
Abstract
Introduction
Innovation
Proteins of the base excision repair (BER) pathway help maintain the stability of the genome by repairing deoxyribonucleic acid (DNA) base damage and DNA single-strand breaks that may arise from oxidative stress and cellular metabolism. Recent studies have suggested that one or more BER proteins may also play a role in epigenetic regulation of gene expression. To shed light on this novel role of BER, this review focuses on the repair of oxidative lesions in nuclear DNA that are induced during histone demethylation. Further, we highlight current studies suggesting a role for BER proteins in DNA demethylation.
The genome of more than 350 species has been sequenced (34), yet the gap between our understanding of the genotype and phenotype is still considerable. Through a series of spatiotemporal developmental steps, stem and progenitor cells of a multicellular organism are differentiated into different cell types with unique gene expression profiles to perform their specific functions (11,16,46,68). It is now quite clear that among the many biological and genetic alterations that define cellular function, the pattern of DNA and histone modification plays a main role to define the cellular phenotypes (54). The balance between the DNA methylation/demethylation and histone modification status can impact the structure of chromatin, define the on/off switch for many genes, and eventually change the physiologic outcome. For example, the fate of a honeybee (Apis mellifera) as either a worker or a queen is determined by its DNA methylation pattern (82). Approximately 3%–4% of genomic cytosine in a typical mammalian cell is methylated to 5-methylcytosine (5mC) as an epigenetic mark (24). These marks generally occur at the dinucleotide CpG of the promoter region of a gene. Methylation of cytosine to 5mC at the CpG sites is an important step in epigenetic transcriptional regulation related to transcriptional repression, X-chromosome inactivation, imprinting, and suppression of parasitic sequences (13,60,83). Abnormal DNA methylation patterns at the CpG sites are often observed in disease states such as global genome hypomethylation and tumor suppressor gene hypermethylation in cancer (28,31).
In addition, because the DNA is wrapped around histone proteins packed into the chromatin structure, post-translational modification of histone proteins is an important method for controlling DNA access and transcriptional regulation (14,41). The tails of H3 and H4 histones can be covalently modified on several residues by methylation, acetylation, phosphorylation, ubiquitylation, SUMOylation, citrullination, and ADP-ribosylation (19,41,64). Based on the status of these histone modifications, the expression of the modified gene might be upregulated or silenced.
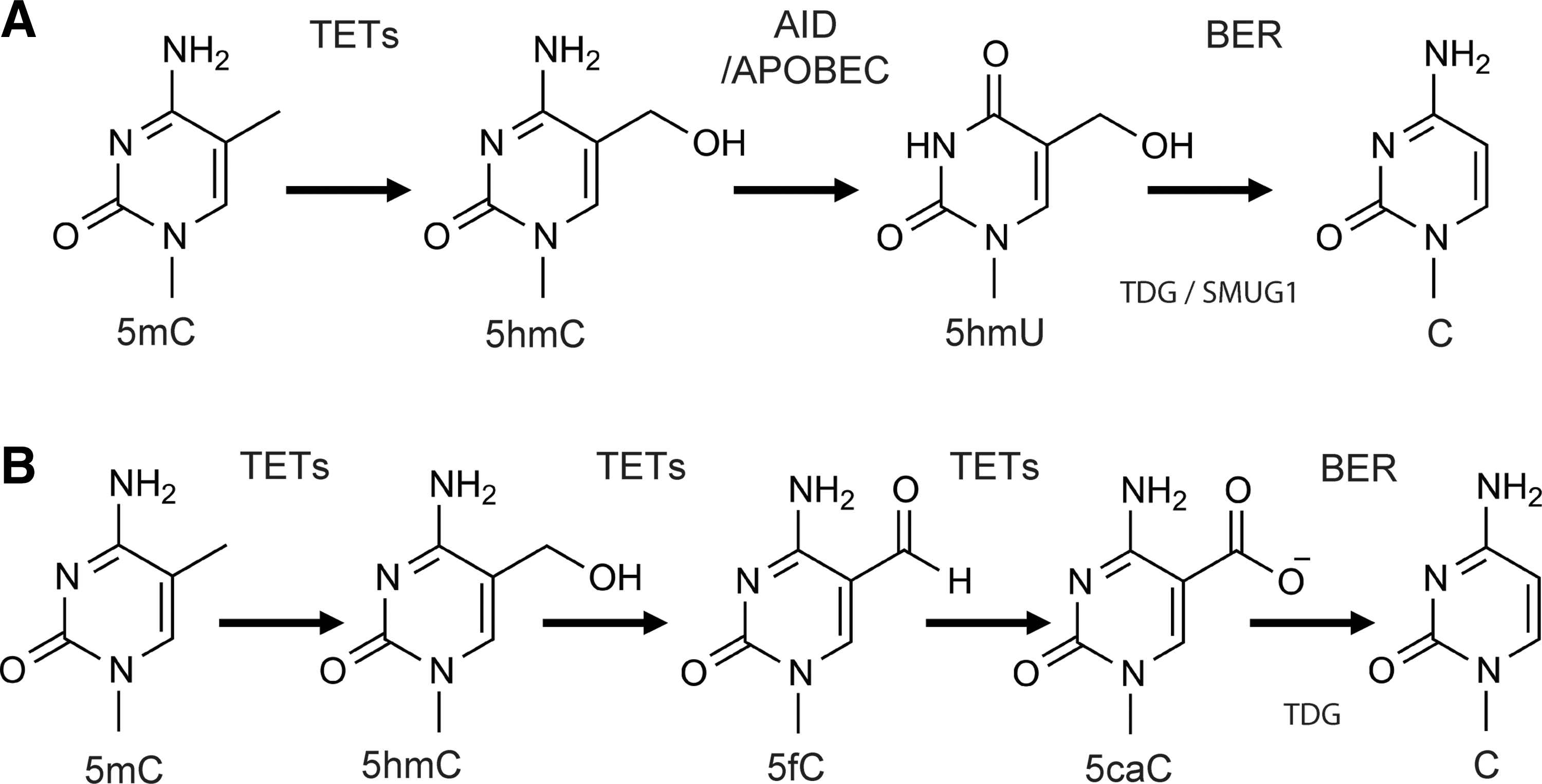
Recent studies have demonstrated that BER may be involved or coupled with both active DNA demethylation (BCADD) and lysine-specific demethylase 1 (LSD1)-mediated histone demethylation (LC-BER) (40,76). In BCADD, three families of enzymes are involved: the Ten–eleven translocation (Tet) protein family, activation-induced deaminase (AID)/apolipoprotein B mRNA-editing enzyme complex (APOBEC), and proteins from the BER pathway. The TET proteins are responsible for the hydroxylation of 5mC to yield 5-hydroxy-methyl-cytosine (5hmC), which can be further oxidized to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). AID/APOBEC proteins can deaminate 5mC (or 5hmC) to form thymine or 5-hydroxymethyluracil (5hmU). In the latter case, the resulting modified base would be mispaired with guanine. Once these modified bases are formed, the BER pathway is initiated by TDG to remove base modifications such as 5fC, 5caC, or 5hmU (22).
