
Abstract
Introduction
T
The reduction potential of redox-active drugs determines whether they could be readily coupled (oxidized and/or reduced) with redox-active biological targets. As long as the compound produces beneficial therapeutic effects, it may not be utterly important to distinguish between the RS scavenged or pathways affected, and to describe the compound strictly as either SOD mimic or peroxynitrite or any other RS scavenger. We are far away from singling out the reactive species and/or other biological targets being involved in the in vivo actions of SOD mimics—many and diverse have already been identified as indicated here and in several other articles published in this Forum. Genetic approaches are essential for the correct conclusions on the type of species/pathways involved in oxidative damage. However, our present understanding based on numerous studies allows us to claim with a fair certainty that more potent the SOD mimic is—the closer its E1/2 to that of SOD enzyme (+300 mV vs. normal hydrogen electrode [NHE])—the more biocompatible it is with cellular redox-based pathways, and, thus, the more easily it could shuttle electrons among reactive species and signaling proteins by normalizing the cellular redox environment. Thus far, our studies indicate that the kcat for the catalysis of O2·− dismutation is a reliable measure of the therapeutic potential of redox-active compounds. Therefore, striving for the most potent SOD mimic may still be the most promising drug design approach.
The targeting of the cellular redox sensitive pathways—redoxome—is still an unusual therapeutic strategy, at least from the point of view of medical audience and pharmaceutical companies. However, cellular metabolism is dominated by redox-based processes: mitochondrial respiration, glycolysis, microsomal electron transport chain, detoxification by cyt P450 enzymes, nitric oxide synthesis, and so on. It is very unlikely that a single drug targeting a single target in a cell, where redundant systems are common, would become a potent therapeutic for a pathological condition with perturbed cellular redox status. Multi-drug strategies are, thus, becoming increasingly common in treating pathological conditions. Since normal and cancer cells differ with regard to their redox status, we have anticipated, and it is becoming increasingly true, that SOD mimics have a differential impact on their metabolism: heal a normal cell and kill a cancer cell. As our knowledge increases, and the impact of redox biology on the cell metabolism becomes exceedingly obvious, the Pharma and the medical researchers start recognizing the advantage of redox-biocompatible therapeutics and promote their development. Indeed, some time ago, NIH offered a funding opportunity for the exploration of “Metals in Medicine.”
Besides its redox activity, the other major factor contributing to the drug efficacy is its bioavailability. Organ distribution, extra- and intracellular levels, and their subcellular localization will impact the final therapeutic outcome. The remarkable in vivo effects of SOD mimics are driving the ongoing studies.
The synthesis, isolation, and purification of cationic metalloporphyrins (MPs) have been challenges. We have learned and provided sufficient evidence that it is of utmost importance to check the quality and identity of drugs used in preclinical models to avoid misinterpretations and loss of time and resources. Comprehensive pharmacokinetic (PK) studies are often lacking, but are essential for drug development toward clinics. For example, with no earlier comprehensive PK studies, curcumin entered and failed the Clinical Trials in Alzheimer's patients (36). It was subsequently shown that it is neither sufficiently bioavailable (not much gets into the blood stream, as it undergoes glucuronidation) nor crosses the blood brain barrier (BBB), a property essential for neurodegenerative diseases; both sets of data should have been a prerequisite for conducting Clinical Trials.
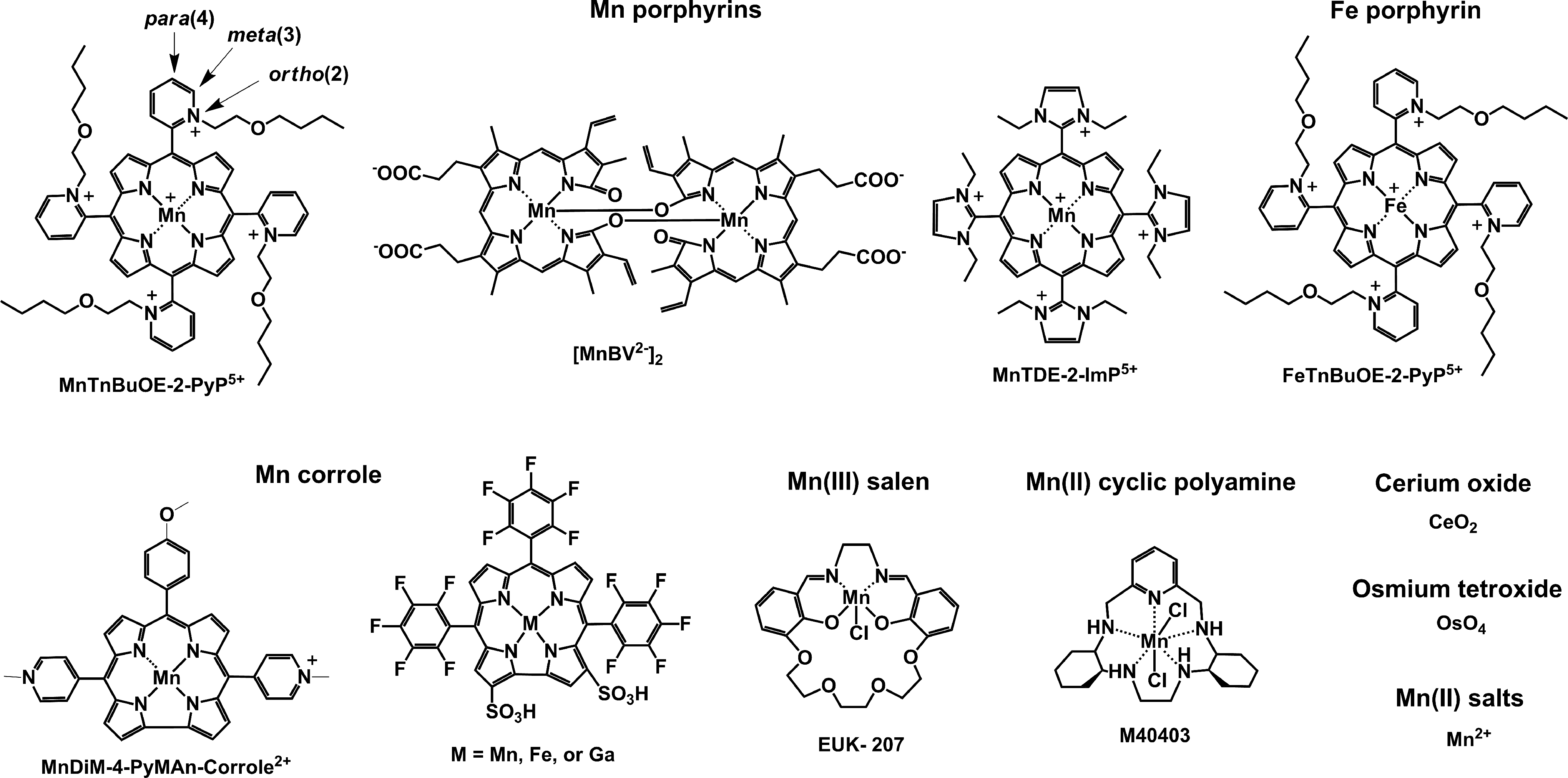
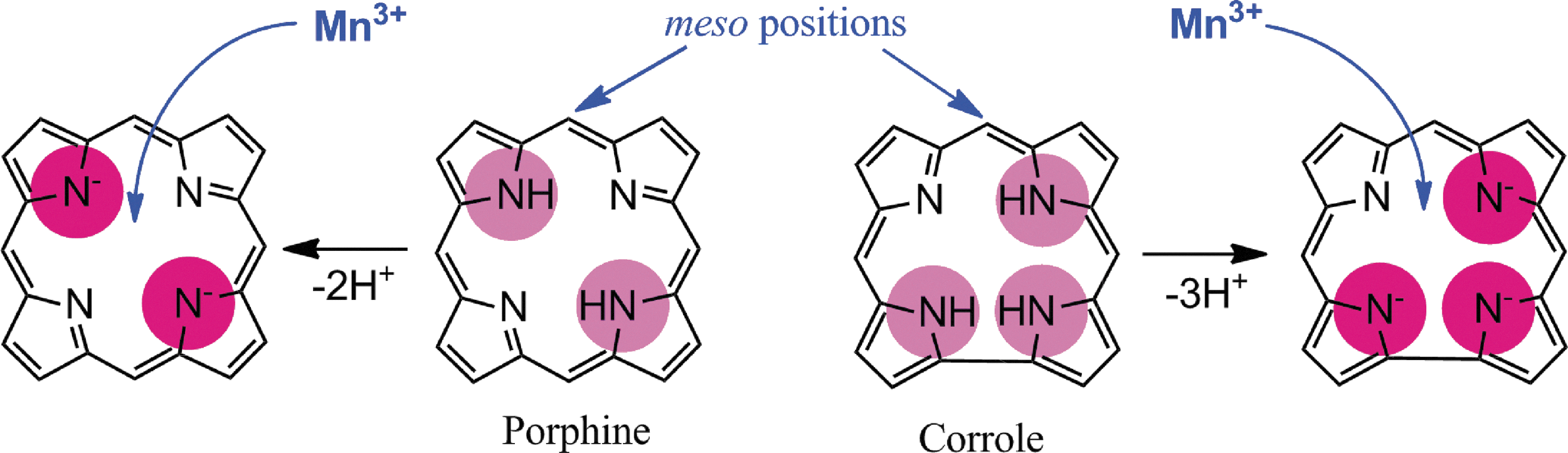
By definition, an SOD mimic/therapeutic is the one that catalyzes the oxidation and reduction of O2·− (Eqs. [1] and [2]). As it happens, only a few compounds are “true” SOD mimics and are discussed in great detail here (ones that are not, are addressed only briefly for the sake of discussion or comparison). The true SOD mimics are MPs, Mn(III) biliverdins, metallocorroles (MCs), Mn(III) salens, Mn(II) cyclic polyamines, and metal oxides; their representatives are shown in Figure 1. MPs can either act as true SOD mimics (Eqs. [1] and [2]) or couple with cellular reductants (Eq. [2]), acting as superoxide reductase [alike enzymes in some organisms (57)]. The removal of O2·− can also be coupled to the reduction of ONOO−, where SOD mimic would act as peroxynitrite/superoxide reducto-oxidase (147, 148, 241). The proportionality of log kcat(O2·−) versus log kred(ONOO−) proves that powerful SOD mimics are also powerful reductants of ONOO− (28, 84, 86). None of the potential therapeutics under investigation and none of the biomarkers available can readily distinguish between O2·−, H2O2, and ONOO−. Modest specificity toward ONOO− relative to O2·−, but not toward H2O2, has been achieved with boron-based reagents (58, 119, 293).
