •− in this liver injury pathway has been established. Here, we demonstrate that a nearly two-fold increase in total liver SOD activity resulting from an IP injection of the specific SOD mimic, GC4401, was capable of protecting the livers of SIRT3−/− animals from radiation-induced injury, 3-nitrotyrosine formation, loss of DHE and MTG staining, and decreases in ETC complex activities. Since GC4401 has highly specific activity toward O2 •− dismutation (3, 27), the data support the hypothesis that removal of O2 •− can prevent liver injury in SIRT3−/− animals resulting from whole body radiation exposure at doses relevant to those exposures commonly used in bone marrow transplantation. These results also continue to support the hypothesis that SIRT3 is a global regulator of mitochondrial oxidative metabolism which is highly relevant to stress-induced normal tissue injury processes after exposure to radiation.
In agreement with previous studies that have shown protection from radiation-induced pulmonary injury in MnSOD-deficient mice as a result of MnSOD liposomal delivery (10), and numerous studies demonstrating that MnSOD improves radiation resistance in vitro (12, 14, 17, 18, 21, 22), the current study extends the critical role of O2 •− metabolism in preventing acute radiation-induced liver injury. Furthermore, the current work is consistent with a previous study showing that bolus injections of CuZnSOD in mice could also protect against radiation-induced lethality from doses that induced injury to the hematopoietic system (25). A similar compound to GC4401, GC4403, has also been shown to demonstrate significant protective effects after lethal whole-body doses of radiation given to mice (41). These previous studies and the data presented in the current report strongly support the hypothesis that removal of O2 •− is important to the survival of mammalian cells after radiation exposure and that SIRT3 is a critical regulator of mitochondrial superoxide metabolism which significantly contributes to radiation response.
The current data measuring ETC chain activities (Fig. 5) also suggest that there may be O2 •− mediated disruption of mitochondrial metabolism within the SIRT3−/− liver cells as indicated by the basal deficits in ETC complex II, complex III, and aconitase activities which are ameliorated by GC4401 in the absence of detectable changes in ETC immuno-reactive protein concentrations. Since all these ETC complexes utilize superoxide sensitive Fe-S clusters for optimal activity, these results support the hypothesis that superoxide may be mediating the inactivation of these complexes in SIRT3−/− liver mitochondria, relative to WT. It is known that Fe2+ formed by the reaction of O2 •− with Fe3+ can undergo Fenton chemistry in the presence of H2O2; however, it has also been shown that significant oxidative damage can take place in the presence of Fe2+ and O2 even without H2O2 (26, 33). This may be particularly important in liver mitochondria where both heme and ETC protein levels are high, and, thus, the potential for labile Fe to react with O2 •− is also high. Published studies have also shown that during ischemia/reperfusion injury, chelation of metals is capable of significantly reducing free radical-mediated injury in cardiovascular systems which are also high in mitochondrial Fe content (37). Fe is known to be an important source of free radicals in aging-associated liver-free radical production (5). These previous studies and the ETC activity data presented in Figure 5 indirectly support the hypothesis that disruptions of O2 •− mediated alterations in Fe metabolism in ETCs may be an important source of damage in the SIRT3−/− livers which may be abrogated by treatment with GC4401.
Another noteworthy finding, presented in Figure 6, is the involvement of p53 in this radiation-induced injury pathway. A relationship between p53, O2 •−, and MnSOD governing p53 activation and proliferative responses has been demonstrated in vitro (31, 32, 43). These studies determined that p53 translocation to the nucleus was preceded by p53 translocation to the mitochondria where MnSOD was negatively impacted by p53. Similar to our data, the application of an SOD mimetic to remove O2 •− prevented the effects of p53 observed in the mitochondria. Another study demonstrated that Protandim, a dietary supplement known to increase cellular antioxidants, was able to prevent p53 translocation to mitochondria by virtue of its induction of MnSOD (28). SIRT3 is already known to support cell survival by increasing degradation of p53 present in the mitochondria, but the decrease in mitochondrially localized p53 on treatment with GC4401 shown in the current article supports the hypothesis that mitochondrial O2 •− plays a significant role in p53 trans-localization to the mitochondria in SIRT3−/− liver cells, independent of SIRT3-mediated deacetylation of p53. Our results also allow for the speculation that increased O2 •− may be an integral signal for p53 translocation to the mitochondria, so that it can be acted on by SIRT3 in the mitochondria of SIRT3 competent cells. Therefore, the absence of SIRT3 may play two roles in regulating p53 in the SIRT3−/− model, that is, causing increased O2 •− production, increasing p53 translocation to the mitochondria, and also failing to initiate p53 degradation through deacetylation of the p53 protein.