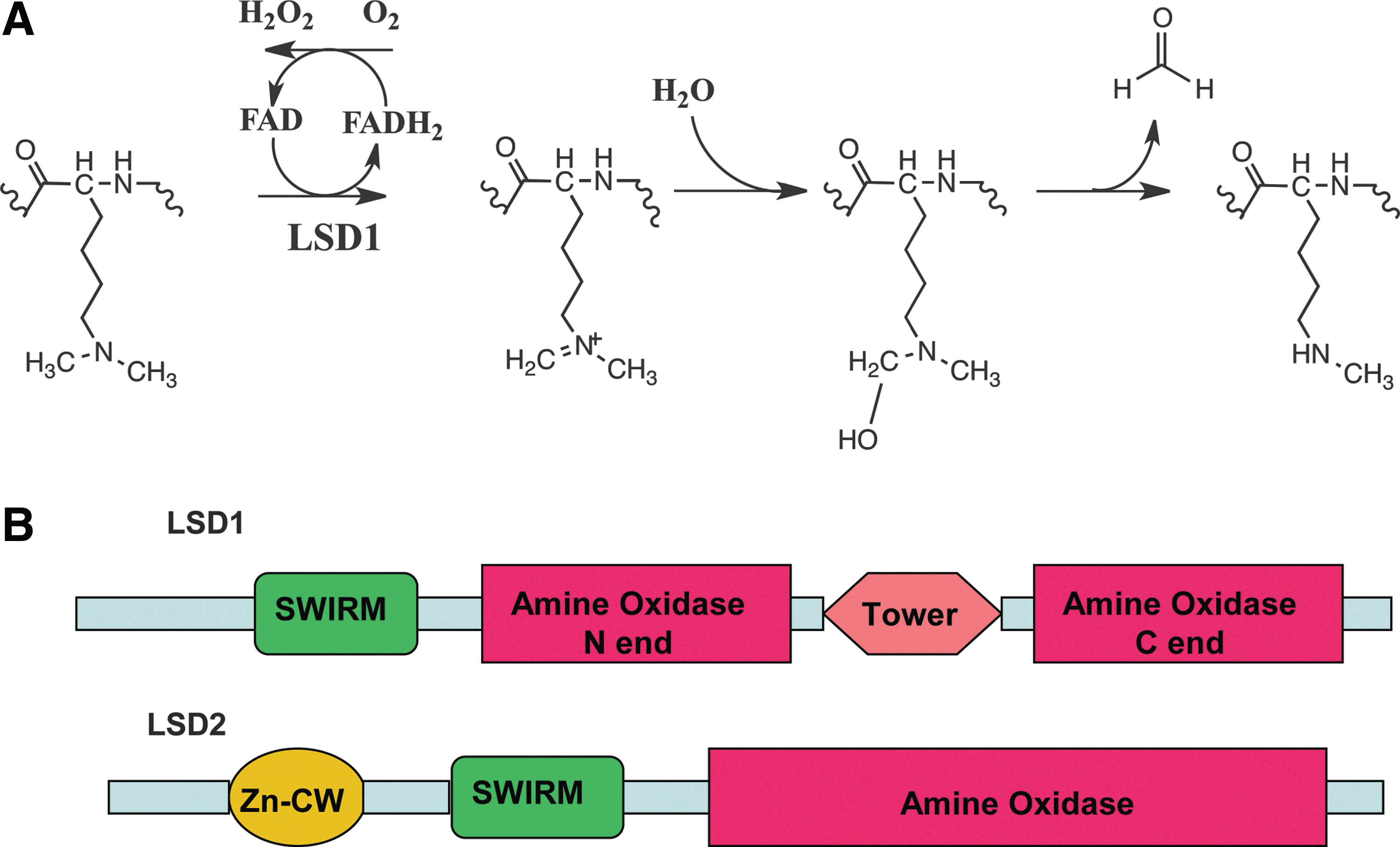
With regard to histone modification, BER is suggested to be coupled to LSD1-mediated histone demethylation to repair base lesions induced by hydrogen peroxide (H2O2) as a byproduct of the LSD1 (or LSD2) enzymatic activity (LC-BER). LSD1 is the first protein demethylase discovered that converts histone H3K4me2 to H3K4me1 or H3K4me0 through a flavin adenine dinucleotide (FAD)-dependent oxidative reaction. In the amine oxidase-mediated demethylation reaction, the cofactor FAD is reduced to FADH2 and then reoxidized to FAD by oxygen with the generation of H2O2 (Fig. 2). The DNA damaged by H2O2 from LSD1-mediated demethylation triggers the recruitment of the BER machinery to the promoter and regulatory response sites. It is suggested that it is the repair of the LSD1-induced DNA damage that facilitates and enhances transcription initiation (78). Therefore, it is likely that the BER machinery is not only critical for preventing reactive oxygen species (ROS)-induced genome instability, but it is also possible that BER is an essential component of ROS-mediated transcriptional regulation. In this review, the two main epigenetic modifications, DNA demethylation and histone demethylation, are discussed in detail with an emphasis on the roles of BER in transcriptional regulation.