Here, we have addressed SOD mimics with regard to their (i) rational design and structure-activity relationships (SARs); (ii) reactivity toward reactive species other than O2·−; (iii) impact on the cellular signaling pathways in various models of oxidative stress injuries; (iv) bioavailability; and (v) therapeutic effects related to the radiation and cancer. For injuries of central nervous systems, see review from Warner and Sheng groups (236); while for inflammation and immunity disorders, see contribution from Piganelli group (208). For the corrole-based SOD mimics, see references from Gross's group (103, 104).
Drug Design
The vital and indispensable role of SOD enzymes in all living organisms has been a driving force in the search for their mimics (see articles in Forum on SOD enzymes in ARS 20/10, and in this Forum on SOD therapeutics). In addition to MPs, different classes of SOD mimics have been developed: Mn salens, Mn corroles, and Mn cyclic polyamines (28). Over the years, reactivities other than toward O2·− for all SOD mimics have been demonstrated. Importantly, the proportionality between the log kcat (O2·−) and their therapeutiuc efficacy has been demonstrated (see below under Drug reactivity section for the basis of such relation). Consequently, the design of SOD mimics has been and remains an excellent strategy in designing drugs for oxidative stress injuries. The rational approach in the design of a good SOD mimic has been to mimic the kinetics and thermodynamics of the enzymatic catalysis of O2·− dismutation: to (i) tune the metal-centered reduction potential around the midpoint (∼+300 mV vs. NHE) between the potential for the oxidation (−180 mV vs. NHE) and the reduction of superoxide (+890 mV vs. NHE) and to (ii) provide favorable electrostatics for the approach of negatively charged O2·− molecule to metal site (81, 137, 280). Such reduction potential at ∼+300 mV versus NHE would provide equal thermodynamics for both steps of the dismutation process (Eqs. [1] and [2]).
In the case of SOD enzyme, the similar kinetics results in identical kred(O2·−) and the kox(O2·−) for the catalysis of O2·− dismutation of ∼109 M −1s−1 (71, 81, 97, 137, 280). The porphyrin structure allows limitless possibilities of modifications. We have elaborated our design strategies in detail elsewhere (26 –28, 33, 177, 271). Briefly, starting from the nonsubstituted meso-phenyl and meso-pyridyl porphyrins, different substituents were attached to adjust the metal-centered reduction potential for MnIII/MnII redox couple. Nonsubstituted Mn(III) porphyrins have E1/2 of ∼−200 to −300 mV versus NHE, way out of the range for a successful catalytic reaction with O2·−; at such E1/2 Mn(III) is stabilized in +3 oxidation state and cannot be reduced to Mn(II)P in a 1st step in order to subsequently reduce O2·−.
The superoxide dismutation by either enzyme or mimic has an antioxidative impact only if H2O2 is efficiently removed; under pathological conditions in which peroxide-removing systems may be suppressed, the O2·− dismutation may result in cytotoxic effects. Such therapeutic effects have been seen with SOD enzymes and their mimics in cancer [see below and in Ref. (177)]. In order to increase the reducibility of the metal center, that is, to increase the electron deficiency of the metal site, the porphyrin structure was modified with electron-withdrawing groups. The breakthrough step in our design strategies was the substitution (quaternization) of pyridyl and imidazolyl nitrogens in the closest (“ortho”) position toward the metal center, where the positive charges exert the strongest thermodynamic and electrostatic effects (Fig. 2). Incidentally, such a pentacationic electrophilic (electron-deficient) molecule also provides the favorable electrostatics for the oxidation and reduction of anionic (electron-rich) nucleophile, O2·−. The first lead compound was identified as Mn(III) meso tetrakis (N-ethylpyridinium-2-yl)porphyrin, MnTE-2-PyP5+. With E1/2 of +228 mV versus NHE (∼+300 mV vs. NHE for SOD enzyme), MnTE-2-PyP5+ reduces and oxidizes O2·− with nearly identical rate constants as does the SOD (31, 32). Such a design later led to imidazolyl analog, MnTDE-2-ImP5+ (26 –28, 131, 211, 225, 239), and to a series of Fe porphyrins (FePs) (30, 146, 172, 173, 193, 207, 214, 215, 244, 259, 273) (see also under Drug Design section). Further, the increased bioavailability and reduced toxicity was achieved through modifications of the substituents in ortho positions (216, 274). The exploration of differently meso and beta substituted MPs led to the very first structure-activity relationship (SAR) not only for porphyrinic compounds but in general as well (30). It establishes the relationship between the thermodynamic property of a catalyst—the E1/2 which describes the likelihood that the reaction will occur—and the kinetic property, log kcat(O2·−), indicating how fast the reaction will occur and is governed by factors such as sterics and electrostatics. Those compounds with favorable E1/2 (>0 mV vs. NHE) have been subsequently tested in a simple superoxide-specific assay where SOD-deficient Escherichia coli grows aerobically as well as wild type only if supplied by true SOD mimics (270). The SOD-deficient yeast, Saccharomyces cerevisiae has been recently established as an additional model (270).
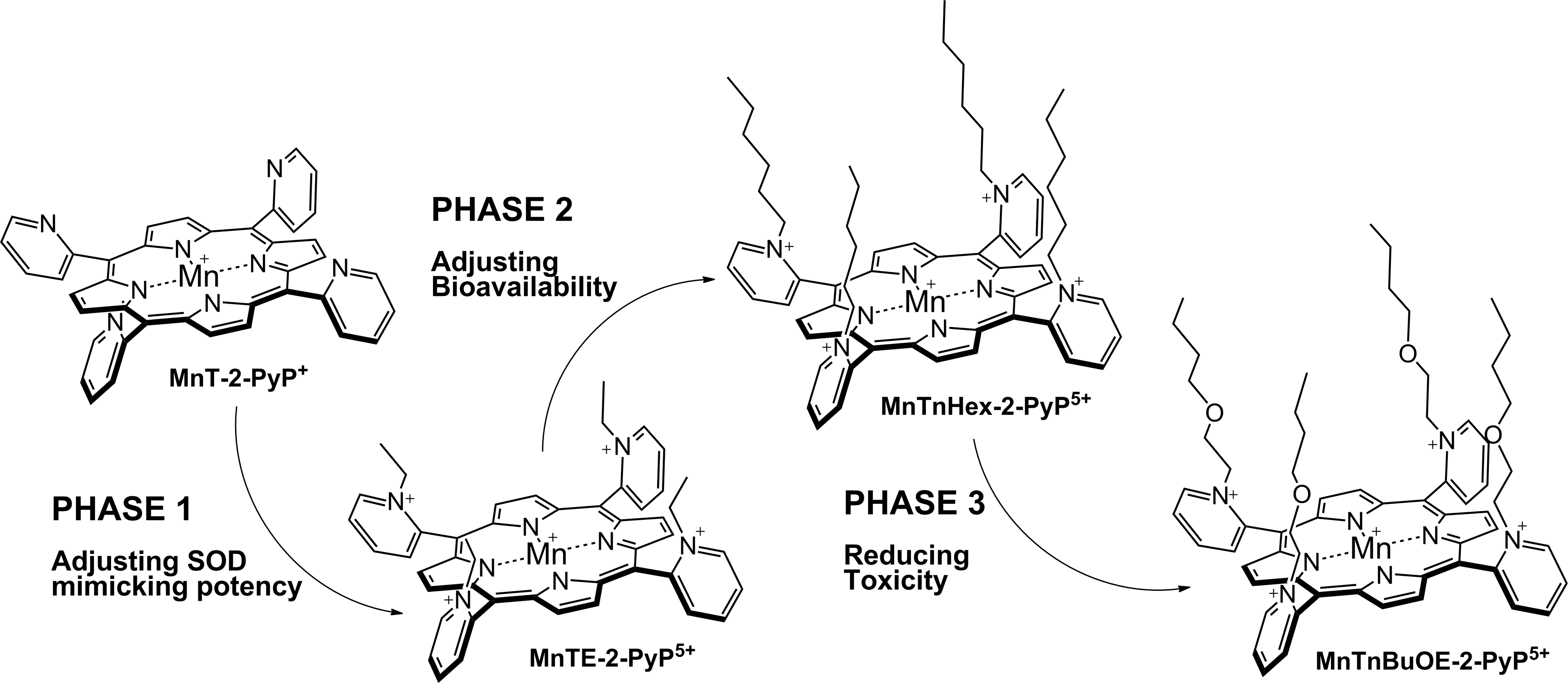
Most of the published work on Mn porphyrins (MnPs) relates to our first lead, MnTE-2-PyP5+, which continues to be widely used for therapeutic and mechanistic purposes. With the small porphyrin structure, we have achieved the potency near or similar to that of SOD enzymes (23, 28, 30 –32, 69) (Fig. 2). The differential sterics, however, affords differential specificity; the SOD enzymes react with ONOO−, thiols, and other species at much lower rates than MnPs. The impact of electrostatics on MnP potency, as dramatic as with SOD enzymes, has been clearly demonstrated (26 –28, 32, 271). The cationic compounds have approximately two to three orders of magnitude higher kcat(O2·−) relative to neutral and anionic MnPs. Based on the ortho effect, the di-ortho imidazolyl derivatives were subsequently synthesized and showed potency in a number of animal models of oxidative stress (26 –28, 131, 211, 225, 239). In E. coli model, the Mn(III) meso-tetrakis(N,N′-diethylimidazolium-2-yl)porphyrin, MnTDE-2-ImP5+(AEOL10150) is inferior relative to porphyrins bearing N-alkylpyridyl substituents (32, 195, 270). The N,N′-dialkylimidazolyl substituents are positioned both above and below the porphyrin plane; in turn, the N,N′-dialkylimidazolylporphyrins are much bulkier than N-alkylpyridyl analogs (Fig. 1). The bulkiness reduces their biodistribution and, in turn, their efficacy. However, such a disadvantage has at least, in part, been outbalanced by the reduced interactions of MnTDE-2-ImP5+with biological molecules and, in turn, may contribute to lower toxicity and could be utilized at higher doses for the same efficacy as MnTE-2-PyP5+ (237). MnTDE-2-ImP5+ is presently under development by Aeolus Pharmaceuticals as a radioprotector (122, 198, 212).