Another interesting observation was the loss of MTG staining seen in SIRT3−/− livers (but not WT) after irradiation, despite no evidence of loss of mitochondrial proteins. MTG stains mitochondria independently of membrane potential through a chloromethyl moiety reactive with mitochondria-specific free thiols facilitating dye localization. This allows for the speculation that MTG staining decreases in irradiated SIRT3−/− liver, because mitochondrial protein thiol oxidation is increased in SIRT3−/− liver after exposure, limiting the free thiols available for binding by the dye. We have also measured serum levels of the liver enzymes, aspartate aminotransferase and alanine aminotransferase, and determined the presence of significant levels of these enzymes in the serum of irradiated SIRT3−/− mice, indicating significant structural damage to hepatocytes. This damage may also prevent the retention of MTG and DHE during these sensitive microscopic tissue assay techniques, leading to a decreased staining with these dyes.
Though the liver is not considered a dose-limiting organ with high sensitivity to radiation, liver injury as a result of whole-body exposure is potentially important to bone marrow transplant patients and other patients receiving abdominal radiation exposures as a part of their treatment. In addition, the common combination of chemotherapeutics and radiation for any number of cancer treatments that involve irradiating the abdomen also presents patients with a potential risk of unexpected liver toxicity as a result of O2 •− generation. During conditions of radiation-induced stress to the abdomen, our data suggest that SOD mimics may afford protection from acute liver injury.
In conclusion, the current data show that radiation-induced, acute liver injury in SIRT3−/− mice is mechanistically linked to O2 •− generation. The results presented here also show that the SOD mimic, GC4401, is capable of mitigating this injury process and suggest that this compound is a good candidate for further development as a radioprotector.
Materials and Methods
Animals and radiation conditions
National Institutes of Health mice #1476, a C57B/6 background strain, lacking SIRT3 expression after neomycin-based disruption of the SIR3 gene, were maintained in accordance with ACURF approval #1104080 at the University of Iowa Animal Care facility as previously described (40). Mice were maintained on normal diets and bred using pairs of SIRT3+/− mice as well as WT×WT and SIRT3−/−×SIRT3−/− breeding pairs, with no outwardly apparent differences in mice produced by either breeding scheme.
GC4401 administration and SOD activity measurements
GC4401 (Galera Therapeutics) was prepared at a concentration of 250 μM in 0.1% saline with 26 mM sodium bicarbonate, pH 7.4 in order to deliver an intra-peritoneal injection of 2 mg/kg to each mouse 30 min before each Cs-137 exposure. GC4401 is a small-molecular-weight SOD mimic with a rate constant comparable to the native enzyme (1.2×109 mol−1·s−1) that is capable of freely entering cells in vivo (3). In order to confirm an increase in liver SOD activity in vivo, total SOD activity was measured by the method of Spitz and Oberley using whole homogenates prepared from livers 30 min after an injection of GC4401 (38).
Liver histological and immunohistochemical scoring
Liver sections were fixed in 10% neutral-buffered formalin. Fixed tissues were processed, paraffin embedded, sectioned at 4 μm, and stained with hematoxylin and eosin. Slides were manually scored in a blinded fashion for degree of hepatocellular vacuolation. The vacuolar differences, when present, were primarily from periportal to midzonal hepatocytes; thus, scoring was based on the average of three periportal images per liver section. For each image, 100 hepatocytes were counted and given a score corresponding to the severity of the vacuolation: low, medium, or high. Low vacuolation was characterized by mild cytoplasmic vacuolation consistent with glycogen accumulation. Medium to high vacuolar scores corresponded to hepatocytes that were moderately to markedly enlarged due to dilation of the cytoplasm by clear space and poorly defined clear vacuoles.