Gene Expression Regulation by Histone Acetylation and Methylation
The basic unit of chromatin consists of 146 base pairs (bp) of DNA wrapped around a histone octamer, which is composed of two copies of each of the four core histones: H2A, H2B, H3, and H4. Thus, post-translational modifications of histones alter the interactions between DNA and histones and modulate DNA access by transcription factors or other regulatory proteins. These alterations result in chromatin structure condensation or relaxation, which leads to the regulation of the expression of a targeted gene (6). The two most common histone modifications are acetylation and methylation. Histone acetylation and deacetylation are highly regulated dynamic processes typically catalyzed by enzymes with a histone acetyltransferase (HAT) or histone deacetylase (HDAC) activity. Acetylation of the (normally) positively charged lysine residue results in an uncharged lysine residue, causing a decreased interaction between the histone and DNA that is generally associated with active transcription. Histone acetylation and deacetylation are usually coregulated with histone methylation and demethylation to facilitate regulation of gene expression. Unlike histone acetylation, methylation of histones does not influence the net charge of the newly modified lysine residues, and hence has no effect on the DNA–histone interaction. However, the histone methylation status is important for the interaction between chromatin and regulatory proteins and therefore can impact transcription. The presence of epsilon-N-methyl-lysine in calf thymus histones was first demonstrated in 1964, and it was suggested that the methyl group might come from methionine (73). In the same year, Allfrey and colleagues reported the possible role of histone methylation and acetylation in regulating RNA synthesis (1). Decades of work on histone modification and regulation of gene expression have developed into a histone-code hypothesis that links the function of histone modifications in chromatin with transcriptional activation or suppression states of related genes (55,87,95). Histone methylation is usually thought to occur at the N-terminal domains of H3 and H4. Histone lysine methyl transferases (HKMTs) can mainly be classified into two families: Su(var)3–9/Enhancer of zeste/Trithorax (SET) domain-containing proteins and the DOT1-like proteins without a SET domain (41). Proteins from both families can transfer a methyl group from S-adenosylmethionine to the epsilon-amino-group of lysine, resulting in the formation of S-adenosylhomocysteine and methyl-lysine. Most HKMTs belong to the SET family and use the SET domain as their catalytic core and almost exclusively act near the N-termini of the histone proteins. On the other hand, DOT1/DOT1L proteins target the lysine tail region of the histone, and DOT1 is the only enzyme known to methylate a lysine residue in the globular core of the histone (103).
Histone Demethylation and Oxidization of DNA
For a long time, histone methylation was considered a permanent and irreversible histone modification and therefore used as an epigenetic mark because of the high thermodynamic stability of the N–CH3 bond (93). The first histone demethylase, LSD1, discovered in 2004 by Shi and coworkers, brought a completely new field to light that suggested a dynamically controlled balance between HKMTs and demethylases in response to the extracellular or intracellular signals needed for transcriptional regulation (81). LSD1 is often upregulated in various tumors and hence is considered a promising oncogenic target. In line with this thought, the inhibition of LSD1 activity has been reported to reduce cancer cell proliferation or block tumor metastasis (52). LSD1 also plays an important role in embryo development, demonstrated by the observation that a null mutation of the mouse isoform of LSD1 (Kdm1a) causes embryonic lethality (100). LSD1 demethylates its histone substrate via an FAD-dependent amine oxidase reaction (Fig. 2A). Because demethylation of trimethylated lysine residues requires a protonated methyl ammonium group for LSD1-catalyzed oxidation, LSD1 is unable to demethylate trimethylated lysines (81). Subsequently, the other histone demethylase family that contains the Jumonji (JmjC) catalytic domain was identified and characterized and shown to demethylate trimethylated lysines (20,62,94,101,104). The JmjC-driven demethylase reaction is compatible with demethylation of mono-, di-, and trimethylated lysines and has a substrate preference for trimethylated lysine demethylation (23,75). Unlike the LSD1 family proteins, JmjC proteins catalyze lysine demethylation of histones through an oxidative reaction that requires Fe(II) and α-ketoglutarate (α-KG) as cofactors. This demethylation mechanism will not result in DNA damage and therefore does not invoke the BER system. As such, these demethylases are not the focus of this review. The details on the JmjC family of demethylases have been reviewed elsewhere (80).
Based on the proposal that amine oxidases might remove the methyl group from histones via an oxidation reaction (7) and the highly homologous sequences between LSD1 and amine oxidases, Shi et al. 81 demonstrated that LSD1 catalyzes the removal of the methyl group from both H3K4me2 and H3K4me1, but not H3K4me3 in vitro via an FAD-dependent oxidization reaction (Fig. 2A). Forneris et al. then demonstrated that molecular oxygen was utilized as the electron acceptor. After exposure of the reduced LSD1, which was generated from the demethylation of H3K4Me2 under anaerobic conditions, to air for a few minutes, the flavin cofactor of LSD1 was reoxidized by oxygen. The reoxidized LSD1 has the same absorption spectrum as the native enzyme (35). In LSD1-catalyzed histone demethylation, the amino-group of the methylated lysine is oxidized, presumably to generate an imine intermediate that will spontaneously hydrolyze to produce formaldehyde and the corresponding amine residue. Substrate oxidation leads to the