Once the SOD-like potency of MPs was optimized, we were challenged by medical audience on their whereabouts in the body, within cells, and particularly in mitochondria. In addition, transport across the BBB and porphyrin oral availability was questioned. Thus, we embarked on a PK journey. HPLC/fluorescence and later LCMS/MS methods were developed for each individual compound (140, 177, 238, 250, 252, 254, 288), which supported the comprehensive biodistribution studies. We have also expanded our synthetic efforts to increase porphyrin bioavailability. Compounds with longer alkyl substituents were synthesized, with MnTnHex-2-PyP5+ among others. This molecule has properties of surfactants (polar cationic nitrogens and hydrophobic alkyl tails), and is, thus, toxic at high doses. However, due to higher bioavailability, submiligram daily doses produce sufficient efficacy in animal models, which allows for a sufficiently wide therapeutic window (28, 210). In a 3rd phase of drug development, oxygen atoms were introduced into the pyridyl N-substituents to disrupt porphyrin surfactant character—the design comparable to the esterification of sodium dodecyl sulfate in order to decrease skin irritability. The insertion of polar oxygens into the alkyl chains disrupts the surfactant property. The resulting molecule, MnTnBuOE-2-PyP5+ (216) is equally lipophilic as MnTnHex-2-PyP5+, but ∼4–5-less toxic to a mouse (Figs. 1 –3). The alkyl analogs, MnTnHex-2-PyP5+ and MnTnHep-2-PyP5+, cause mouse death at 5 and 2.5 mg/kg, respectively; while no toxicity was observed with 5 mg/kg of MnTnBuOE-2-PyP5+ (Fig. 3). Further, in a S. cerevisae assay, MnTnHex-2-PyP5+ and MnTnHep-2-PyP5+—but not MnTnBuOE-2-PyP5+—became toxic at 30 and 5 μM, respectively (Fig. 3). Due to superior properties, MnTnBuOE-2-PyP5+ is presently under aggressive clinical development by BioMimetix Pharmaceutical, Inc.; its GMP scale-up is completed and safety/toxicity studies are underway. All three porphyrin-based lead candidates (MnTE-2-PyP5+, MnTnHex-2-PyP5+, and MnTnBuOE-2-PyP5+) are excellent tools to explore the factors that determine drug bioavailability, efficacy, and toxicity. They are also indispensable for mechanistic studies.

The design of metallocorrole (MC)-based class of SOD mimics is briefly discussed next (104). The design of Mn(II) cyclic polyamines, which resulted in an optimized molecule, M40403 has been extensively covered by Riley's group (14, 167, 226 –228, 232). No modification in Mn salen core structure affected the SOD-like activity of the basic EUK-8 structure (73, 74). Under high stomach acidity, the Mn salen derivatives lose Mn (229). Their insufficient metal/ligand stability was significantly enhanced in EUK-207 structure (Fig. 1) via derivatization with crown ether (229), as it lowers the loss of the metal when given orally (229).
SARs for Different Redox-Active Compounds
Comprehensive SAR-related studies were conducted on MnPs (18, 27, 28, 30, 32, 271). Recently, Gross' group has established limited SAR for MCs (196). Here, we showed for the first time that SAR established originally for MPs (28, 30, 32) is fairly valid for many redox-active compounds that are aimed at being SOD mimics (Fig. 4); the largest deviation is demonstrated for nonmetal-based nitroxides; however, their reduction potential relates to an irreversible oxoammonium cation/nitroxide redox couple (14, 167, 228). The scattering of the data reflects the impact of factors other than E1/2 such as electrostatic and electronic effects, shape, size, and bulkiness of the molecule (27, 28, 221, 223). With compounds that are both anionic and distorted (the octabromosulfonato, MnIIIBr8TSPP3− and octabromocarboxylato MnIIIBr8TBAP3−), the access of superoxide to the cationic Mn center is hindered as opposed to the anionic planar MnTSPP3− and MnTBAP3−. Steric hindrance is compensated to some extent by favorable thermodynamics as a result of the strong electro-withdrawing effect of eight bromines. However, those compounds still exhibit the largest deviation from the SAR. The compounds with too negative values of E1/2 (stabilized in higher +3 Mn oxidation state) cannot be reduced by O2·− to Mn +2 in a 1st step of O2·− dismutation process. The compounds with highly positive values of E1/2 cannot be easily oxidized by O2·− to Mn +3 in a 1st step. Among them are porphyrins which contain Mn in its +2 oxidation state, and are, thus, not very stable, that is, lose Mn readily (23, 69): MnPs [MnIIBr8TM-3(or 4)-PyP4+, MnIICl5TE-2-PyP4+], and Mn(II) cyclic polyamines, such as M40403. Among them are also very stable electron-rich metal complexes, Mn(III) corroles, and Mn(III) biliverdins, which undergo facile oxidation by O2·− to a higher Mn +4 oxidation state in a 1st step of a dismutation process.
A closer look at the thermodynamic and kinetic data indicates that perhaps two SARs can be constructed: one for compounds utilizing MIII/MII couple (red line) and the other one for those compounds employing MIV/MIII couple (green line) (Fig. 4). The latter are Mn(III) biliverdin and its analogs and Mn(III) corroles. The optimal potentials for those SARs are ∼300 mV away from each other.

Mn(III) biliverdins
The exploration of Mn(III) biliverdin derivatives and metal(III) corroles taught us that the magnitude of their reduction potential is a key factor in O2·− dismutation, with a minor role of the nature of the redox couple involved (thick curve in Fig. 4). In other words, superoxide does not care much with whom it exchanges the electrons as long as it occurs at the potential where O2·− could be easily reduced or oxidized. We have shown that Mn biliverdin dismutes O2·− employing MnIV/MnIII redox couple (Eqs. [3] and [4]) as efficiently as MnP employing MnIII/MnII redox couple. The E1/2 for MnIV/MnIII redox couple is shifted ∼130 mV more positive than E1/2 of MnTE-2-PyP5+ for MnIII/MnII redox couple.
However, Mn(III) biliverdin exerts no electrostatic facilitation for the approach of superoxide in either step of dismutation. Thus, the thermodynamics is solely the responsible factor for the fairly high kcat(O2·−). According to Marcus equation for an outer-sphere electron transfer, for each increase in E1/2 of 120 mV, the rate constant increases 10-fold (30). We have shown that neutral Mn(III) porphyrins have ∼100-fold lower kcat(O2·−) than cationic MnPs of the same E1/2. Were the E1/2=+228 mV versus NHE for anionic (MnBV2−), the log kcat(O2 •−) would be ∼5. Based on the SAR (160), a 232 mV increase in E1/2 from +228 (MnTE-2-PyP5+) to +460 mV ([MnBV2−]2), would allow for ∼100-fold increase in log kcat(O2·−) from ∼5 to 7.4.
Mn(III) corroles
Gross's group reported that MCs employ MnIVC/MnIIIC redox couple for O2·− dismutation (Eqs. [5] and [6]) (80, 196). The reason for that is the stabilization of a higher Mn +4 oxidation state. The redox cycling via MIIIC/MIIC occurs at too negative potentials to be of biological relevance (162). Consequently, metallocoroles could not readily oxidize cellular reductants, thiols, or ascorbate in a first step of their redox cycling. Rather, MCs would act as reductants of reactive species, such as O2·−, H2O2, or ONOO− in a 1st step, while undergoing oxidation from MIIIC to MIVC. In a subsequent step of re-reduction, MCs would behave as strong oxidants; the coupling with cellular reductants in that step is likely, but has not yet been reported. Once reaching a cell, though, MnPs would prompty behave as oxidants of ascorbate, simple thiols, or protein thiols, undergoing reduction from MnIIIP to MnIIP. Both metalocorroles and MPs are efficacious in treating oxidative stress injuries. Is some common pathway operative for both classes of redox-active compounds, affecting the cellular redoxome, that we could not have yet forseen?
Mn is coordinated to trianionic (biliverdin and corrole), while porphyrins are dianionic ligands (see Figs. 1 and 10). Trianionic coordination in Mn(III) biliverdin dimer is assured by the coordination of Mn of one monomer to oxygen of another monomer (Fig. 1). Mn(III) corroles do not require oxygen binding to stabilize Mn in higher +4 oxidation state, whereas Mn in O=MnIVP is stabilized in +4 oxidation state with oxygen (80). Axial protonation equilibria for corroles have not been reported.
The first generation of Mn corroles has E1/2 ∼+1000 mV versus NHE, which is out of range for the O2·− reduction, that is, disfavors Mn oxidation. Consequently, the marginal SOD-like activity was reported with log kcat(O2·−) ∼6, which is only slightly above the value for O2·− self-dismutation [log kcat(O2·−) ∼5.7] [log kcat(O2·−) for SOD enzymes is in a range 8.84–9.2] (80). Mn corroles were subsequently modified with electron-donating groups, which decreased the E1/2 (196). Based on the data in Refs. (78, 194) and the similarity of E1/2 of Mn corroles in acetonitrile and in an aqueous system, the values of E1/2 in aqueous medium for several corroles plotted in SAR (Fig. 4) were estimated (80, 104, 196) (Fig. 1). Cytochrome c assay was used by Gross's group to determine the kcat(O2·−). Preliminary stopped-flow data support cyt c data (Gross et al., unpublished).
