
Abstract
Biological Chemistry of Nitric Oxide
Nitric oxide (NO) is a reactive molecule produced in almost all type of organisms, ranging from bacteria to plants, fungi, and animal cells. In mammals, including humans, low concentrations of NO regulate activities such as vascular resistance, oxygen supply, wound healing, angiogenesis, and memory formation, and at higher concentrations kill microorganisms and modulates inflammation (65, 76). NO is an endogenously synthesized free radical, first characterized as an endothelial-derived relaxation factor (116). The many physiological actions of NO are a function of the broad distribution of sites of production, combined with its facile reactions with metalloproteins and oxygen radical species (65, 67). The site, rate, and amount of NO production together determine the specific physiological outcome of a given NO signaling pathway (100). The involvement of NO in redox and free radical processes results in NO having a physiological impact that extends well beyond its recognized ability to mediate signal transduction through its interaction with soluble guanylate cyclase, a heterodimeric heme protein that catalyzes cyclic guanosine monophosphate (cGMP) formation after NO binding at the heme center (66, 76).
NO Synthesis
NO formation via the five-electron oxidation of the terminal guanidino nitrogen atom of L-arginine is a reaction catalyzed by the dimeric heme enzyme NO synthase (NOS) (114, 115, 117). NOS contains both flavin adenine dinucleotide and flavin mononucleotide cofactors (92, 94, 118). NOS-mediated production of NO requires nicotinamide adenine dinucleotide phosphate and molecular oxygen as cosubstrates, and utilizes a heme complex, reduced glutathione (GSH), and tetrahydrobiopterin (Fig. 1) (60, 91, 113). NOS exists as two main isoforms, the constitutive enzyme identified in endothelial (eNOS) and neuronal cells (nNOS), and the inducible enzyme (iNOS) present in smooth muscle cells, neutrophils, and macrophages (Fig. 2) (57, 58, 79, 80). Constitutive NOS activity is under the regulatory control of Ca2+ and calmodulin (21, 97). Once activated, constitutive NOS generates low levels of NO until Ca2+ levels decrease. This way, NOS can rapidly affect vascular tone via NO, and regulate the release of neurotransmitters by causing the influx of Ca2+ into the cells (85, 97, 122). iNOS is regulated at the transcriptional level; therefore, it requires several hours to exert a physiological response (13, 156). In terms of NO generation (per mole of enzyme per minute), the iNOS is substantially more effective than constitutive NOS, producing potentially cytotoxic levels of NO (86, 154). In summary, eNOS and nNOS enzyme activity is primarily regulated by intracellular calcium concentration, while iNOS is not dependent on intracellular calcium. The enzymatic activity of iNOS is primarily regulated by transcriptional regulation of its expression. Consequently, nNOS and eNOS constitutively produce minute amounts of NO in the nM range, in contrast to iNOS, which following an inducible latent period can produce NO in the μM range for an extended period of time (26).


Properties of NO
NO, as a free radical gas, has a short half-life in physiological conditions (7, 13, 55). Once formed, the free radical NO rapidly diffuses across biological membranes where it may react with oxygen, hemoglobin (Hb), redox metals, and superoxide anion (152). Thus, protection of the NO molecule by mechanisms to store and transport NO are required to accomplish its biological effects far from its source (68, 133). Reaction products of unprotected NO include higher nitrogen oxides, tyrosine nitrations, heme oxidation or reduction, and various S-nitrosations (S-nitroso, SNO). Although NO does not directly react with the functional groups of biological molecules, oxidized derivatives of NO have a nitrosating capacity. Specifically, NO reacts with NO2 to form the nitrosating species N2O3 with a rate constant that is near diffusion-limited (74). N2O3 can undergo hydrolysis to nitrite and the nitrosating agent NO+ at a slow rate; therefore, N2O3 becomes important for nitrosation of thiols under physiological conditions (136). N2O3 reacts with thiols in the intracellular milieu where GSH concentrations are in the millimolar range. The formation of SNOs also occurs out of cells under physiological conditions. The parent thiol can either be a low-molecular-weight species such as GSH or a cysteinyl side chain of a protein (136, 138). SNOs exist as the predominant form of NO in plasma and further serve as a circulating pool of bioavailable NO (30, 134, 158).
The formation of SNOs from direct reaction of NO with thiols in biological systems requires the presence of oxygen, as their reaction produces a radical intermediate, R-S-N·-O-H, that in the presence of an electron acceptor, such as oxygen, is converted to SNO by the reduction of the acceptor (134, 139). The formation of SNOs in biological systems is influenced by the relative concentrations of NO and thiols, as well as transition metal ions and oxygen-derived free radicals in the surrounding milieu (62, 121). Moreover, SNO formation in many cases is ultimately due to the interaction of a thiol moiety with an oxidized derivative of NO with nitrosating capability, such as NO+ derived from the dissociation of N2O3 (25, 48, 81).