two-electron reduction of the cofactor FAD, which is reoxidized by molecular oxygen to produce H2O2, which was initially detected with a peroxidase-coupled assay. Because of the formation of the imine intermediate, LSD1-mediated demethylation is critically dependent on the protonation of the nitrogen. Thus, this enzyme can only catalyze demethylation of mono- and dimethylated lysines. In 2009, Karytinos and colleagues discovered a second FAD-dependent H3 lysine demethylase, LSD2. LSD2 was demonstrated to be specific for demethylation of H3K4me1 and H3K4me2 via a mechanism similar to LSD1 (58). Both proteins have a C-terminal amine oxidase domain hosting the FAD cofactor for catalytic activity that is preceded by a SWIRM domain, a six-α-helical structural module frequently found in chromatin-associated proteins and important to LSD1 protein stability (Fig. 2B). The main difference between LSD1 and LSD2 is in the N-terminus. Unlike the N-terminal sequence of LSD2 that forms a CW-type zinc-finger domain (residues 130–200), the first 150 amino acids of LSD1 are predicted to be disordered (58). Another difference is that LSD2 does not have a Tower domain that in LSD1 is important for CoRest binding, which therefore precludes the possibility of LSD2/CoRest complex formation (58). In addition, the N-terminal CW-type zinc-finger domain found only in LSD2 suggests the possibility of an interaction between LSD2 and nucleosomal DNA (58). The sequence and structural differences between LSD1 and LSD2 may foreshadow functional specificities of the two demethylases. Recently, Huang and colleagues showed that inhibition of LSD1 activity by the monoamine oxidase inhibitor pargyline or reduction of LSD1 expression by small interfering RNA (siRNA) resulted in increased acetylation of H3K9 (AcH3K9), an epigenetic mark for transcriptionally active chromatin. Further, the reactivation of many tumor suppressor genes resulted in growth inhibition of breast cancer cells. However, the reduction of LSD2 expression by siRNA-mediated knockdown did not change the level of AcH3K9. This result suggests that LSD2 activity may not functionally complement the HDAC activity of LSD1 (52). In 2010, van Essen and colleagues demonstrated that LSD2 was required for the removal of the methyl group from H3K9me2 at the Mdc and Il12b promoters when stimulus-induced NF-κB was recruited to the promoter region for activating those genes (97). In addition, Fang et al. reported that LSD2 mainly binds to the gene bodies (but not promoters) to demethylate H3K4me2 and may be associated with elongation factors to regulate gene transcription after initiation (29). In both the LSD1- and LSD2-mediated demethylation reaction, the H2O2 product generated oxidative damage on the nearby DNA (21). It has been reported that when LSD1-mediated H3K4 demethylation occurred, the percentage of cells labeled with a fluorescein-tagged 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG)-binding protein increased from 5% at a basal level to 74%, close to the percentage (84%) that resulted from the treatment of H2O2 as a positive control (21). Although only the oxidatively modified form of deoxyguanosine (8-oxodG) was reported after LSD1-mediated histone demethylation (4,78), all four deoxynucleosides (deoxyadenosine, deoxyguanosine, thymidine, and deoxycytidine) and the methylated form of deoxycytidine (5-methyl-deoxycytidine) can be oxidatively damaged (88). The most common and intensively studied oxidatively damaged base is 8-oxodG (59). Other major oxidative lesions include 2,6-diamino-4-hydroxy-5-formamidopyrimidine. With the exception of DNA polymerase iota (ι), DNA replication in mammalian cells is not significantly blocked by 8-oxodG damage, and thus the 8-oxodG DNA lesion is minimally cytotoxic and in fact is mostly mutagenic (106). Since 8-oxodG potentially mispairs with A, either DNA replication or DNA repair-mediated DNA synthesis opposite the 8-oxodG lesion yields deoxyadenosine monophosphate (dAMP) insertion (47,65,67,85,107). Eventually, this results in a G⇒T substitution mutation (85). In general, the 8-oxodG lesion is mainly repaired by the BER pathway, and it has been suggested that the repair (BER) synthesis mediated by Pol-lambda (λ) preferentially inserts deoxycytidine triphosphate (dCTP) opposite 8-oxodG (69,70,98,99). Similarly, BER synthesis mediated by Polβ prefers insertion of dCTP (by a factor of 2:1) over insertion of the mutagenic dAMP base (71). If not repaired, the 8-oxodG lesion can be further oxidized to yield several mutagenic base lesions, including guanidinohydantoin and spiroiminodihydantoin (44,74).
Deoxyadenosine can be oxidized into two major products: 8-oxo-7,8-dihydro-2′-deoxyadenosine (8-oxodA) and 4,6-diamino-5-formamidopyrimidine (FapydA) (12). The FapydA lesion was first detected in both normal and cancerous tissues (77) and is the most abundant of the adenine lesions induced by γ-radiation (18). Both lesions are weakly mutagenic (57,92), and tandem 8-oxodA lesions can be induced by hydroxyl radicals. BER is the major pathway to repair such DNA lesions. The major 2′-deoxycytidine-oxidized product is 5-hydroxy-2′-deoxycytidine (OH5dC). Inaccurate replication across the OH5dC lesion incorporated into DNA tends to generate the C⇒T transition mutations (30). Thymine can be oxidized to thymine glycol (Tg). The Tg lesion is formed by a hydroxyl radical attack on the double bond of thymine at C5 or C6 or by hydrogen abstraction from the methyl group (56). Tg was originally identified in purified DNA after oxidation with ionizing radiation (36). Although the Tg lesion blocks human replicative DNA polymerases, the lesion only results in a minimal increase in mutations, since several translesion DNA polymerases such as DNA polymerase η, κ, ν, β, and λ readily bypass the lesion (10,33,66,91). Additional oxidized thymine analogs include 5,6-dihydro-thymine and 5-hydroxy-5,6-dihydro-thymine. Further details on the repair of oxidative DNA base lesions have been reviewed previously (88).