Fe(III) porphyrins
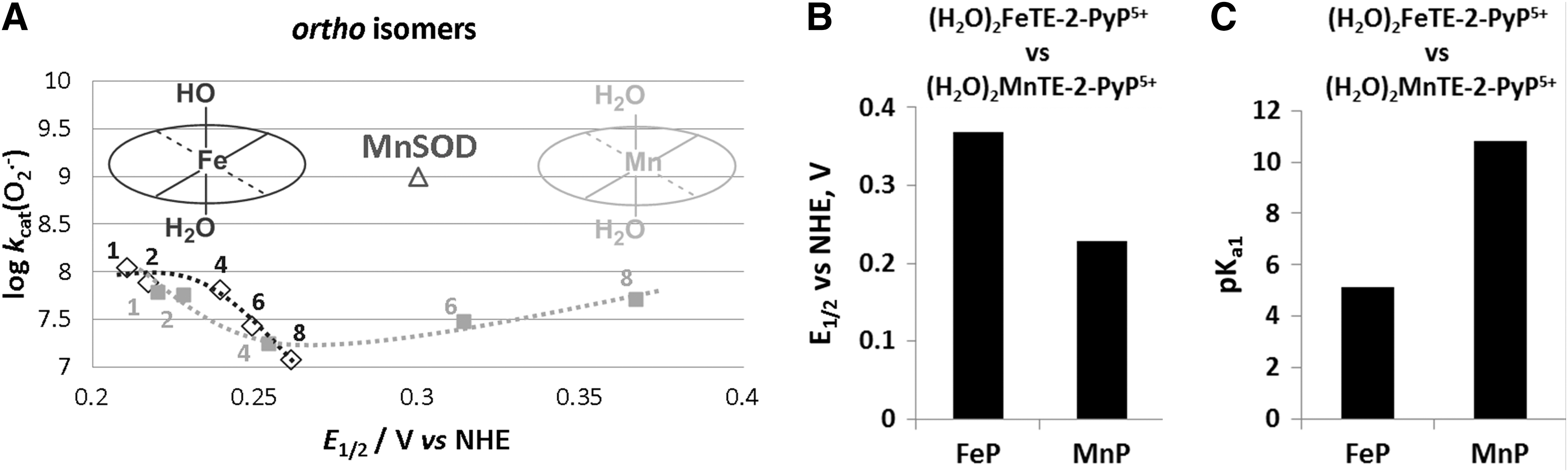
We have originally developed SAR for both Mn and Fe complexes (30). Recently, we synthesized a series of ortho and meta Fe complexes to understand the intriguing differential in vivo behavior of Fe and MnPs (273). FePs have also been often used in cellular and animal models of oxidative stress (146, 172, 173, 193, 207, 214, 215, 244, 259). While MnP employs (H2O)2MnIIIP5+/(H2O)MnIIP4+, the FeP utilizes (OH)(H2O)FeIIIP4+/(H2O)FeIIP4+ redox couple for O2·− dismutation. Consequently, the dismutation catalyzed by FePs involves MP axial protonation equilibria (Eqs. [7] and [8]) (273). Thus, the oxidized and reduced FePs have the same total charge at pH 7.8, whereas Mn species do not; the consequence of such difference on in vivo interactions has not yet been investigated. Both Fe-OH and Mn-H2O centers operate at very similar E1/2 and, thus, exhibit similar kcat(O2·−) values (see Eqs. [1], [2], [7], and [8]) (Figs. 5 and 6) (see also FePs vs. MnPs below):

Cerium dioxide, CeO2
Nanoparticles of CeO2 (nanoceria), with a unique electronic structure similar to nitrone spin traps and of mixed valence state, are reportedly very potent SOD mimics. The equations [9] and [10] account for their remarkable SOD-like activity, which is somewhat higher than for SOD enzyme (138), log kcat=9.55. The E1/2 for CeIV/CeIII redox couple varies with medium, and in deionized water, it is ∼+400 mV versus NHE (261). Neither charge nor reduction potential easily justifies such high kcat(O2·−) values.
When the fraction of CeIV increases over CeIII, the SOD-like activity gets reduced (113). As a result of simultaneous existence of CeIII and CeIV in nanoparticles, cerium dioxide forms oxygen vacancies or defects in the lattice structure by the loss of oxygen and/or its electrons. Catalysis can occur at the same cerium atom or independently at different oxygen vacancy sites.
Osmium tetroxide, OsO4
An aqueous solution of OsO4 has a SOD-like potency which is comparable to that of cerium dioxide. However, it is a very toxic compound. Its high oxidizing power has been employed in the treatment of diseased arthritic knee (99). It is widely used for biological staining, as it binds to phospholipids. The log kcat(O2·−) is pH independent in the pH range 5.1 to 8.7. The following equations for superoxide dismutation have been proposed (99):
Osmium(VII) disproportionates to OsVIII and OsVI:
The log kcat(O2·−)=9.15 is described by equations [11] and [12]. The OsVI/OsVII redox couple could cycle with O2·− also (Eq. [14]) with log kcat=8.98:
Therefore, with hardly any electrostatics, the only explanation for a high kcat(O2·−) may be a very positive reduction potential for OsVIII/OsVII redox couple (not reported).
Manganese salts
Solvated Mn2+ is a fairly strong SOD mimic (phosphate buffer, pH 7.8) (Table 1). The log kcat(O2·−)>6.30 was reported. The Eoxid for MnII/MnIII oxidation is +850 mV versus NHE (17, 247, 249). Other salts, in particular Mn lactate [only 65-fold less potent than SOD enzyme (11)], are even better (28). The human body contains ∼10 mg of manganese, most of which is found in liver, bones, and kidneys. Chronic exposure to Mn levels can lead to a variety of psychiatric and motor disturbances, known as manganism (170). This is a likely reason for the lack of the use of manganese-containing drugs in medicine (15, 118, 230). Based on a number of studies, disturbed iron metabolism could underlie the neurotoxic action of manganese, resembling Parkinson's disease. Chronic exposure to Mn causes its accumulation in the nervous system, predominantly in basal ganglia, inducing a decrease in dopamine levels leading to cell death. Martins et al. reported that treatment of mice with MnCl2 in drinking water resulted in a 2.5–5-fold increase in catalase and SOD activities and was more pronounced in cortex and cerebellum than in hippocampus and striatum (170). The highest lipid and glutathione oxidation was observed in hippocampus. The high toxicity of Mn requires a thorough removal of “free” Mn from its formulations. We have addressed levels of free Mn in MnP preparations, left from the metallation of the porphyrin ligand (222, 224). Under in vivo conditions, no significant release of Mn is expected from stable porphyrins and corrole complexes; based on reported stability in aqueous solutions, an extensive loss of Mn from polyamines and salens is anticipated. The data on Mn loss from Mn complexes and its in vivo consequences has not been explored. Thus far, no metal-free porphyrin was detected in biological tissue (253).
For comparison, the values for some other compounds are also given. All Mn porphyrins are diaqua species, while Fe porphyrins are monohydroxo monoaqua species at pH 7.8. Water molecules are not indicated in the Table. The compounds are technically not SOD mimics if they disproportionate O2·− with a rate constant close to self-dismutation k=5×105 M −1s−1.
E1/2 is determined in 0.05 M phosphate buffer (pH 7.8, 0.1 M NaCl); bkcat in M−1s−1 was determined by cytochrome c assay in 0.05 M potassium phosphate buffer (pH 7.8, at 25±1 0C); cp.r., pulse radiolysis; dE1/2 data associated with the MnIV/MnIII reduction potential. eTovmasyan et al., unpublished; fvalue estimated based on cyt c assay for EUK-8 (249); IC50 ∼0.48 μM (NBT assay); gin acetonitrile, Mn(II) complexes were 1 mM, 0.1 M tetrabutylammoniumhexafluorophosphate (TBAPF6) as supporting electrolyte, Ref. (167); hRef. (28), the one-electron reduction potential refers to RNO+/RNO redox couple. idata in mV versus NHE, they are based on the data obtained in acetonitrile versus Ag/AgCl for MIV/MIII redox couple with 0.3 M tetrabutylammonium perchlorate (for MnDiM-4-PyMAn-Corrole3+) or 0.1 M TBAP/TBAPF6 (for FeTrF5Ph-β(SO3)2-Corrole2−) as electrolyte (80, 196), and the similarity of E1/2 in aqueous medium and acetonitrile (80), the E1/2 in mV versus NHE in aqeous medium was estimated; such data are plotted in Figure 4; jE1/2 for MnTrF5Ph-β(SO3)2-Corrole2− is converted to mV versus NHE from the value given in mV versus Ag/AgCl in 0.01 M phosphate buffer, pH 7.4, 3 M KCl as electrolyte, (80); kE1/2 for CeIV/CeIII (261). lOxidation potential only, MnIII/MnII redox couple is irreversible; When references are not indicated, the data are taken from Refs. (26 –28).
E1/2, half-wave reduction potential; M40403, cyclic polyamine; NHE, normal hydrogen electrode; SOD, superoxide dismutase.
FePs Versus MnPs
The key importance of the metal site in SOD enzymes has been extensively studied. Elegant work by Anne-Frances Miller and colleagues clearly describes how the metal coordination sphere is significantly more specific to the type of metal in enzymes than in their mimics (280, 281, 300). When Fe in FeSOD gets replaced by Mn (and vice versa), the coordination sphere, originally designed for Fe, is too rigid to allow the amino-acid residues to adopt the appropriate configuration around an incoming Mn. Consequently, the engineered enzyme has dramatically modified E1/2, and, therefore, its ability to catalyze O2·− dismutation is lost (Fig. 5).
However, in MPs, when Mn replaces Fe, axial coordination around Mn is readily re-adjusted due to the absence of amino-acid residues, which complicates the aqua/hydroxo protonation equilibrium of the metal center. The simple Fe-OH versus Mn-H2O axial modification provides appropriate reduction potential to maintain similarly high SOD-like activity of Fe and MnPs (Fig. 5). Apart from SOD activity, however, such a different axial coordination of the metal site results in a large difference between the aqueous chemistry of these two classes of MPs and, thus, influences their entire chemistry and biology as discussed next (273). The (OH)FeTM-4-PyP4+ was the first porphyrin reported to have SOD-like activity with log kcat(O2·−)=7.20 (27, 203). It is frequently used in vivo due to its commercial availability (12, 47, 77, 102, 135, 157, 188-190, 200, 256, 257).