Endothelial signal transduction by NO
Over 20 years of research strongly implicates a major role for NO in endothelial signaling (10). NO is clearly an important regulatory determinant of vascular tissue homeostasis (151). Traditionally, NO was thought to signal exclusively via its stimulation of guanylyl cyclase, inducing an increase in intracellular cGMP levels (66, 76). However, the effects of cGMP occur through three main groups of cellular targets: cGMP-dependent protein kinases (PKGs), cGMP-gated cation channels, and phosphodiesterase (PDEs) (37). cGMP binding activates PKG, which phosphorylates serines and threonines on many cellular proteins, frequently resulting in changes in activity or function, subcellular localization, or regulatory features. The proteins that are so modified by PKG commonly regulate calcium homeostasis, calcium sensitivity of cellular proteins, platelet activation and adhesion, smooth muscle contraction, cardiac function, gene expression, feedback of the NO-signaling pathway, and other processes. cGMP-dependent effects are mediated by other proteins whose activities are allosterically modified by cGMP, such as PDE and ion channels, and via cross-activation of cAMP-dependent kinase (protein kinase A) (37, 110).
SNO and tyrosine nitrosation signaling
Recent work has revealed that some of NOs signaling effects can also be transduced by SNO and tyrosine nitrosation (9, 35, 71). Smooth muscle has been considered to be the prototypical NO target tissue, since NO lowers basal calcium levels and counters agonist-mediated increases in intracellular calcium (4, 53). This NO-mediated lowering of calcium promotes relaxation by changes in cytosolic calcium levels and in the net flux of calcium between the cytosol, plasma membrane, and endoplasmic reticulum L-type voltage-gated calcium channels, which permit the inward flow of calcium when cells become depolarized (78, 142). Based on these arguments, increases in NO promote vasorelaxation, but the lowering of NO levels does not directly induce constriction. The low levels of NO limit the inhibition of physiological vasoconstrictors (e.g., inhibits angiotensin II, endothelin-1, and sympathetic vasoconstriction), which in parallel with NO vasorelaxing effects determine vascular tone.
NO and immune activation
Activation of the immune system is important for defense against invading organisms as well as tumor cells, but can also lead to damage of host tissues after prolonged stimulation (3). NO belongs to the nonspecific immune defense system (34, 119). It induces the killing of invading microorganisms and also contributes to tumor cell destruction by inducing apoptotic cell death of tumor cells (64, 69, 155). NO-mediated cytotoxicity seems to be mainly caused by inhibition of energy metabolism via interaction with mitochondrial respiration, and further interference with glycolysis and the citric acid cycle (11). NO has been shown to inhibit mitochondrial respiration in bacteria and eukaryotic cells reversibly by binding to the oxygen-binding site of cytochrome oxidase in competition with oxygen (14, 132). In addition, several studies suggest that the effects of NO on energy metabolism are mainly due to the formation of peroxynitrite (99). Thus, the reaction of NO with superoxide leading to the generation of peroxynitrite may enhance the cytotoxic potential of NO. Peroxynitrite reacts with a variety of biomolecules, including proteins, lipids, and DNA. Peroxynitrite cytotoxicity includes multiple pathways from lipid peroxidation, direct inhibition of mitochondrial respiratory chain enzymes, inactivation of glyceroldehyde-3-phosphate dehydrogenase, inhibition of membrane Na/K adenosine triphosphate (ATP)-ase activity, inactivation of membrane Na channels, and other protein modifications. Long-term peroxynitrite breaks DNA strands, with subsequent activation of the nuclear enzymes and eventually resulting in necrosis (141).
However, under conditions where there is excessive production of NO through the prolonged induction of iNOS, there is the prospect for clinically deleterious consequences in part due to NO-initiated apoptosis of immune-competent cells, resulting in a reduced immune response. Both exogenous and endogenous NO has also been shown to be capable of pro- and anti-apoptotic activity (140, 149). These seemingly opposing effects of NO are a clear biphasic dose-dependent phenomenon. Thus, under physiological conditions, low levels of NO may inhibit apoptosis of immune-competent cells, thereby supporting the immune defense, whereas high levels may induce cell death, thus limiting the inflammatory process with different thresholds for different tissues (24, 54, 84, 144). In practical terms, for the immune defense system, the beneficial effects are typically overwhelmed when the mediator is released for prolonged time periods in high concentrations, and apoptosis is produced by long-lasting, high NO levels or synthesis that leads to tissue destruction (33, 87, 144).
Low-grade inflammation of the vascular endothelium as in endothelial dysfunction (ED) is associated with decreased production of NO via the eNOS pathway (3, 143). In vivo studies have yet to determine the actual role of NO in the progression of vascular ED (27, 146). The impaired eNOS and signaling associated with ED is implicated in atherosclerosis, hypertension, reperfusion injury, and diabetic angiopathy (31). Paradoxically, high levels of NO produced by iNOS have been implicated in viral myocarditis, myocardial infarction, and cardiac allograft rejection (96). It also appears that the massive iNOS-associated inflammatory cascade resulting in shock secondary to hemorrhage can be abrogated by low levels of NO if generated or introduced in the circulation at the start of the process (59, 104, 150). NO, as a therapeutic agent, offers much promise, but obviously much work is required to better understand how to navigate between the differing NO-induced responses.