Ki67 and 3-nitrotyrosine levels were measured via immunohistochemistry. Slides were retrieved in citrate buffer (pH 6.0) using a Decloaking Chamber (Biocare) for 3-nitrotyrosine and a pressure cooker for Ki-67. Endogenous peroxidase was quenched with 3% hydrogen peroxide for 8 min followed by incubation in 10% goat serum (nitrotyrosine) or Background Buster (Ki-67) (Innovex Biosciences) to block nonspecific binding. Anti-3-nitrotyrosine rabbit polyclonal antibody (Millipore; #06284, 1:2000) was applied for 1 h in Dako (Carpinteria, CA) buffer at room temperature. Anti-Ki-67 rabbit monoclonal antibody (Epitomics; #4203–1, 1:400) was applied overnight at 4°C. Dako Rabbit EnVision™ HRP System reagent was applied for 30 min (3-nitrotyrosine) or 1 h (Ki-67), and then, slides were developed with Dako diaminobenzidine (DAB) plus for 5 min followed by DAB Enhancer for 3 min. Slides were counterstained with hematoxylin. Negative control slides were stained using the same procedure, omitting the primary antibody. Ki67 immunohistochemical staining was quantified by counting positive hepatocyte nuclei per 400×field. A total of 6×400×fields were scored per liver, and means of these scores were calculated for further statistical analysis. 3-Nitrotyrosine immunohistochemical staining was quantified by counting positive cells (cytoplasmic or nuclear staining) per 400×field with the following scoring system: 0 (0 positive cells), 1 (1–10 positive cells), 2 (10–20 positive cells), and 3 (>20 positive cells). A total of 6×400×fields were scored, and means of these scores were calculated for further statistical analysis.
Measurement of O2 •− and mitotracker green staining in liver sections in situ
Assays were adapted from previous studies in cardiovascular models with the method originally developed by Dr. Francis Miller at the University of Iowa (8, 9, 29). Livers were harvested, and a 0.5–1 cm section across the middle of the left lobe of the liver was placed in a cryomold, covered in Optimal Cutting Temperature medium, and frozen on an aluminum block that was partially submerged in liquid nitrogen. Within 2 h after harvest, samples were sectioned at 8 μm via cryotome and kept frozen within the chamber until all slides had been cut. On removal from the chamber, samples were immediately exposed to 5 μM DHE (Invitrogen) and 100 nM MTG (Invitrogen/Life Technologies) in PBS, pH 7.4 and placed in an incubator for 30 min at 37°C. For MitoSOX Red oxidation experiments, 100 nM MitoSOX Red (Invitrogen/Life Technologies) was used in PBS, pH 7.4 and rinsed 1 min after application because of the sensitivity of this reagent. Slides were gently rinsed with PBS, and cover slips were placed without fixation and immediately imaged with a Zeiss 710 confocal microscope. For each of these vital dyes, mean fluorescence intensities of at least three images each from two sections per liver were measured using ImageJ, and the means of these images were used for further statistical analysis.
In order to confirm that O2 •− was responsible for the in situ DHE oxidation, slides were preincubated with 4 μM GC4401 for 10 min before staining with DHE, resulting in prevention of ∼70%–85% of DHE oxidation compared with control slides preincubated with PBS before DHE staining (Fig. 2). The proportion of DHE oxidation that was inhibited by GC4401 did not differ between genotypes of mice or between treatment groups.
Liver mitochondria isolations
All reagents used during isolations and ETC assays are from Sigma-Aldrich, unless otherwise noted. Mitochondria were isolated as previously described (7). Briefly, livers were harvested, centrifuged (200g) in cold homogenizing medium containing 0.25 M sucrose, 5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 0.1 mM EDTA, 0.1% fatty acid-free bovine serum albumin (BSA), and pH 7.25 to rinse blood from the organ. Tissue was placed in 10 ml of the same medium, homogenized on ice using a glass/dounce homogenizer, and centrifuged at 1000g for 10 min at 4°C. Supernatants were transferred to high-speed centrifuge tubes and centrifuged at 10,000g for 10 min at 4°C. Supernatants were then discarded, and crude mitochondrial pellets were resuspended in cold potassium phosphate buffer (pH 7.2).