The acidity of simple “free” ion, hexaaqua species [FeIII(H2O)6]3+ is pKa1 ∼2.2, and of [MnIII(H2O)6]3+ is pKa1 ∼0.1. This indicates that the Mn3+ is more acidic than the Fe3+ hexaaqua species by ∼2 log units; the consequence of this is the lack of stable Mn +3 salts. Such a remarkable difference in pKa1, as a result of different electronic ligand fields of Mn and Fe metal centers, translates into the difference between the metal-centered reduction potentials of MIII/MII redox couples (M=metal), and, eventually, into the major difference between the “free Fe” and “free Mn” biology. Due to a much higher reduction potential of “free” MnIII/MnII (Eo=+1.51 V vs. NHE) than of free FeIII/FeII (Eo=+0.77 V vs. NHE), Mn cannot be easily oxidized with H2O2 to produce •OH radical, which means that Mn does not undergo “Fenton chemistry.” On metal binding to SOD protein or its mimic, its reduction potential changes dramatically, falling between the potential for O2·− oxidation and reduction. Such a change in E1/2 is enabled by the changes in the metal coordination sphere (Fig. 5) (273).
The acidity of the MP axial waters with regard to the free water (pKw ∼14) is increased by ∼8.5 and ∼3 log units on coordination to the pentacationic FeP and MnP moieties, respectively. Such a large difference between the pKa1 values shows that the axially coordinated water in Fe(III) N-alkylpyridyl porphyrins is about 5 log units more acidic (pKa1 ∼5) than in the corresponding MnPs (pKa1 ∼11). This is a reversal relative to the acidity of Mn and Fe hexaaqua species. Consequently, at pH=7.8, the axial water in FePs (but not in MnPs) is deprotonated, giving rise to (OH)(H2O)FeIIIP. The (OH)(H2O)FeIIIP and (H2O)2MnIIIP species have almost identical E1/2. Consequently, their ability to catalyze O2·− dismutation is similar on thermodynamic grounds (Table 1). However, the hydroxo ligand labilizes trans-axial water. It is well known that such a trans-axial effect increases the reactivity of the metal site. Thus, kcat(O2·−) for FePs is larger than for MnPs (Table 1), as is the reactivity toward other molecules such as ascorbate, thiols, and peroxide (30, 272, 273). The difference is smaller with O2·− dismutation, as the process is predominantly outer-sphere (i.e., does not involve axial binding), with only partial inner-sphere character (273). Since axial hydroxo ligand neutralizes a single charge at the metal site, the FePs are tetracationic, while MnPs are pentacationic. We do not yet know the contribution of the lower electrostatics on the kcat(O2·−) of FePs relative to MnPs. Lengthening the alkyl chains from 1 to 8 carbon atoms has a large effect on E1/2 of MnPs, whereas it has a significantly smaller effect on the E1/2 of FePs. The E1/2 of FePs appears to be predominantly determined by the axially bound OH− ligand and not by the peripheral pyridyl substituents (Fig. 6). Such a difference in axial coordination translates into a remarkably different SAR of the isomeric Mn(III) and Fe(III) N-alkylpyridyl porphyrins (Fig. 6, shown only for ortho isomers). The E1/2 is inversely related to the pKa3 of the pyrrolic nitrogens of porphyrin ligand (30) and to the pKa1 values of axial waters as exemplified for (OH)(H2O)FeTE-2-PyP4+ versus (H2O) 2MnTE-2-PyP5+ in Figure 6. In other words, the higher the E1/2, the more electron deficient the metal is and the more strongly it binds the oxygen atom of axial water and pyrrolic nitrogens. Consequently, the protons (of both axial water and pyrrolic niytrogens) leave at lower pH; in turn, the pKa values are lower. Thus, the E1/2 is an excellent measure of the acidity of the metal center.
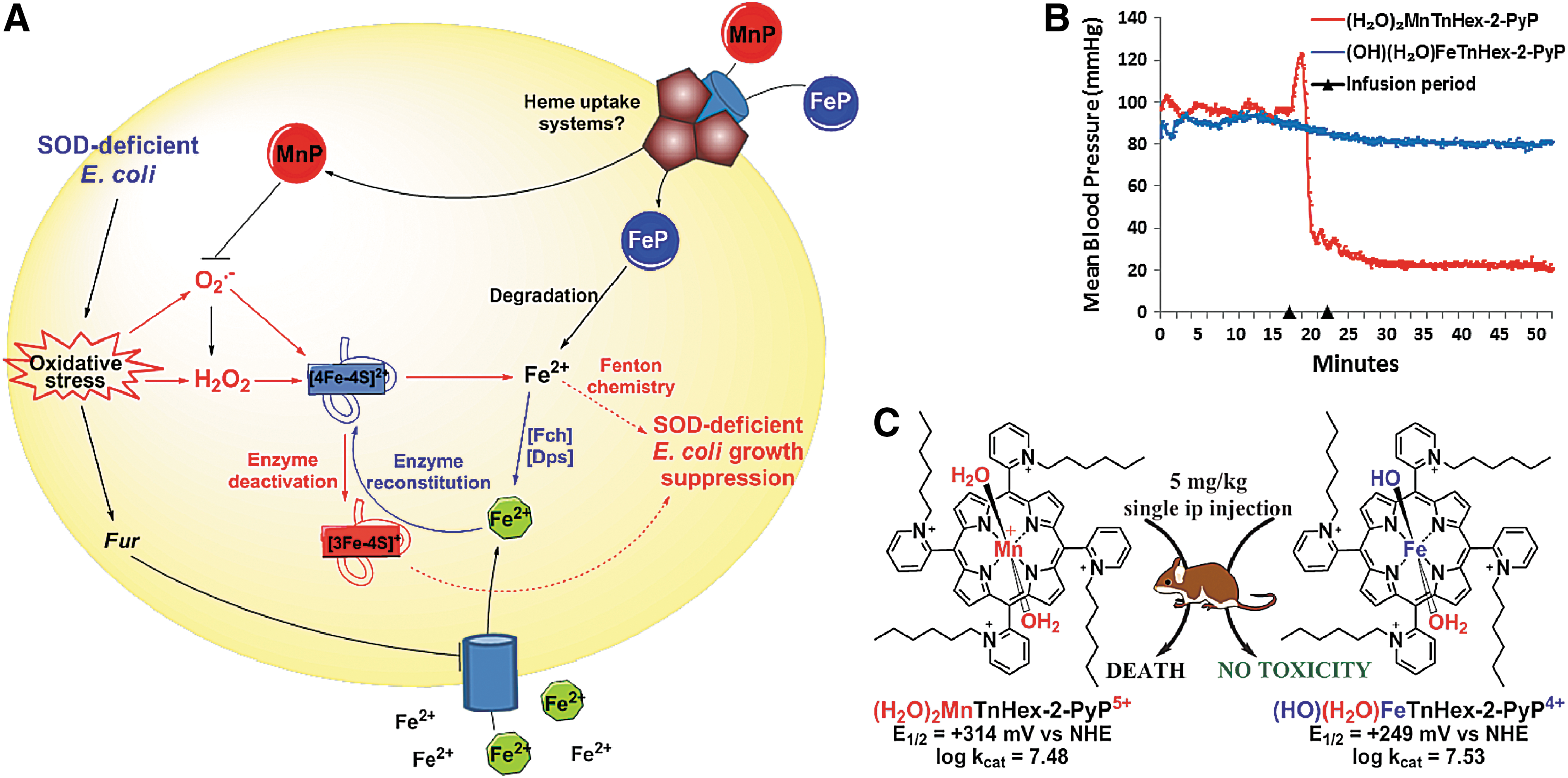
Due to their high SOD-like activity, FePs were evaluated on their ability to protect SOD-deficient E. coli when growing aerobically (Fig. 7) (30). SOD-deficient mutant lacks cytosolic SOD enzymes MnSOD and FeSOD and cannot grow as efficiently as wild type under aerobic conditions and in a restricted medium where the syntheses of branched, aromatic, and sulfur-containing amino acids are catalyzed by superoxide-sensitive enzymes (26, 28). Under identical concentrations (∼20 μM), MnPs were protective, while FePs were toxic (30). However, FePs were efficacious at approximately ∼1000-fold lower, 0.01 μM concentrations (270, 273). Further, the E. coli growth pattern is different with FePs relative to MnPs. Initially, the growth delay was seen with FePs; eventually, the FeP-supported growth surpassed the growth of wild-type E. coli (Fig. 7). Mn and FePs have similar lipophilicities, bulkiness, and kcat(O2·−) values (Table 1). Therefore, had they acted as SOD mimics, they would have been protective under identical conditions as MnPs (Table 1).

Further studies demonstrate that the identical impact on the growth of SOD-deficient E. coli was produced with equal concentrations of Fe(II) citrate, Fe(II) sulfate, and FePs: 0.1 or 1 μM (272, 273). The metal/porphyrin stability studies in aqueous system in the presence of ascorbate, glutathione, or cysteine followed in order to gain insight into the possible degradation of MP (272). Hydrogen peroxide is the product of redox cycling of MPs with the reducing agents, and it degrades (“bleaches”) the FePs more readily than MnPs. Subsequent studies with E. coli clearly showed that FeP, but not MnP, undergoes fast degradation during the first several hours of E. coli growth. At lower levels of free Fe, the E. coli likely uses it to restore 4Fe-4S clusters of Fe-bearing enzymes (such as aconitase). Upon the attack of O2·− these enzymes reversibly lose Fe2+. A recent manuscript by Gu and Imlay substantiates the impact of O2·− on Fe-containing enzymes in SOD-deficient E. coli; at least 3 more enzymes (threonine dehydrogenase, ribulose-5-phosphate 3-epimerase, and peptide deformylase) were clearly identified as undergoing loss of Fe and subsequent inactivation (101). Inactivation is, in part, related to the Zn incorporation at the Fe site. Fur protein also seems to be a candidate (101). At higher levels of Fe, the toxicity is most likely due to the Fenton chemistry-driven·OH production at the metal site of FeP or at “free” low-molecular Fe (273). Were the mechanism of action of FePs versus MnPs indeed as different in mammalian systems as shown in E. coli, our present understanding of the favorable effects often exerted by FePs in animal models of disease would need reconsideration (12, 47, 77, 102, 256). The E. coli data illustrate the complexity of the biology of redox-compatible metal complexes and call for caution when interpreting the in vivo data. Studies on cells other than E. coli are in progress. The scheme that represents differential actions of FeP versus MnP in SOD-deficient E. coli and in mouse is shown in Figure 8.