The Concept of Bioactive Forms of NO
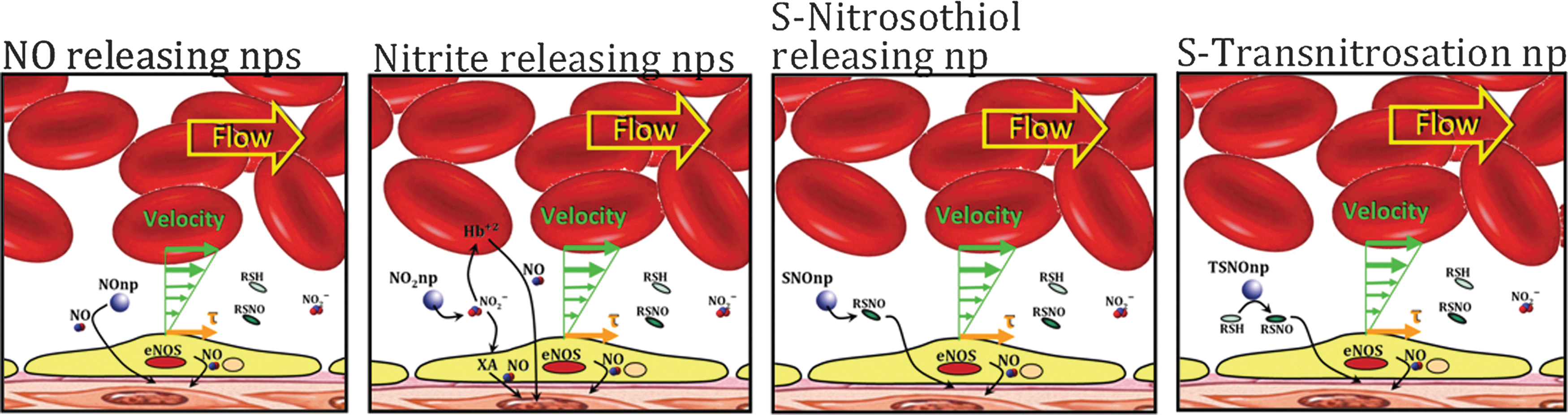
Once formed, NO rapidly diffuses across biological membranes where it can react with oxygen, Hb, redox metals, and superoxide anions (52, 69, 116). As a result, NO itself is relatively short-lived and hence likely to be biologically active only near its site of production (55). There are, however, other NO-related molecular species that are longer lived than free NO but are still capable of evoking many if not all of the physiological responses associated with free NO (156). Along with free NO, these other NO-related species, which are either derived from NO or capable of generating free NO, are termed bioactive NO (34, 40, 153). These long-lived species represent forms of NO that can be transported through the circulation with retention of the biological functioning of free NO. SNO-nitroso thiol (SNO)-containing molecules and the nitrite anion are the two most thoroughly studied members of the bioactive NO family besides free NO (Figure 3) (41, 82, 137). In addition, there are synthetic strategies for producing therapeutically useful molecules that can release NO or other forms of bioactive NO when introduced into the circulation (73, 98). These include organic nitrates that are extensively used to ameliorate transient ischemic cardiac events. These drugs are relatively short lived and require enzymatic activity for activation, which accounts for the progressive onset of the so-called nitroglycerin tolerance phenomenon (38, 102, 119). Recent development of nanoparticle formulations capable of sustained slow release of therapeutically effective levels of either NO (18, 19) or S-nitrosothiols (103) in the circulation offer promise of new clinical strategies based on bioactive forms of NO (Fig. 4).