ETC activity assays
All ETC activity assays were adapted from previously described protocols by Turnbull and colleagues (4). All buffers used were at pH 7.2, and assays were performed on a Beckman DU 800 spectrophotometer using submitochondrial particles generated from crude preparations described earlier to prevent interference by other proteins such as the cytochrome P450 enzymes. For Complex I activity, isolated mitochondria were lysed via a single freeze/thaw. Complex I activity was assayed as the rate of rotenone-inhibitable nicotinamide adenine dinucleotide (NADH) oxidation. Samples were assayed with or without 200 μg/ml rotenone in working buffer containing 25 mM potassium phosphate buffer, 5 mM magnesium chloride, 2 mM potassium cyanide, 2.5 mg/ml BSA, 0.13 mM NADH, 200 μg/ml antimycin A, and 7.5 mM coenzyme Q1. For Complex II activity, isolated mitochondria were lysed with three freeze/thaw cycles, and activity was measured as the rate of reduction of 2,6-dichloroindophenol by coenzyme Q in the presence and absence of 0.2 M succinate. Samples were incubated with or without succinate in 25 mM potassium phosphate buffer, 5 mM magnesium chloride, 2 mM potassium cyanide, and 2.5 mg/ml BSA for 10 min at 30°C. After incubation, 200 μg/ml antimycin A, 200 μg/ml rotenone, 5 mM 2,6-dichloroindophenol, and 7.5 mM coenzyme Q1 were added to each cuvette and incubated for 1 min before reading absorbance rates. Complex III activity was assayed as the rate of cytochrome c reduction by coenzyme Q2. Freshly isolated mitochondria were assayed in 25 mM potassium phosphate buffer, 5 mM magnesium chloride, 2 mM potassium cyanide, 2.5 mg/ml BSA, 0.5 mM n-dodecyl β-maltoside, 200 μg/ml rotenone, 1.5 mM cytochrome c, and 3.5 mM coenzyme Q2. Complex IV activity was assayed as the rate of cytochrome c oxidation in mitochondria lysed via three freeze/thaw cycles. Samples were assayed in 25 mM potassium phosphate buffer, 0.5 mM n-dodecyl β-maltoside, and 1.5 mM reduced cytochrome c. Since O2 •− can interfere with these assays, for example by reducing cytochrome c, and mitochondria from these experiments are presumed to produce different levels of O2 •−, 50 U exogenous CuZnSOD was added to each assay. In order to measure citrate synthase activity, acetyl-CoA (4 mM) and oxaloacetic acid (10 mM) are added to a working solution containing 0.2% Triton X-100 and 50 μg mitochondrial proteins. Reduced CoA (produced by citrate synthase within the sample) will produce thionitrobenzoic acid from dithio-bis-nitrobenzoic acid, and the change in absorbance is measured at 412 nm. Aconitase activity was evaluated by homogenizing samples in 50 mM Tris-HCl, pH 7.4 with 0.6 mM MnCl2 and 5 mM Na-citrate and combining 200 μg protein with 200 μM NADP+ and 2 U/ml isocitrate dehydrogenase in the same buffer. Absorption at 340 nm was monitored until changes in absorption became linear, at which point absorption at 340 was measured every 30 s for 5 min. Activity is expressed as the rate of the absorption change divided by the extinction coefficient for NADPH and then divided by protein added.
Immunoblotting
Western blot analyses were performed as previously described (39). p53 blots were performed using the FL393 antibody (Santa Cruz Biotech), which was a generous gift from Dr. Dawn Quelle at the University of Iowa. Complex I (NDUFB8) and complex II (SDHB) antibodies were from Invitrogen. The aconitase 2 antibody (Abcam) was a generous gift from Dr. Frederick Domann at the University of Iowa. Complex IV (COXIV) antibody was from Abcam. Western blots were imaged and evaluated using a Typhoon FLA 7000 Phosphorimager (GE).
Statistical analyses
Data were analyzed by either one-way (analyses without GC4401) or two-way (with GC4401) ANOVA followed manually by Fisher's Least Significant Difference comparison, and results were considered significant at p<0.05. The number of mice per group demonstrating increased serum aspartate aminotransferase and alanine aminotransferase levels above the normal range were analyzed using a chi-squared calculation. Results are presented as mean±S.D for biochemical and enzymatic assays. Results are presented as mean±S.E. for histologic scoring and microscopic analyses. Statistical analyses were performed using Graphpad Prism software.
Footnotes
Acknowledgments
The authors thank Janis Rodgers and the University of Iowa Comparative Pathology Laboratory for technical assistance. The authors also thank Michael McCormick, Amanda Kalen, and the Radiation and Free Radical Research Core at the University of Iowa for irradiations and access to equipment that is necessary for enzymology and biochemistry experiments. The authors also thank Drs. Dawn Quelle and Frederick Domann for the generous gift of antibodies used in this work, Dr. Francis Miller for helpful advice in setting up the DHE oxidation assays in tissue sections, and Dr. Kyle Brown for helpful discussions about liver injury. The work was supported by a joint Department of Energy Grant/NASA Grant # DE-SC0000830, NIH T32 CA078586, A01CA152601, R01152799, R01168292 and NIH P30 CA086862.
Author Disclosure Statement
Dr. Dennis Riley is the Chief Scientific Officer at Galera Therapeutics, which provided the SOD mimic compounds used in this study. No other author has competing financial interests in this work.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.