In addition to Fe(III) N-alkylpyridylporphyrins, Fe complexes, which bear ortho pyridyl nitrogens quaternized with triethyleneglycols and benzoates, have been explored (18). The nature of the N-pyridyl substituents only marginally affects the magnitude of kcat(O2·−). Though discussed as exclusive ONOO− scavengers, these compounds are as good superoxide scavengers as N-alkylpyridylporphyrins (28, 146, 172, 173, 187, 214, 215, 244, 258). The FeTSPP3− and its mesityl analog FeTMSP7− were also reported as specific ONOO− scavengers (241). FeTM-4-PyP5+ has often been described as a peroxynitrite decomposition catalyst (77, 157, 187). FeTSPP3− and FeTMSP7− are anionic and have fairly low E1/2 (E1/2=+0.023 mV vs. NHE for FeTSPP3−) and irreversible redox, and are thus poor SOD mimics, but can still reduce ONOO− (due to its high oxidizing power), though with modest rate constants (241).
Drug Reactivity
Reactivity toward reactive species other than O2·−
Figure 9 and Table 1 summarize available data on the SOD-like activity of different redox-active compounds toward small molecules; the reactivity toward protein thiols is discussed under Reactivity toward cellular reductants and Reactivity toward signaling proteins sections. What is shown in Table 1, and below (under reactivities toward different small and large reactive species), is perhaps only a small fraction of their reactivities in a complex milieu of a cell. Based on our present knowledge, it is incorrect to use a single redox-active compound such as MnP as a sole tool to specifically prove the involvement of a single reactive species in a certain pathological disorder. The reason for SOD-like activity, as well as for other reactivities, lies in the electron-deficient (electrophilic) nature of Mn(III) N-alkylpyridylporphyrins that favors the reaction and/or binding of electron-rich anionic ligands such as ONOO−, ClO−, HO2 −, RS−, and HA−. Once such a porphyrinic electrophile reaches the cell, the data show that it readily undergoes reduction while oxidizing abundant ascorbate and thiols. In subsequent reoxidation of MnP with O2 or O2·−, and oxidation of ascorbate radical to dehydroascorbate, the peroxide is produced. MnP can employ peroxide and/or GSH to inactivate nuclear factor κB (NF-κB) (82, 125, 127, 238, 240), suppress mitochondrial respiration (124) and glycolysis (70, 124), or induce adaptive responses (52, 75). Such pro-oxidative actions agree well with what Forman et al. recently put forward as the mechanism of action of natural antioxidants (89). The final outcome will largely depend on the colocalization of MnPs with reactive species and their concentration levels. Given the complex cellular mileu, the existence of subcellular fragments, and the complex redox chemistry of redox-biocompatible compounds, it is difficult to impossible to understand the full biological reactivity of MnPs. The details are given next.

Reactivity toward ONOO−
The log kcat(O2·−) is directly proportional to log kred (ONOO−) (28, 84, 86). Thus, potent SOD mimics are potent peroxynitrite scavengers (Eqs. [15] and [16]). We have shown with MnPs that the reason lies in the fact that the most potent SOD mimics are electron-deficient and that such MnPs would favor reactions and/or binding of electron-rich anionic ligands such as ONOO−. The reactivity of pentacationic MnPs toward superoxide [kcat (O2·−)] is higher than toward peroxynitrite [kred (ONOO−)]; pentacationic MnP would prefer O2·− compared with ONOO− if it encounters both species under identical concentrations. The log kcat (O2·−)=7.76 for MnTE-2-PyP5+ at 250C, while its log kred(ONOO−)=7.53 but at 37°C (28). Once oxidized to O=MnIVP with ONOO−, MnP would regenerate itself as a catalyst with cellular reductants acting as electron pools and sparing biological targets from highly oxidizing Mn oxo species (86).
In the reaction with ONOO−, MnPs can cycle either one-electronically via O=MnIVP/MnIIIP redox, producing highly oxidizing radical, ·NO2 (Eq. [15]), or two-electronically via O=MnIVP/MnIIP redox (Eq. [16]), producing benign nitrite NO2 − (80, 196). The MnPs are in vivo maintained in reduced Mn +2 state by cellular reductants. Thus, the 2-electron- is more likely than the one-electron reaction.
The compounds with negative E1/2 (such as MnIIITBAP3−, E1/2=−194 mV vs. NHE) cannot participate in a 1st step of O2·− dismutation (Eq. [1]). However, MnIIITBAP3− can be oxidized to O=MnIVP with ONOO− (and perhaps other strong oxidants ClO−, H2O2, and lipid radicals), which may explain its reported in vivo efficacy (19). The log kred (ONOO−)=5.02 for MnTBAP3− is >100-fold lower than of MnTE-2-PyP5+ (log kred=7.53) (28, 223). ONOO− can also oxidize Mn texaphyrin with even lower kred(ONOO−)=3×104 M −1s−1 (84, 196, 242) and some other MnPs, such as Mn(III) meso-tetracyclohexenylporphyrin as well as biscyclohexano-fused Mn(III) complex of bis(hydroxyphenyl)dipyrromethene (217, 218). The other compounds that cannot be oxidized by O2·− are MitoQH2 (quinol), MitoQH (semiquinone), and nitroxides (177). Once it reaches mitochondria, the MitoQ readily gets reduced to MitoQH2 by components of the electron transport chain (177). MitoQH2can be oxidized by ONOO− to MitoQH, which then dismutes (disproportionates) to MitoQ and MitoQH2. Only MitoQ (quinone) and oxoammonium cation react with O2·− with high rate constants to yield MitoQH and nitroxide, respectively (177). Nitroxides get oxidized with the degradation product of ONOO−, CO3·− and ·NO2, and protein-derived radicals, thiyl and peroxyl radicals giving rise to oxoammnium cation (28). Oxoammonium cation though reacts with O2·− (28). Mn(III) cyclic polyamines are often reported as specific to O2·− (59, 169, 171, 186, 228, 267). The reactivity of Mn cyclic polyamine M40403 toward ·NO has been reported (87). Mn salen derivatives show a wide range of reactivities toward ONOO−, H2O2, ClO−, and ·NO (74, 132, 234). The MCs are reactive toward O2·− and ONOO− (28). The reactivity of cerium oxide (nanoceria) toward O2·−, H2O2, and ·NO has been reported (28). Work in progress shows that potent SOD mimics are also very reactive toward ClO− (109). One can easily envision that many other reactions are possible with each of those compounds within cells. Thus, we can only safely predict which reaction is possible but not which will occur.
MCs employ MIVC/MIIIC or O=MVC/MIIIC to cycle with O2·−, H2O2, and ONOO− (see earlier under SARs for diverse redox-active compounds—Mn(III) corroles). Similar to MnPs, two-electron oxidation with ONOO− would lead to benign NO2 − production (162). Reduction of strong oxidizing high-valent Mn will be achieved at the expense of reductants acting as electron pools.
Reactivity toward ·NO
Cationic Mn(III) N-substituted pyridylporphyrins favor reactions with ·NO; at submicromolar concentrations and at 1:1 ratio, a fairly stable complex is formed, (NO)MnIIP, with Mn in +2 oxidation state (Eq. [18]). The reaction is very slow with t1/2 ∼60 min; the same product is formed much faster if MnIIIP5+ is first reduced with cellular reductants such as thiol or ascorbate (HA− and RS−, monodeprotonated species are major ones at pH 7.8) (Eq. [17]). The oxidation of cysteine (similar to the Eq. [17b] shown for glutathione) by MnIIIP resembles the reported thiol oxidase action of SOD enzyme (294). The rate constant for nitrosylation of MnP (Eq. [18]), estimated by stopped flow, is k ∼106 M
−1s−1. The complex slowly undergoes the oxidation, whereby initial MnIIIP gets restored and nitrate is eventually formed (Eqs. [19] and [20]). MnIIP4+ can undergo oxidation with either oxygen or superoxide (the latter reaction being 2nd step of O2·− dismutation process), depending on their relative levels in cells. The reaction of MnIITE-2-PyP4+ with O2 was estimated to occur with a rate constant of ∼8×104 M
−1s−1 (248). Axially coordinated waters are omitted in the equations for simplicity, as no protonation equilibria are involved at the Mn site:
The ·NO scavenging by MnP may affect cellular signaling pathways. The ·NO binding shifts the reduction potential for the MnIIIP/MnIIP redox couple by +600 mV (133, 184, 276), stabilizing Mn in +2 oxidation state. The reactivity of nitrosylated (NO)MnIIP4+ toward O2·− is presently under investigation.
Reactivity toward H2O2
Hydrogen peroxide (H2O2) is a strong oxidant; thus, very few organic ligand-containing compounds are stable enough in its presence to be efficient catalysts of its dismutation. In vivo H2O2 is produced by different oxidases, such as NADPH- and xanthine oxidases, and is also a product of O2·− dismutation during mitochondrial respiration. Under nM levels, it is a main signaling species due to its long life, lack of charge, and ability to cross cellular membranes (90). It is also a source of other highly oxidizing species, such as·OH radical, alkoxyl (RO·) and peroxyl (RO2·) radicals, and protein thiyl radicals (89, 109).
The preliminary studies on the catalysis of H2O2 dismutation (producing H2O and O2) indicate that the log kcat(H2O2) is in the range of 1–2 log units for several MnPs [MnTE-2(and 3)-PyP5+, MnTBAP3−, and MnTnHex-2(and 3)-PyP5+] (Weitner et al., unpublished). Data agree fairly well with those reported from Fridovich's group for MnTM-4-PyP5+ and MnTBAP3− (66). Due to the excessive bleaching, Mahammed and Gross were not able to assess the kcat(H2O2) for FeTSPP3− (161). Starting with an FeP, containing three mesityl groups on porphyrin meso positions, Nocera's group added the fourth meso pendant group producing structure with kcat(H2O2) increased ∼3000-fold relative to Fe(III) tetrakis-meso(2,4,6-methylphenyl)porphyrin (49). The rate constant for the reduction of H2O2 to H2O with MnTDE-2-ImP5+ at pH ∼7 (128) is low, kred(H2O2) ∼102 M −1s−1, and is reportedly faster with less electron-rich MnTM-2-PyP5+ (30).