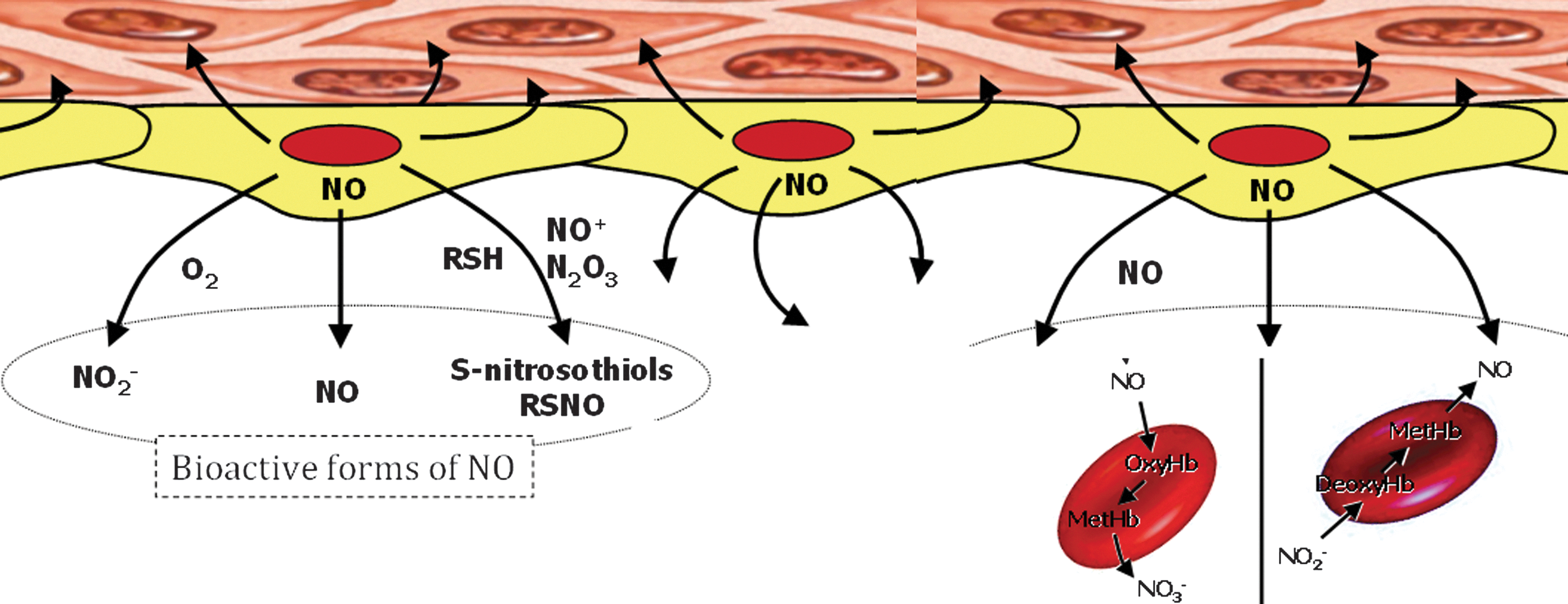
Hb as a Scavenger of Bioactive NO
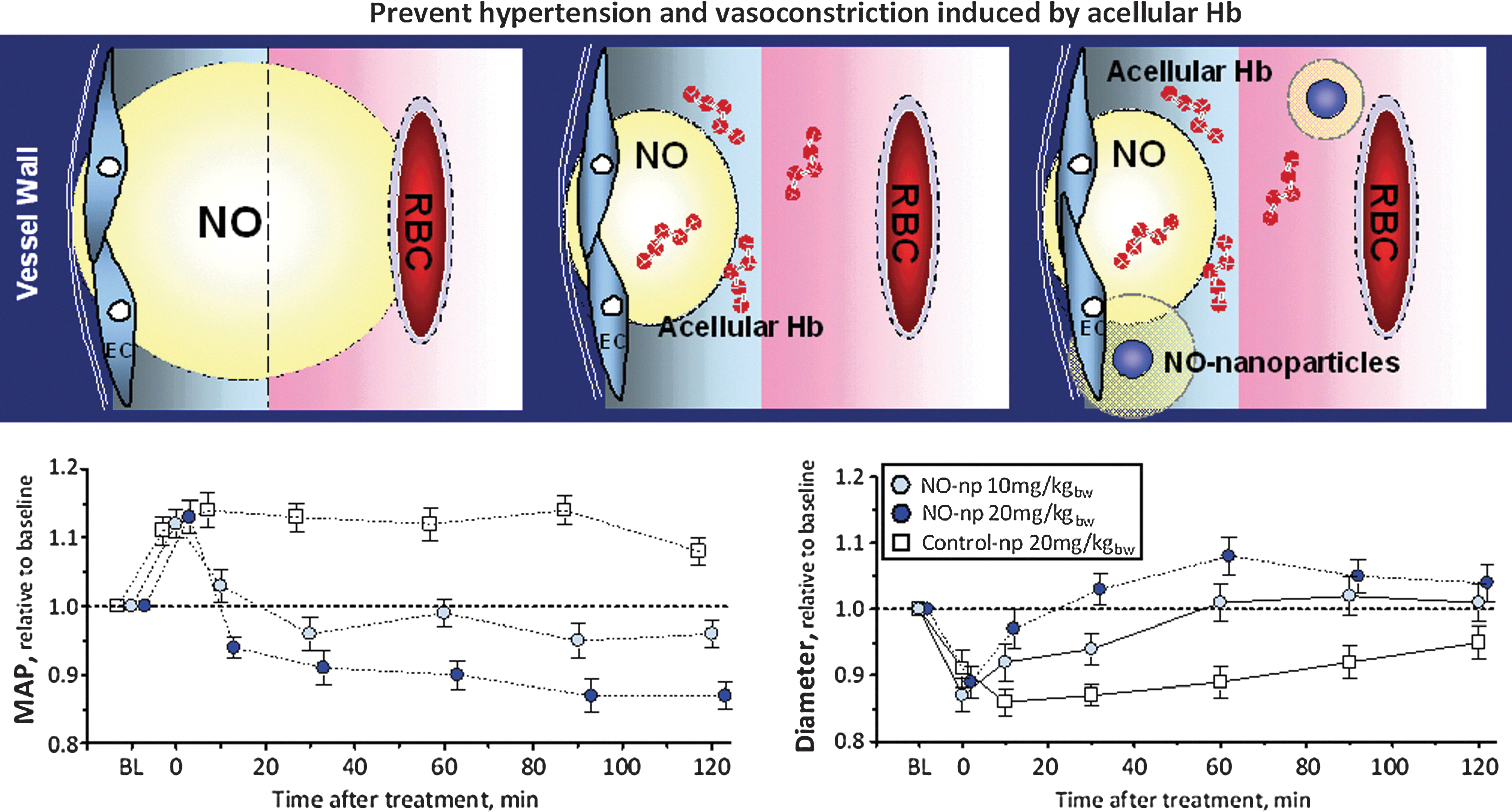
Hb can undergo several reactions that are capable of reducing levels of bioactive NO within the circulation (29, 133). The extent of the physiological consequences of Hb-associated NO scavenging are dependent on whether the Hb is localized within the red blood cell (RBC) or circulates as acellular Hb due to either hemolysis or infusion of Hb-based oxygen carriers (HBOCs) (12, 22, 29, 47, 62, 72, 133). The basis for Hb being initially viewed as a scavenger of NO that can effectively block NO bioactivity arose from studies of the physiological consequences of introducing acellular Hb into the circulation (22, 133). Exogenous acellular oxy- or deoxy-Hb results in vasoconstriction and transient hypertension (15, 111, 130). In the presence of acellular Hb, NO is predominantly quenched through an NO dioxygenation (NOD) reaction and to a lesser degree through the formation of the “dead end” (highly stable and relatively nonreactive) ferrous heme-nitrosyl form, either through the rapid binding of NO to five coordinate ferrous hemes (as in deoxy Hb) or through reductive nitrosylation subsequent to the binding of NO to ferric (as in metHb) heme centers (77, 111, 120, 157). The NOD reaction involves the efficient reaction of NO with oxy-hemes to form met (ferric) heme and nitrate. Therefore, the NO reactions with Hb will depend on the redox and ligation states of the heme iron which are modulated by O2 levels, the nature of the Hb (mutant or recombinant Hbs) and allosteric effectors (111, 120, 159). The scavenging of NO by acellular Hb has been viewed as a major source of HBOC-induced toxicity, which has in part limited the development of these materials for clinical use (5, 15, 111). The relevance of the NOD reaction as a contributor to HBOC-induced toxicity was compellingly demonstrated through the use of recombinant Hbs that were engineered to have reduced levels of NOD activity (32, 93, 112).
Vasoactivity associated with infused glutaraldehyde-polymerized bovine Hb engineered with low oxygen affinity suggested that vasoactivity is a function of molecular size (20). However, the hypertensive effect of the diaspirin crosslinked Hb and of the polymerized diaspirin crosslinked Hb were not different in rats (1). The difference between studies may be due to the fraction of unpolymerized Hb remaining in solution, and the size of the polymers studied (15, 130). The variation in degree