BER Pathway and the Repair of Oxidatively Damaged DNA
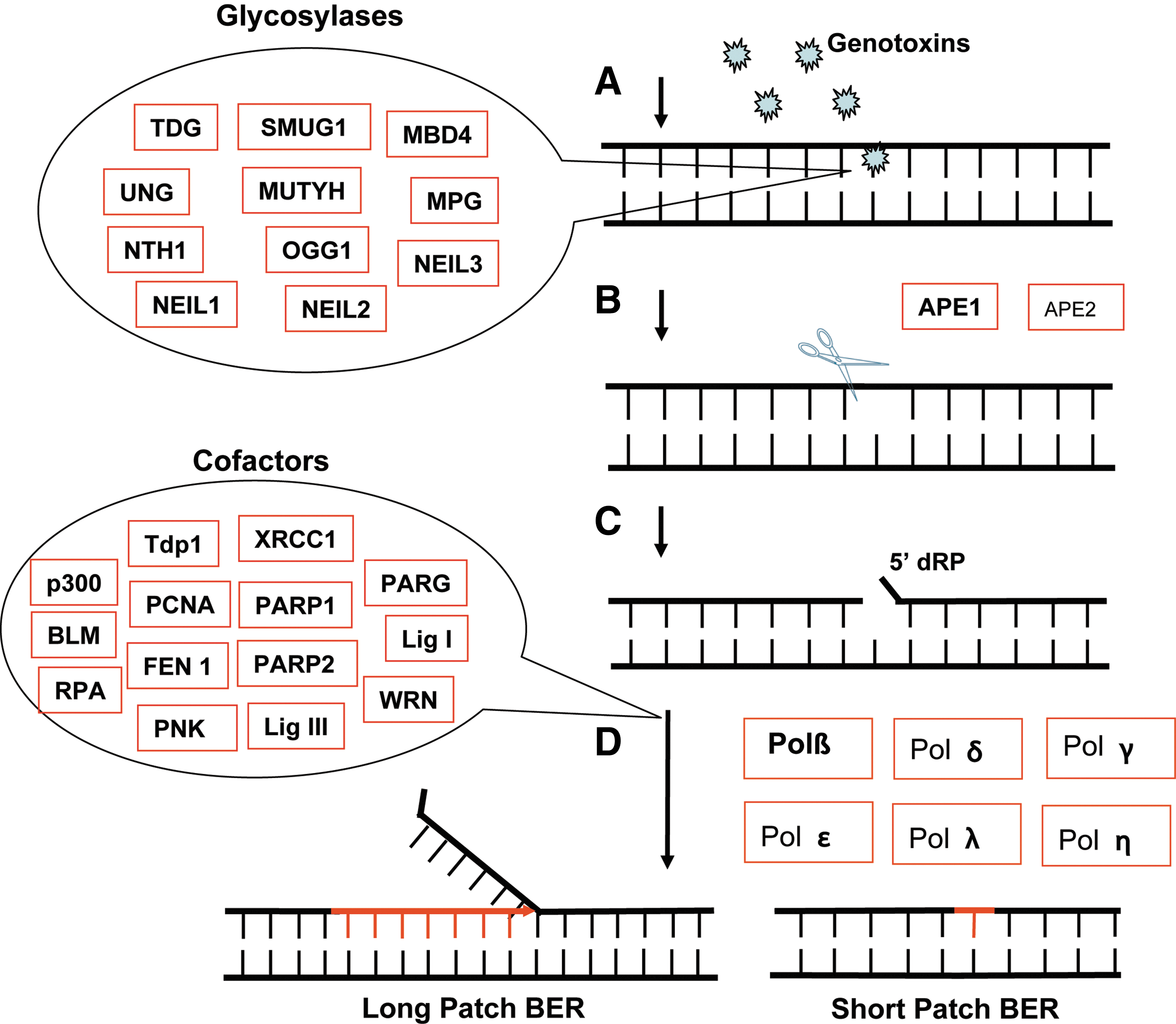
Both environmental and endogenous factors can induce DNA base damage, requiring repair by multiple DNA repair pathways to maintain genome stability and to prevent cell malignant transformation. The BER pathway is the predominant DNA repair pathway for repairing oxidatively damaged DNA. The BER pathway consists of three functional steps: (i) lesion recognition/strand scission, (ii) gap tailoring, and (iii) DNA synthesis/ligation (3,88). The initial step of recognition and strand scission involves the removal of the oxidatively damaged DNA lesion by one of several DNA glycosylases (Fig. 1) (88). This is followed by hydrolysis of the DNA backbone primarily by APE1, an AP endonuclease. Gap tailoring can be conducted by several enzymes depending on the initial lesion, and finally DNA synthesis allows the insertion of the correct DNA base followed by ligation to seal the DNA backbone (3). There are several classes of DNA glycosylases, monofunctional and bifunctional glycosylases, which differ based on function. Monofunctional glycosylases hydrolyze the N-glycosidic bond to remove the damaged base. This class includes UNG, SMUG1 TDG, MBD4, MPG, and MUTYH. Monofunctional DNA glycosylases such as SMUG1 and MYH use an activated water molecule to cleave the N-glycosidic bond. After monofunctional glycosylases remove the damaged base, an AP endonuclease (APE1 or APE2) is required to hydrolyze the phosphodiester bond of DNA, creating a single-strand break with a 5′deoxyribose-phosphate (5′dRP) group that is ultimately gap-tailored by Polβ (86). In contrast, bifunctional glycosylases possess an AP lyase activity in addition to the glycosylase activity. OGG1, NTHL1, and NEIL3 are bifunctional DNA glycosylases that have an associated β-elimination activity, whereas NEIL1 and NEIL2 are bifunctional DNA glycosylases that have an associated β,δ-elimination activity. In human cells, NTHL1 (NTH1), NEIL1, or NEIL2 is usually involved in removing oxidative pyrimidine lesions while OGG1 is primarily responsible for the removal of oxidatively modified purine bases. SMUG1 excises a subset of oxidative base damage, including 5-hydroxyuracil, 5hmU, and 5-formyluracil. MUTYH (MYH) can remove the normal A-base when misincorporated opposite the template 8-oxoguanine (8-oxodG) during DNA replication or repair synthesis. Interestingly, it has been reported that MYH mutations are related to a colorectal adenoma syndrome (MYH-associated polyposis) and high colorectal cancer risk (17). Bifunctional DNA glycosylases (NTH1, NEIL1, NEIL2, and OGG1) use Lys or Pro for direct attack on sugar C1’ to hydrolyze the N-glycosidic bond. Based on the initiating lesion and the mechanism of base removal, the BER pathway can be classified into either a long-patch BER subpathway (Fig. 1) or three short-patch subpathways, including a monofunctional DNA glycosylase-initiated subpathway, bifunctional DNA glycosylases with associated β-elimination-initiated subpathway, and bifunctional DNA glycosylases with associated β,δ-elimination-initiated subpathway (88). The monofunctional DNA glycosylase-initiated subpathway uses the classic short-patch BER mechanism starting with hydrolysis of the N-glycosidic bond to form an abasic site (3). The gap margins with a 3′-OH and a 5′-dRP group are tailored by the 5′-dRP lyase activity of Polβ, followed by Polβ-mediated gap filling (DNA synthesis). Either DNA ligase I (LigI) or a complex of DNA ligase III (LigIII) and XRCC1 seals the DNA chain (3). Recent evidence indicating that LigIII is not required for nuclear BER may suggest that LigI is the preferred ligase in BER (37,84). Strand-break repair and signaling are mediated by XRCC1 together with PARP1, PARP2, and PARG. XRCC1 acts as a scaffold for protein complex formation (2,3).