Corrole is a trianionic ligand, while porphyrin is dianionic; as a result, the metal site is more electron rich in MCs, giving rise to more stable complexes in reduced metal +3 oxidation state with regard to the loss of metal (Fig. 10). Further, after the reaction with H2O2, metal in MCs is stabilized much more in higher metal +5 oxidation state relative to MPs; consequently, Fe corroles are more efficient catalysts of H2O2 dismutation (161). The log kcat(H2O2) of 3.8 was reported for Fe corrole-bearing pentafluorophenyl groups on three meso positions and two sulfonato groups at neighboring pyrrolic rings (161). Although possessing only ∼0.6% of enzyme activity [assuming log kcat ∼6 for enzyme (109)], the Fe corrole may still have the highest kcat(H2O2) among the synthetic redox-active compounds. Whether and how this affects its therapeutic efficacy is not clear. When an aqueous solution of a compound was given orally at 20 mg/kg/day for 7 weeks, it prevented cataract incidents, favorably affected kidney function, and decreased serum cholesterol and triglyceride levels. Such a study suggests sufficient stability of that compound toward the proton-dependent loss of metal. As much as 300 mg/kg caused only a mild adverse effect (103).
Under physiological conditions, H2O2 is maintained at nM steady-state levels by abundant peroxide-removing enzymes. Under pathological conditions, particularly in cancer, some of the enzymes are reportedly down-regulated (8, 93, 191, 233, 235, 245). The reaction of MnP with oxygen (Eq. [21]), superoxide (Eq. [2]) or cycling with ascorbate (Eq. [17a]) or thiols (Eq. [17b]) would result in increased levels of H2O2. Subsequently, MnP can utilize H2O2 to glutathionylate thiols of subunits of critical anti-apoptotic transcription factor, NF-κB (124). Tome's group demonstrated that the glutathionylation occurs only in the presence of H2O2 and GSH and could be best presented with reactions [22] through [24], where MnP acts as glutathione peroxidase, GPx. This agrees with reported GPx-like activity of para isomer, MnTM-4-PyP5+ (9). A reaction given by equation [22] is proposed by Jin et al. for MnTDE-2-ImP5+, which is similar in reactivities to MnTE-2-PyP5+ (128). The reaction described by Eq. [24] presents the disulfide interchange reaction (109):
The anticancer radiation or corticosteroid-based therapy gives rise to high levels of H2O2. When administered along with MnP, the metal complex would catalyze glutathionylation of p65 subunit of NF-κB by H2O2 and GSH. Use of the redox-active metal complex along with the source of H2O2 has been proposed by us and others (268, 299) as a prospective anticancer treatment.
The Mn salen derivatives have reportedly the advantage because of their dual SOD and catalase-like activities (74). While the first is fair [log kcat(H2O2) ∼6, Table 1] (34, 74, 249), the catalase-like activity is, however, insignificant, representing only ∼7×10−5% of the enzyme activity (1, 73, 74, 98, 132, 202). Besides unfavorable thermodynamics, the low metal/ligand stability of Mn salens disfavors high catalase-like activity.
Reactivity toward cellular reductants
Due to the biocompatible E1/2 and favorable electrostatics (26, 28, 177), the pentacationic electrophilic Mn(III) N-substituted pyridylporphyrins rapidly react with anionic deprotonated forms of cellular reductants: ascorbate, glutathione, cysteine, and protein thiols (29, 33, 299). In such reactions, the MnPs act as oxidants. When undergoing oxidation with strong oxidants (such as ONOO− or H2O2), the highly oxidizing Mn in +4 and +5 oxidation state is formed. It gets reduced back to either Mn +3 or +2 oxidation states while oxidizing reductants (instead of biological targets) that serve as suppliers of electrons (Eq. [25]) (38, 84, 86, 277).
These reductants also likely couple with MnP in removing O2·− in vivo; consequently, the MnP may act as superoxide reductase rather than as SOD (Eq. [17] as a first step and Eq. [2] as a second step). The antioxidative effect of MnP in removing O2·− is assured only with sufficient physiological levels of enzymes that are capable of removing the H2O2. Otherwise, similar to cancer cells in which such enzymes are frequently down-regulated, the redox cycling of MnP with ascorbate or glutathione or cysteine would result in the accumulation of peroxide and its involvement in the oxidation of biological targets.
Ascorbate
The antitumor potential of ascorbate was demonstrated in a number of tumor cell lines and in animal models only when given intravenously (iv) or intraperitoneally (ip) (44 –46, 82, 116, 151, 199). Supplementation of ascorbate to an immunodeficient mouse with rapidly spreading 9l glioblastoma reduced tumor growth and weight by 41%–53%; in 30% cases, cancer spreads to other organs, while no dissemination of cancer was seen in ascorbate-treated mice (46). Ascorbate antitumor action was assigned to its oxidation and subsequent cytoxic H2O2 formation, catalyzed by endogenous metalloproteins (116). A much higher yield of peroxide may be achieved if catalysts are optimized for ascorbate oxidation. Such are isomeric Mn(III) N-substituted pyridylporphyrins; their H2O2-producing potency has already been shown by us and others (82, 220, 273, 299) (Fig. 11).

Ascorbate distributes into cells via SVCT1 and SVCT2 transporters, whereas GLUT transporters are responsible for the dehydroascorbate uptake (55). Decreased tumor ascorbate levels in endometrial cancer have been associated with high hypoxia inducible factor-1α (HIF-1α), vascular endothelial growth factor (VEGF), and GLUT favoring tumor progression (141).
We were the first to show, using several tumor cell lines, that the bio-compatible pentacationic electron-deficient ortho isomeric MnPs catalyze ascorbate oxidation while producing high amounts of peroxide and exerting cytotoxic effects [Roberts et al., unpublished (299) (Fig. 12) (see also Reactivity toward signaling proteins section). With two normal cell lines, either none or lower cytotoxicity relative to tumor cell was demonstrated (Fig. 12). A normal cell is well equipped with multiple H2O2-removing systems such as catalases, glutathione peroxidases, glutathione transferases, peroxyredoxins, and thioredoxins. Tumor cells, however, with a lower buffering capacity, are much more sensitive to any further increase in oxidative stress (Fig. 12) (8, 35, 93, 142, 177, 191, 233, 235, 245). In a cell culture, the MnP/ascorbate showed promise as potential treatment of inflammatory breast cancer; the noncaspase-, but AIF- and NF-κB-based apoptosis was promoted (82). Even when the SOD enzyme was up-regulated, the suppression of tumorigenesis was demonstrated by Tome et al., which is likely due to the down-regulation of peroxide-removing enzymes (40). The rapamycin increased levels of reactive species in a cellular model of mantle cell lymphoma (Grant519 and NCEB1) via inhibition of mTORC2 signaling. These cells presumably have insufficient levels of MnSOD, which would have otherwise reduced tumor progression. The increased levels of reactive species consequently up-regulate the MnSOD enzyme. The up-regulation of MnSOD increases the levels of reactive species, which suppresses the tumor cell growth (41).
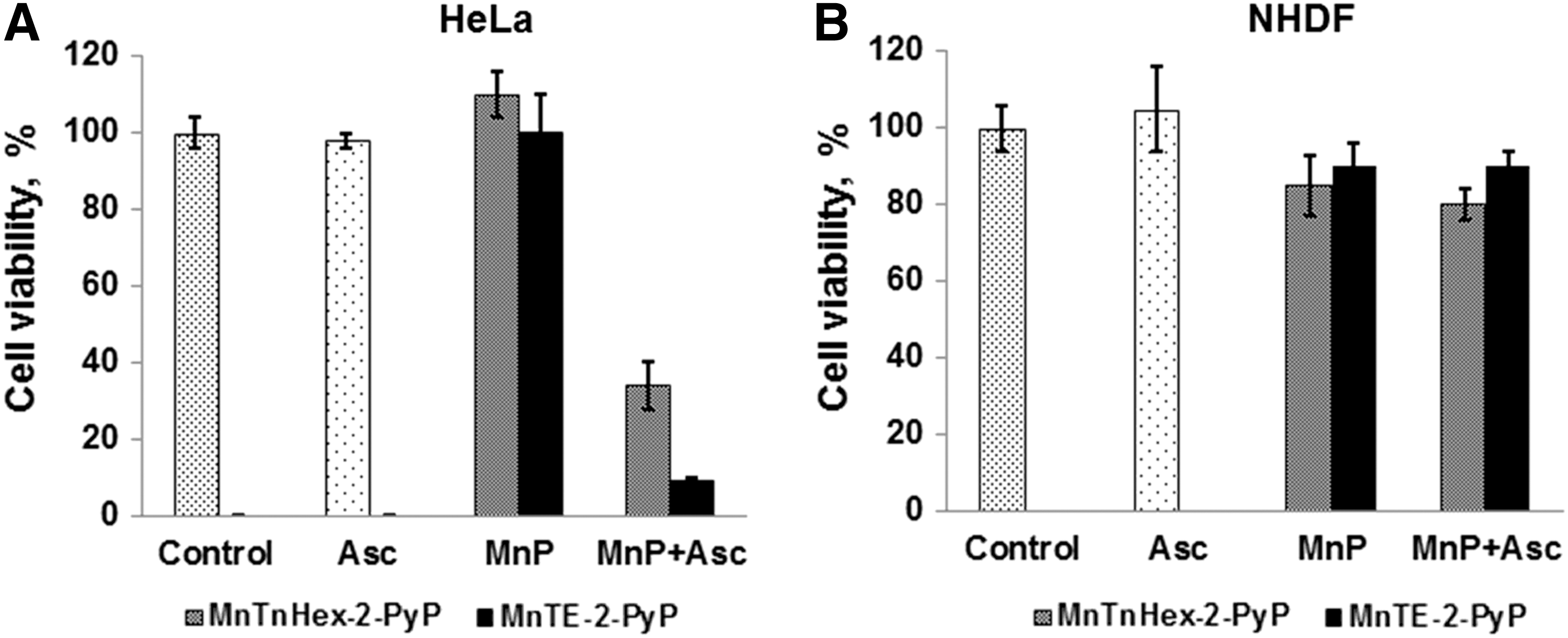
The para isomeric MnPs, such as MnTM-4-PyP5+, could be a better catalyst for ascorbate oxidation than their ortho analogs when judged solely on a thermodynamic basis. The E1/2 of +60 mV versus NHE shows that, once reduced, the MnTM-4-PyP5+ more readily reoxidizes, thus producing H2O2. The ortho analog MnTE-2-PyP5+ with 162 mV more positive E1/2 is more electron deficient and prefers residing in a reduced state. Further, the para compounds are planar and, thus, favor intercalation within groves of nucleic acids, which may prevent their action and/or cause toxicity to normal tissue (18, 220). Also, it would undergo oxidative degradation faster then ortho analog (30, 128). Rawal et al just reported the tumor suppression in sc nude mouse xenograft model of human pancreatic cell line (220).