of acellular Hb-induced vasoactivity has often been attributed to size-dependent differences in the degree of NO scavenging due to differences in the extent to which the acellular Hbs extravasate through the endothelial lining of the vasculature (130). This concept seems to be supported by results on very large polymeric Hbs such as the one produced through the zero-link procedure where the carboxyl groups on the surface of a molecule are activated with carbodiimide so as to make them able to form covalent pseudopeptide bonds with the amino groups on the surface of an adjacent molecule. It is called zero-link because no reactive groups are left behind in the obtained polymers (56). This treatment generated extensive polymerization of bovine Hb molecules (ZL-HbBv) with average size near 25 MDa (29). Residual small-size polymers, below 1000 kDa, and nonreacted Hb are eliminated by diafiltration over a 300 kDa nominal molecular weight (NMW) membrane (29, 36). ZL-HbBv does not extravasate and, in the models where it has been tested, no increase in blood pressure was observed (10, 30). However, there are problems with the extravasation concept. Hb extravasation results in the placement of Hb between the major NO source, the endothelium, and smooth muscle, which could presumably create a sink for NO at its major target site, resulting in an increase of vascular tone. However, the logic of the extravasation process is not clear, because the quantities of Hb molecules that can be located in the interstitium between endothelium and smooth muscle should be small compared to the blood compartment (20). Furthermore, the amount of unreacted Hb present in the interstitium would be rapidly exhausted, and converted to methemoglobin (metHb), thus hindering vascular tone changes. Most significantly, in a study that compared the perivascular NO levels as a function of added acellular Hbs, very similar NO levels were observed for both PEGylated and poly-ethylene glycol (PEG)-free acellular Hbs (148). PEGylation limits extravasation; hence, the results imply a comparable degree of NO scavenging for these Hbs, none of which are size-enhanced to the degree of the zero-link polymeric Hb (148). If extravasation per se is not a critical factor, it may be that the size enhancement associated with the zero-link polymeric Hb limits scavenging due to hydrodynamic factors that limit the proximity of the polymeric Hb to the endothelium, much the same way that RBC streaming creates a cell free zone in the vasculature.