The BER subpathway initiated by bifunctional DNA glycosylases associated with β-elimination is the predominant BER mechanism for the removal of oxidatively damaged DNA. A bifunctional DNA glycosylase such as OGG1, NTHL1, or NEIL3 hydrolyzes the DNA backbone, 3′ to the incised base, leaving a 3′-unsaturated aldehyde after β-elimination and a 5′-phosphate at the termini of the repair gap. NEIL3 mainly is used for nuclear DNA repair, whereas OGG1 and NTHL1 can repair oxidatively damaged DNA in both the nucleus and the mitochondria. The gap tailoring is performed by the 3′-phosphodiesterase activity of APE1 (3). Next, gap filling (new DNA synthesis) is mediated by Polβ, and the DNA backbone is sealed by the XRCC1/LigIII heterodimer or LigI (3). In some cases, replicative DNA polymerases (δ and ɛ) or low-fidelity DNA polymerases may insert a wrong base opposite many oxidative lesions. To prevent the accumulation and the eventual onset of G⇒T substitution mutations, failure to repair the 8-oxodG lesion also triggers repair of the A-base opposite the 8-oxodG lesion by the MYH glycosylase (26,85).
Another BER subpathway is initiated by bifunctional DNA glycosylases with an associated β,δ-elimination (NEIL1 and NEIL2). These DNA glycosylases are followed by an APE1-independent repair mechanism. Binding of NEIL2 to the lesion recruits XRCC1 to the DNA (15). It has not yet been established if the same process of XRCC1 recruitment is mediated by NEIL1. NEIL1 or NEIL2 generates a 5′- and 3′-phosphate at the ends of the DNA in the single-base gap, resulting from hydrolysis of the glycosidic bond to release the base, cleavage of the DNA-3′ to the abasic site via β-elimination, and then cleavage of the DNA-5′ to the abasic site via δ-elimination, releasing the trans-4-hydroxy-2,4-pentadienal (25,102). The phosphatase PNKP is subsequently recruited to the site to remove the 3′-phosphate in the gap and complete the gap-tailoring step. The repair is finalized by DNA synthesis with Polβ and ligation with XRCC1/LigIII (25,102).
BER and Epigenetic Regulation via Induction and Repair of 8-oxodG
In almost all types of cells, a detectable low basal level of oxidative DNA modifications (e.g., 8-oxodG) is observed, likely due to a steady state between continuous generation of these and related DNA modifications by ROS and simultaneous repair mainly by BER mechanisms (51). The endogenous oxidative nuclear DNA damage is traditionally thought to arise from ROS generated via the mitochondrial electron transport chain. This concept was challenged by Hoffmann and colleagues in 2004 (51). The authors reported that a reduction in mitochondrial ROS production by depletion of mitochondrial DNA or an increase in mitochondrial ROS production via interfering with the mitochondrial electron transport chain did not change the density of nuclear DNA damage (51). The observation implied there may be additional sources of ROS that contribute to the steady-state level of endogenously induced nuclear DNA base modifications. Moreover, Ziel et al. proposed that the oxidative DNA modifications might be utilized as signals for transcriptional regulation (109). Here, the authors observed DNA oxidative modifications enriched at hypoxic-response elements located in the promoter region of the VEGF gene. Moreover, compared to the reporter gene with wild-type hypoxic elements, luciferase activity increased with AP-site-modified hypoxic elements. As described above, the AP sites are repair intermediates formed after glycosylase-mediated base lesion removal (Fig. 1). Because AP sites are repaired by the BER pathway, these results suggested that the BER pathway may play a role in transcriptional regulation (109). More recent studies directly demonstrated that the BER pathway may participate in estrogen-induced target gene expression and Myc-related transcriptional regulation (see below).
BER and Estrogen-Induced Epigenetic Regulation
The first direct evidence of LC-BER and the involvement of BER in transcriptional regulation was reported by Perillo et al. in a study of estrogen-induced target gene expression mediated by LSD1 (78). As shown in Figure 3, the authors used a model system (estrogen receptor [ER]-positive MCF7 cells treated with 17β-estradiol [E2]), demonstrating that the ER was recruited to the promoter and estrogen-response elements (EREs) and enhancers (Fig. 3A). Binding of activated ER by E2 to both the B-cell CLL/lymphoma 2 (bcl-2) promoter and the ERE enhancer regions, which are 1.5-kb apart, formed a chromatin loop between the promoter and the ERE enhancer (Fig. 3B). The chromatin looping lasted for 60 minutes after E2 addition and then disappeared. Assembly of the transcription complex occurred between 30 and 45 minutes after E2 addition. Meanwhile, H3K9me2 demethylation occurred at the promoter and enhancer sites of the bcl-2 or pS2 genes. Both chromatin looping and estrogen-induced transcription were prevented when the LSD1 activity was inhibited by the free radical-scavenging drug N-acetyl-