Despite the so-called cell-free zone phenomenon as well as the NO diffusion barrier associated with the RBC, the RBCs still represent a potentially large sink for endothelial-generated NO (12, 81, 83, 95). This potential of the RBCs acting as an NO sink raises the question as to how NO bioactivity is maintained in the circulation (83). A similar possible paradox emerges from the study showing comparable degrees of NO scavenging by different acellular Hbs, but the degree of vasoactivity among these acellular Hbs substantially differs (128, 130, 148). There have been several studies that implicate RBCs in the phenomenon of hypoxic vasodilation with Hb seemingly playing a key role in the process. Furthermore, there are acellular Hb preparations that are not merely inactive with respect to inducing vasoconstriction but are actually vasodilatory. Such findings suggest a mechanism where Hb under the right conditions or with right modifications can undergo reactions that compensate for NO scavenging by generating some form of bioactive NO (Fig. 5).
Hb as a Source of Bioactive NO
Several studies show that NO bioactivity can be exported from the site of NO production through circulating RBCs or preserved in stable mobile stores (35, 39, 49, 88, 138). Oxygenation-dependent phenomena such as hypoxic vasodilation imply that the RBC-dependent export of NO bioactivity may involve the allosteric protein Hb (23, 75). There are several reactions that can potentially account for how Hb participates not only in RBC-associated NO bioactivity but also in the mechanism for vasodilatory activity on the part of certain HBOCs (5, 15, 20, 123).
The potential role of Hb as a source of bioactive NO started with the proposal that Hb could transport and deliver NO in an allosterically controlled manner through the formation of SNO-Hb (49, 50, 157). SNO-Hb is a derivative of Hb in which the reactive thiols associated with the two β93 Cys undergo SNO (35, 121, 129). Both the formation of the β93 Cys SNOs and the efficacy for the transfer or release of these SNO-associated NOs were proposed to be sensitive to the quaternary structure of the Hb tetramer (36). Although SNO-Hb has been observed in vivo (2), its role remains controversial (70). There are studies that show that transport of NO bioactivity by RBCs including hypoxic vasodilation does not require SNO-Hb participation (75, 135, 147). New mouse models that exclusively express either human wild-type Hb or human Hb in which the beta-Cys-93 residue is replaced with alanine have been used to assess the role of SNO-Hb in hypoxic vasodilation (70). The substitution of this residue, precluding Hb SNO, did not affect vasodilator effects of Hb during hypoxic conditions (70). In addition, vasoinactive maleimide modification-based PEGylated Hbs lack the reactive thiols and are thus not capable of forming SNO-Hb (89, 90). Thus, SNO-Hb may function to facilitate RBC-generated NO bioactivity but is not essential (28, 46). Since SNO-containing molecules are likely to be the basis for the long-lived transportable NO bioactivity, the more intriguing and as yet unanswered question is through what reactions can Hb create reactive species capable of SNO of reactive thiols on either Hb or other thiol containing molecules?
Hb Nitrite Reductase as a Source of Bioactive NO
Nitrite appears to be an either essential or important reagent in the production of bioactive NO through Hb both in the RBC and as an acellular component in the circulation. The role of nitrite is dramatically seen in results which show that reversal of L-NAME (L-NG-Nitroarginine methyl ester)-induced vasoconstriction by vasodilatory HBOCs requires the addition of nitrite at levels where the nitrite alone has no impact (unpublished observations). L-NAME impedes endothelial production of NO, thereby dramatically reducing both NO and nitrite levels in the circulation. Nitrite levels and NO levels are directly correlated under physiological conditions.
A central role for nitrite in NO production from Hb emerged when it was shown (22, 42, 45, 105) that deoxy Hb can catalyze the conversion of nitrite to NO though a nitrite reductase reaction. In this reaction, five-coordinate ferrous heme reacts with nitrite to produce NO and ferric heme (8). Furthermore, the rate of the reaction is allosterically controlled through interactions such as ligand binding and pH that influence the equilibrium between the T and R quaternary states of the Hb tetramer. Studies on chemically modified Hbs (43, 44), sol-gel encapsulated Hbs (125), and Hb dimers bound to haptoglobin (124) all indicate the R state deoxy hemes manifest a faster initial rate for this reaction by a factor of ∼10 over the corresponding T state deoxy hemes (124, 152). The redox potential and quaternary structure (R or T state) determine nitrite reductase reaction (51, 127). Therefore, the higher redox potential (and T-state) shows a lower rate of NO release, where as a lower redox potential (R-state) gives a faster rate (127). This allosterically responsive behavior links this reaction with hypoxic vasodilation. As part of the mechanism, a half-oxygenated R state Hb would have the maximum NO-generating efficacy since it represents a compromise between accessible reactive deoxy heme sites and the higher reactivity associated with the R state: Too much deoxygenation results in T state formation and too much oxygenation results in too few sites available for the reaction with nitrite (8, 22, 23, 152). The importance of this reaction is also apparent from HBOC studies. Those Hbs with the highest rates for nitrite reductase typically show the lowest levels of induced vasoconstriction. Furthermore, those Hbs with the highest rates for the NR reaction are most able to reverse L-NAME-induced vasoconstriction in the presence of low levels of added nitrite (levels insufficient to elicit activity in the absence of Hb) (23).
Nitrite Anhydrase
Although the nitrite reductase activity of Hb is a suggestive candidate for the biochemical/biophysical basis for Hb-mediated production of bioactive NO, there are limitations that indicate that it may be only part of a bigger picture (23). As discussed previously, the production of free NO per se may not be adequate for long-distance transfer of NO reactivity since it is readily scavenged by Hb and has a limited capacity to survive long-distance diffusion (8, 46, 63, 131). The paradox of experimentally observed NO generation and release despite robust NO-scavenging reactions inside RBCs has led investigators to probe the deoxy Hb nitrite reaction for additional related mechanisms that can generate stable intermediate NOx species that could avoid heme scavenging (8, 106). The formation of ferric heme as a product of either the nitrite reductase reaction or the NOD reaction raises the possibility that an metHb-associated species could play a role. In particular, NO-metHb and nitrite-metHb have been the focus in this line of investigation.
NO ferric heme can exist in equilibrium with a ferrous-NO+ species. The nitrosonium ion can nitrostate thiols, hence the interest in this species. Rifkind and colleagues have recently reported an electron paramagnetic resonance (EPR)-silent NO-modified Hb intermediate detected by reductive chemiluminescence (105, 106), supporting the hypothesis that intermediates result from NO–metHb (Fe3+–NO) stabilized by electron delocalization between the NO, heme, and possibly β-cysteine 93, which could ultimately nitrosate thiols and thus allow for red cell export of bioactive NO. Although NO–metHb is certainly a potential source for a transient intermediate in the nitrite reductase reaction, the relatively high rates of both NO dissociation (88) and reductive nitrosylation (145) associated with this species pose limits on its potential contribution to the production of long bioactive species. In the absence of additional reactants, it does not appear likely that NO-metHb is a major source of long-lived bioactive NO. The missing role and understanding on the nitrite anhydrase are the reaction of met-Hb with nitrite when NO is present to yield N2O3, a potent S-nitrosating agent.
Nitrite-metHb has been proposed to play a major role in generating bioactive NO through a reaction described as nitrite anhydrase, whereby NO reacts with the bound nitrite to produce N2O3 coordinated to a ferrous heme (8, 145). N2O3 is a potent S-nitrosating agent that is relatively long lived in hydrophobic environments such as lipid membranes. Experiments in which the met nitrite derivative of Hb is prepared using a large excess of nitrite embedded within a trehalose-based glassy film show that upon exposure of the glassy film to NO gas, the very slow diffusion of the NO into the protein results in the formation of a species having a distinct visible spectrum that resembles that of the ferrous NO derivative, but with both of the two Q band peaks blue shifted slightly by a few nanometers (108, 109). A similar spectrum has been generated from sol-gel encapsulated met nitrite Hb bathed in a buffer with a large excess of nitrite when small aliquots of NO-containing buffer are added (109, 126). Under oxygen-free conditions, the species associated with this spectrum causes an increase in the fluorescence spectrum of diaminofluorescein (DAF) consistent with the formation of N2O3 (Fig. 6) (108, 109). Additionally, this species is stable for several hours. It has been proposed that this species is a nitrite anhydrase intermediate in which N2O3 is bound to a ferrous heme (8, 126). In these experiments, nonphysiological high concentrations of nitrite were used to generate this intermediate. Under physiological conditions, the much lower concentration of nitrite raises legitimate questions as to the relevance of this mechanism. Nitrite has an even higher rate of dissociation from ferric heme than NO, which poses the issue as to whether under physiological conditions the formation of ferric nitrite is a plausible initial step for this reaction since ferric NO heme should dominate under most circumstances. An additional complication associated with this mechanism is that if in fact the ferric NO derivative dominates a relatively rapid formation of the dead-end ferrous NO heme species through reductive nitrosylation could dominate over the proposed nitrite anhydrase reaction (145). A recent simulation (61) showed that from a quantum chemical perspective the N2O3 intermediate could be formed through either of two pathways: (i) nitrite reacting with ferric heme bound NO and (ii) NO reacting with ferric heme bound nitrite. Although this study addresses the actual sequence in the proposed mechanism, there remains the issue that, at low physiological concentrations of nitrite and NO, it is not clear how the nitrite anhydrase reaction can compete with dissociation processes and reductive nitrosylation (105).

Most recently (our unpublished observations), we have shown that this same intermediate can be formed under conditions that approximate physiological conditions with respect to nitrite and NO (from NONOates). Most significant is that these recent studies also address the seeming sequence flaws in the proposed nitrite anhydrase mechanism (8, 126). It is observed that the same intermediate that is generated when NO is added to metHb in the presence of large excesses of nitrite can also rapidly form when very low levels of nitrite are added to NO-metHb. This study supports the prediction that the intermediate can form rapidly regardless of whether NO or nitrite bind first, and furthermore that the affinity for nitrite when the NO is bound to the met heme becomes sufficiently enhanced that formation of the intermediate can under certain conditions out compete the reductive nitrosylation reaction (108, 126). The species associated with the intermediate spectrum appears to readily react with GSH to form S-nitrosoglutathione, which is consistent with the claim that this intermediate species is the heme-bound N2O3 intermediate. The role of this process in hypoxic vasodilation is supported by our recent unpublished observation that, under the same solution pH conditions, the branching between reductive nitrosylation and nitrite anhydrase is controlled by the quaternary structure and/or redox potential. High redox potential species (e.g., T state metHb and metHbE) primarily undergo reductive nitrosylation, whereas low redox potential species (R state metHb) form the intermediate.