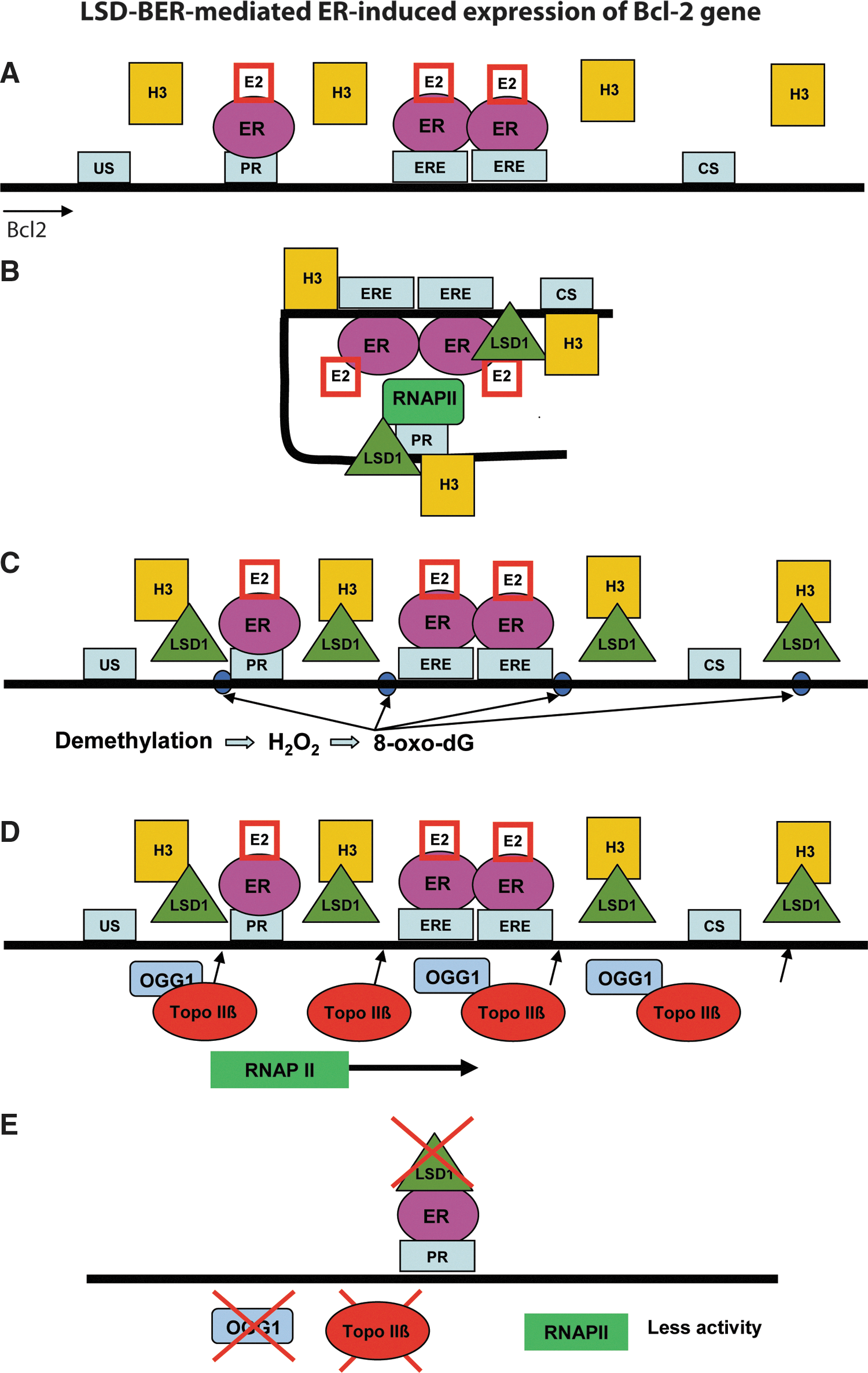
The proposed mechanism of histone demethylation coupling with BER (LC-BER) for transcriptional regulation is as follows: LSD1 demethylation results in the accumulation of 8-oxodG lesions via the generation of H2O2 at the sites of demethylation. The 8-oxodG lesions are repaired by OGG1, ultimately generating DNA single-strand breaks during BER that are recognized by TopoIIb to facilitate chromatin relaxation and formation of the complex process of transcription initiation. As such, H3K9me2 demethylation produced temporally limited and localized H2O2 at the promoter and enhancer regions, providing a local signal for the assembly of transcription initiation complex proteins mediated in part by BER proteins such as OGG1 and APE1. Overall, this study demonstrated that LSD1-mediated histone demethylation is coupled with BER to initiate hormone-dependent gene expression (78).
BER and Myc-Induced Epigenetic Regulation
More recently, the same group observed LSD1-BER-mediated transcriptional regulation in Myc-activated transcription of the Myc-target genes Ncl and CAD (4,5). Myc is one of the most common activators of cell proliferation used by cancer cells to drive disease progression. Because the E-box sequence (CACGTG) for Myc binding was estimated to exist in 15% of the all promoter regions in the human genome (32,42,105), the involvement of BER in the mechanism of Myc-induced transcriptional regulation will have broad significance for cancer therapy research. Amente and colleagues demonstrated that LSD1-BER-coupled epigenetic regulation via demethylation of H3K4Me2 by LSD1 at the promoter and E-box sites drives Myc-mediated gene expression (Fig. 4) (4). In this study, the activation of Myc with tamoxifen (OHT) was set as the start point (Fig. 4A). About 30–60 minutes after Myc activation, Myc was recruited mainly on the E-boxes of both the Ncl and CAD genes and stably accumulated at the E-box until the end point (240 minutes) (Fig. 4B). LSD1 was recruited to the transcription start site (TSS) and the coding region (CR) of both genes shortly after Myc activation (Fig. 4C). The authors suggested that the recruitment of LSD1 to the promoters is an early and transient event, because the existence of LSD1 on both the TSS and the E-box overlapped with H3K4me2 demethylation on those sites only in the period from 30 to 60 minutes. Ncl and Cad mRNA accumulation was observed 1 hour after Myc induction. Four hours later, not only the methylation status of both sites was back to normal but also the BER proteins were no longer present at the sites. Stable acetylated histone H4 accumulation was detected on the TSS and CR of both the genes with a peak level at 240 minutes after Myc activation. During the demethylation of H3K4me2, the fluorescent signal for detection of 8-oxodG was significantly increased in Myc-activated cells (Fig. 4C), similar to the level of cells exposed to H2O2, but not in the Myc-null cells. In separate experiments, addition of siRNA specific for Myc or addition of pargyline (an LSD1 inhibitor) abolished the signal for 8-oxodG detection. It was also demonstrated that two BER enzymes (OGG1 and APE1) were engaged at the TSS and E-boxes of the Ncl and CAD genes after Myc activation (4). OGG1 appears to be required for the removal of the 8-oxodG lesion, and APE1 was required to complete repair (Fig. 4D). However, no additional BER proteins were evaluated in this initial report. The functional role of BER for transcriptional control was confirmed by the loss of expression of either OGG1 or APE1 via targeted siRNA treatment, dramatically reducing the expression of both the Ncl and CAD genes after Myc activation (Fig. 4E). The proposed mechanism for Myc-induced transcription is that Myc recruits LSD1 to the target E-box site and initiates histone H3 demethylation, which is then linked to oxidation and DNA repair (LC-BER) to promote the assembly of the transcript initiation complex.

Epigenetic Regulation via BCADD
In 1998, Um and colleagues reported that TDG directly interacted with the retinoic acid receptor and the retinoid X receptor (RXR) in a ligand-independent manner (96). Overexpression of TDG induced a fourfold increase of the β-galactosidase reporter gene expression controlled by a retinoic acid-responsive promoter (96). Subsequently, using chicken 5-methylcytosine DNA glycosylase (5-MCDG), a homolog of human TDG, Zhu and colleagues discovered the upregulation of reporter gene expression induced by a similar 5-MCDG-RXR complex that was the result of DNA demethylation of the downstream ecdysone–retinoic acid-responsive enhancer (108). In a nonmammalian model, participation of BER in DNA demethylation is clearly documented. In Arabidopsis, the BER proteins Demeter and ROS1 directly remove 5mC via their glycosylase activities (39,72). In Xenopus, growth arrest and DNA damage-inducible protein 45-alpha (Gadd45a) is the major protein to initiate DNA demethylation, and the interaction of Gadd45a with the nucleotide excision repair protein XPG is suggested (8). In zebrafish embryos, 5mC is converted to thymine by AID, and then the G:T mismatch is removed by the zebrafish thymine glycosylase MBD4, associating Gadd45 with the BER pathway (79). In a mammalian model, the participation of BER in active demethylation has recently been documented using mouse embryos during genome-wide epigenetic reprogramming. In 2010, Hajkova reported the initiation of epigenetic reprogramming at embryonic day 7.25 (E7.25), followed by genome-wide DNA demethylation. Involvement of BER was proposed after an increase in expression of APE1, XRCC1, and the formation of PAR, suggesting the involvement of BER in mouse primordial germ cells, but not the neighboring somatic cells (45). The mechanism of participation of BER proteins in mammalian demethylation was not clear until the discovery of the TET protein family (see Fig. 5) (90). TET protein family members are α-KG and Fe(II)-dependent dioxygenases that include the isoforms TET1, TET2, and TET3. TET1 is mainly expressed in embryonic stem cells (ESCs), whereas TET2 and TET3 are more ubiquitously expressed (90). All TETs can oxidize 5mC to 5hmC (22,48,89,90) (Fig. 5A, B). TET2 mutations were linked to many types of myeloid malignancies and resulted in a significant decrease in the level of 5hmC in bone marrow cells isolated from patients with acute myeloid leukemia as compared to healthy individuals (63). BER was proposed to participate in TET-mediated DNA demethylation by repairing the 5hmU that is formed from 5hmC by the AID/APOBEC family of cytidine deaminases (43) (Fig. 5A). In addition, recent studies in mammalian cells demonstrated two new cytosine modifications, formylcytosine and carboxylcytosine, which are generated by two successive oxidation reactions of 5hmC catalyzed by the TET proteins (22,53) (Fig. 5B). It has recently been reported that the existence of 5fC or 5caC in a DNA template dramatically reduced the RNA Pol II elongation efficiency compared to the DNA templates that had unmodified C, 5mC, or 5hmC bases. It was demonstrated that the DNA templates with either 5fC or 5caC resulted in much lower GTP incorporation efficiencies than the others. In addition, 5fC- and 5caC-modified bases also caused a significant level of RNA Pol II complex backtracking, further suggesting that RNA Pol II shifted from an active state to a paused state. In addition, 5fC greatly reduced the fidelity of nucleotide incorporation (61). TDG was shown to be the glycosylase responsible for removal of the carboxylcytosine base that was then followed by complete BER processing to restore the normal cytosine (Fig. 5B). A depletion of TDG leads to accumulation of carboxylcytosine in mouse ESCs (48). Moreover, a loss of TDG function is lethal to mouse embryos in an early developmental stage associated with epigenetic aberrations affecting the expression of developmental genes (22).
Further, a recent report demonstrated that another BER protein, PARP1, is involved in the regulation of the process required for reprogramming somatic cells into pluripotent stem cells (iPSCs) when using the pluripotency factors
Summary
Gene expression in eukaryotic cells is regulated at multiple levels of transcriptional control to allow response to growth factors, cellular stress, DNA damage, or development. The primary control comes from genetic information encoded in the DNA sequence, which defines protein sequences, signal sequences, and noncoding regulatory elements for transcription factors, enhancers, or silencers. A secondary, but critical, level of control of gene expression is at the epigenetic level via DNA methylation and histone modification. The BER pathway has recently been demonstrated to be necessary for both DNA methylation- and histone modification-mediated epigenetic regulation separate from its main function in maintaining genome stability. Impaired BER can also have significant effects on the cellular DNA methylation status. For example, knockout of TDG resulted in mass DNA methylation changes in many gene promoter regions in mouse embryos and resulted in early embryonic lethality (22). The discovery of the function of the TET family proteins suggests that the dynamic regulation of DNA methylation by active demethylation requires the BER pathway as the final effector. Two possible TET-BER-regulated active demethylations were proposed. Cortellino et al. proposed that 5hmC may first be deaminated by the AID/APOBEC family of cytidine deaminases to generate 5hmU. BER is then engaged to repair the 5hmU:G mismatch, because both TDG and SMUG1 were demonstrated to effectively repair 5hmU in the 5hmU:G mispair in dsDNA, yielding the correct C:G base pair (22). He and colleagues proposed another active DNA demethylation pathway in which the 5mC lesion is iteratively oxidized by TET proteins into 5hmC, 5fC, and 5caC, followed by a conversion back to an unmethylated cytosine via TDG-initiated BER (48). Further studies are needed to determine the condition for activating these two pathways. BER also plays an important role in transcriptional regulation at sites of histone demethylation. As we discussed above, defects in BER directly reduced estrogen- or Myc-induced target gene expression. The 8-oxodG lesion, produced from H2O2, generated by LSD1 or LSD2 during the process of histone demethylation, serves as the signal to recruit BER enzymes to hydrolyze and relax high GC-content promoter regions, facilitating and enhancing transcription initiation and elongation (5).
Appropriately controlled epigenetic regulation is critical for the normal development and health of an organism. Misregulation of epigenetic control, regarding either DNA methylation or histone methylation, has been associated with cancer, chromosomal instability syndromes, and mental retardation. Recent reports show that BER is indispensable for epigenetic events such as hormone-modulated gene expression and iPSC reprogramming. Such efforts to address the role of BER proteins in epigenetic regulation could broaden cancer therapeutic strategies to include epigenetic modifiers combined with BER inhibitors.
Footnotes
Acknowledgments
This work was supported by grants from National Institutes of Health (GM087798, CA148629, ES019498, ES021116, and GM099213) to R.W.S.
Author Disclosure Statement
R.W.S. is a scientific consultant for Trevigen, Inc. The remaining authors state that there is no conflict of interest.