Strategies for the Design of HBOC Formulations
The core function of infused HBOCs is typically to enhance tissue oxygenation. Very often the conditions that trigger the need for HBOCs to enhance tissue oxygenation are also accompanied by either pro-inflammatory factors or overt inflammation that can counter the efficacy of HBOC-facilitated oxygen delivery. As a consequence, HBOC formulation design must accommodate the potential complications arising from infusing HBOCs into a circulation where the endothelium may be dysfunctional. ED is associated with biochemical and physiological changes that result in eNOS no longer functioning as a source of endothelial-generated NO. In contrast, under such conditions, eNOS becomes a generator of reactive oxygen species (ROS) which further exacerbates the inflammation and its consequences. Given that NO supplementation at low to moderate levels in vasculature can counter inflammation, there is added importance in the design of HBOCs with respect to both minimizing the scavenging of NO and generating bioactive NO.
Limiting NO scavenging can be addressed by either of two strategies: (i) creation of sufficiently large polymeric Hbs that have flow properties similar to RBCs, and (ii) creation of recombinant Hbs with reduced rates for the NOD reaction. As noted above, reducing the scavenging may not be sufficient in the context of inflammation. The design of Hbs with enhanced capacity to generate bioactive NO requires heme sites with low redox potentials. Low redox potentials can be achieved through chemical or mutagenic strategies. To date, these strategies have focused on modifications, including some PEGylation platforms that favor an equilibrium shift in favor of the R state quaternary structure (89, 90). Along these lines, the strong complex between haptoglobin and the αβ dimer of HbA has been shown to have an enhanced capacity for generating NO from nitrite consistent with its nonallosteric R-like oxygen binding properties (124). It is not clear at this time as to whether PEGylation and other size enhancement strategies go beyond merely shifting the allosteric equilibrium or can actually tune the redox potential through direct physicochemical interactions. The problem with many of these strategies based on engineering modifications that enhance bioactive NO production is that they typically reduce oxygen delivery by virtue of enhancing oxygen affinity. It may be that the NO production related effects can compensate by increasing tissue perfusion.
Studies using HBOC nitrite reductase activity to generate NO and N2O3 to offset the NO scavenging in swine found an increase in the risk of pulmonary complications in a dose-dependent fashion (101). In this study the swine underwent uncontrolled liver hemorrhage before receiving up to three 10 min 10 ml/kg infusions of HBOC with or without concurrent NaNO2. As expected, nitrite temporarily reduced systemic and pulmonary blood pressure increases from HBOC in a dose-dependent fashion. There was no significant effect between groups in indices of tissue oxygenation or survival. In this model, the transient nature of nitrite in offsetting HBOC-induced vasoconstriction makes it less clinically promising than anticipated and the combination of nitrite and HBOC appear to be more harmful than beneficial (101).
Clinical trials on HBOCs have not provided information on the mechanism of toxicity, although all commercial formulations have shown to be harmful (17, 107). HBOC mediated a vascular dysfunction that has been limited to hypertension and vasoconstriction, although they also induce inflammation and oxidative stress (6). Specifically, it will be important to reconcile the different hypotheses of Hb toxicity (i.e., NO depletion or peroxidative reactions) in disease-state animal models so that the molecular basis of Hb toxicity can be better understood. HBOC-induced oxidative stress is based on general assumptions of Hb oxidative chemistry with little mechanistic insight available from biological systems. An integrative approach might be beneficial to extract specific determinants of HBOC toxicity that could then be targeted by rational HBOC design strategies. Lastly, animal models of ED combined with more human-like antioxidant plasma and tissue environments as well as more specific markers of extracellular Hb toxicity could improve the predictive value of preclinical studies and facilitate the development of safe and effective HBOCs (16).
In some sense, many of the promising high oxygen affinity HBOC products are more suitably labeled perfusion enhancers, since it does not appear that they function through enhanced oxygen unloading. Work on the HbE (127), a naturally occurring mutant of human Hb with significant clinical consequence that contains a modification at the α1β1 interface, shows that it may be possible to tune the redox potential independent of the allosteric properties. An alternative approach is to create a formulation that (i) addresses oxygen transport and ROS-related toxicity through HBOCs that are designed to have oxygen transport and delivery properties that match the physiological demands of the clinical setting as well as stability properties that limit loss of heme and ROS production, and (ii) address the inflammation issues by providing sustained delivery of NO or SNO through biocompatible nanoparticle platforms that have extended circulation times (18, 19, 103, 104). It is clear that much progress has been made and much more progress is needed.
Footnotes
Acknowledgments
This work was supported by the National Institutes of Health (NIH) under Program project P01-HL071064, and grants R01-HL52684, R01-HL62354, and R01-HL62318.