It is important to note that the reduction of MnIIIP5+ with ascorbate reduces a total charge of MnP from penta- to tetracationic and, thus, enhances lipopilicity and cellular and tissue accumulation (251, 270). In cellular studies, the MnP/ascorbate was added exogenously into medium where it produced peroxide outside of the cell. Thus, under such conditions, no impact of the magnitude of MnP lipophilicity on its therapeutic efficacy was observed (299). In an E. coli model, however, the accumulation of ascorbate-reduced tetracationic MnP is greatly enhanced (251, 270). Thus, in addition to appropriate thermodynamics and kinetics of MnP/ascorbate redox system, the bioavailability of MnP would play a role in in vivo studies.
Welsh et al. just completed Phase I Clinical Trials in pancreatic cancer, where ascorbate was administered along with gemcitabine (291). Pancreatic cancer is the fourth leading cause of death in the United States, with 80% of mortality. The data obtained from Phase I are enthusiastic and warrant Phase II Clinical trials. As compared with an earlier report by Monti et al. (181), Welsh et al. infused ascorbate to achieve at least 20 mM levels in plasma, which would assure the antitumoral effect of ascorbate based on an earlier study of Du et al. (78). While Monti's study lasted only 8 weeks, to assure the safety and tolerability, the Welsh et al. study was several months long. In the absence of dose-limiting toxicity, the treatment was continued until progression (defined by Response Evaluation Criteria in Solid Tumors, RECIST). Even with only 8 weeks of ascorbate administration, the reduction in tumor volume was observed in 8 out of 9 patients in the Monti et al. study (181). The progression-free survival and overall survival was 12.7 weeks and 6 months with Monti et al. and 26 weeks and 12 months with Welsh et al. (181, 291). It is important to note that the high levels of ascorbate and peroxide did not induce systemic oxidative stress, as levels of F2-isoprostanes remained the same or were reduced. The same was true for blood cells glutathione, which was unchanged as was the half-cell reduction potential. The ascorbate radicals were undetectable before treatment, while they were markedly increased in treated patients.
Buettner's group has also reported that ascorbate acts as a radiosensitizer in pancreatic cancer via production of reactive species (4, 290). The group is further exploring the role of transition metals and their porphyrin complexes on ascorbate-induced cytotoxicity in cancer (220).
Thiols
Reactivity toward glutathione and N-acetylcysteine has been addressed by Ferrer–Sueta et al. and by us (33, 85, 272). Reactivity of different MnPs (varying in total charge and E1/2) toward cysteine (cys-62) residue of p50 subunit of NF-κB protein in a cell-free system (thus GSH free), resulting presumably in disulfide formation, has been demonstrated by Tse et al. (33, 278) (See under Reactivity towards signaling proteins section). The S-glutathionylation of another p65 subunit of NF-κB has been demonstrated in a cellular lymphoma study by Jaramillo et al. (125). The opposite was recently demonstrated by Fridovich–Keil's group, where MnTE-2-PyP5+ prevented the protein glutathionylation in a galactose transferase Drosophila melanogaster knock-out when growing in the presence of galactose (130). In the absence of galactose no effect on glutathionylated proteins of GALT-null, but increase in their levels in controls were seen. When catalyzing the glutathionylation, the MnP acts as a pro-oxidant, producing the GS· radical. The GS· then combines with another GS·, giving rise to GSSG; or, following the Winterbourn electron sink pathway, the GS·may react with GSH, giving rise to GSSG·radical. The GSSG·radical, by oxidation with O2, forms GSSG and O2·− (292). The GSSG can exchange glutathione spontaneously with protein thiol whereby glutathionylated protein, protein-S-S-G will be formed. However, this may be prevented if GSSG is safely removed by glutathione reductase. Tome's group has reported that H2O2 and GSH are essential for glutathionylation to occur (124). The cancer cells and a genetically modified GALT-null Drosophila, due to differential levels of peroxide, would exhibit a differential outcome: one, higher and the other, lower levels of protein glutathionylation, respectively.
Reactivity toward signaling proteins
The investigations of the effect of MnPs on the up-regulation of numerous cytokines (33) made it clear that either direct reactions with reactive species (which supposedly signal the start of the cellular transcription) or direct reactions with signaling proteins may be involved. The driving force for understanding such observations was the fact that MnPs were as efficient in reducing mouse or rat infarct volume when given at early (6 min) or at late time points (90 min or 6 h) after reperfusion (238 –240). Our knowledge at the time when the observation was made would predict that only immediate infusion of porphyrin into the brain at the time of reperfusion (the moment of peak production of reactive species) would ameliorate the primary oxidative damage. The protection demonstrated with delayed treatment, however, suggested that MnP must have suppressed the cellular transcription which would have otherwise perpetuated the oxidative stress (Fig. 13). Further exploration indicated that at least one major transcription factor, NF-κB, has been inactivated by the action of MnP (240). Similarly, the radioprotective effect of MnP was demonstrated when it was given for the duration of two weeks starting at any time postradiation from 2 h up to 8 weeks (96). HIF-1α and the genes it controls (VEGF, TGF-β) have been involved (95, 96). In cancer studies, the impact of MnP on activator protein-1 (AP-1) has been shown (304). In diabetes again, NF-κB and SP-1 have been identified as targets of MnPs (33, 278). Based on earlier and ongoing studies and our growing knowledge on MnPs, there is more to be identified.
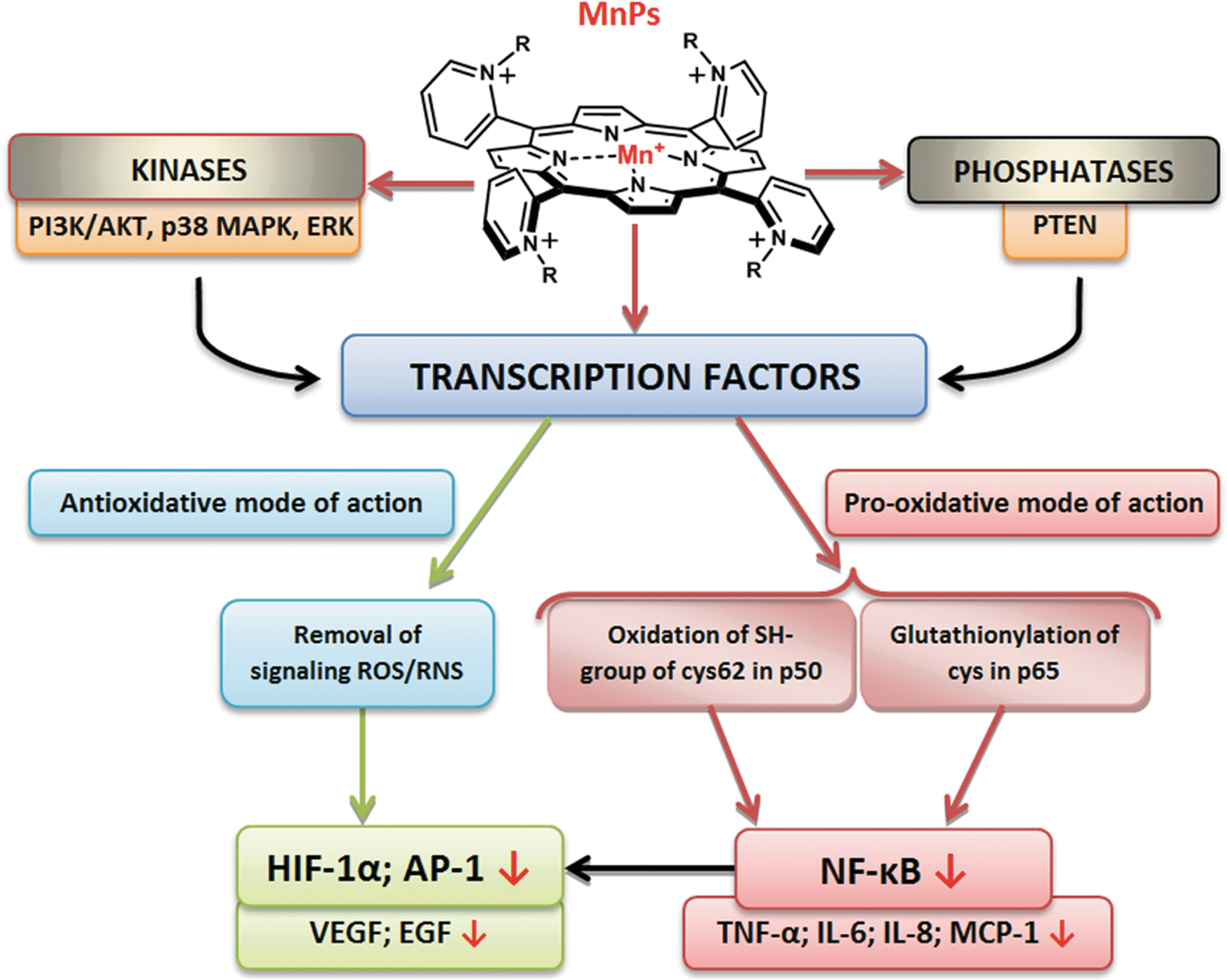
The mechanism of MnP-transcription factor interactions is not fully understood. We have initially assigned the effects of MnP almost exclusively to the removal of signaling reactive species (O2·−, ONOO−), which resulted in the suppression of the cellular transcriptional activity. However, the most recent data suggest the direct and peroxide-driven oxidation of signaling proteins as a prevailing action of MnPs; depending on the cell type, the therapeutic outcome may be anti- or pro-oxidative (Fig. 13).
NF-κB—diabetes-and stroke-related cellular and animal studies
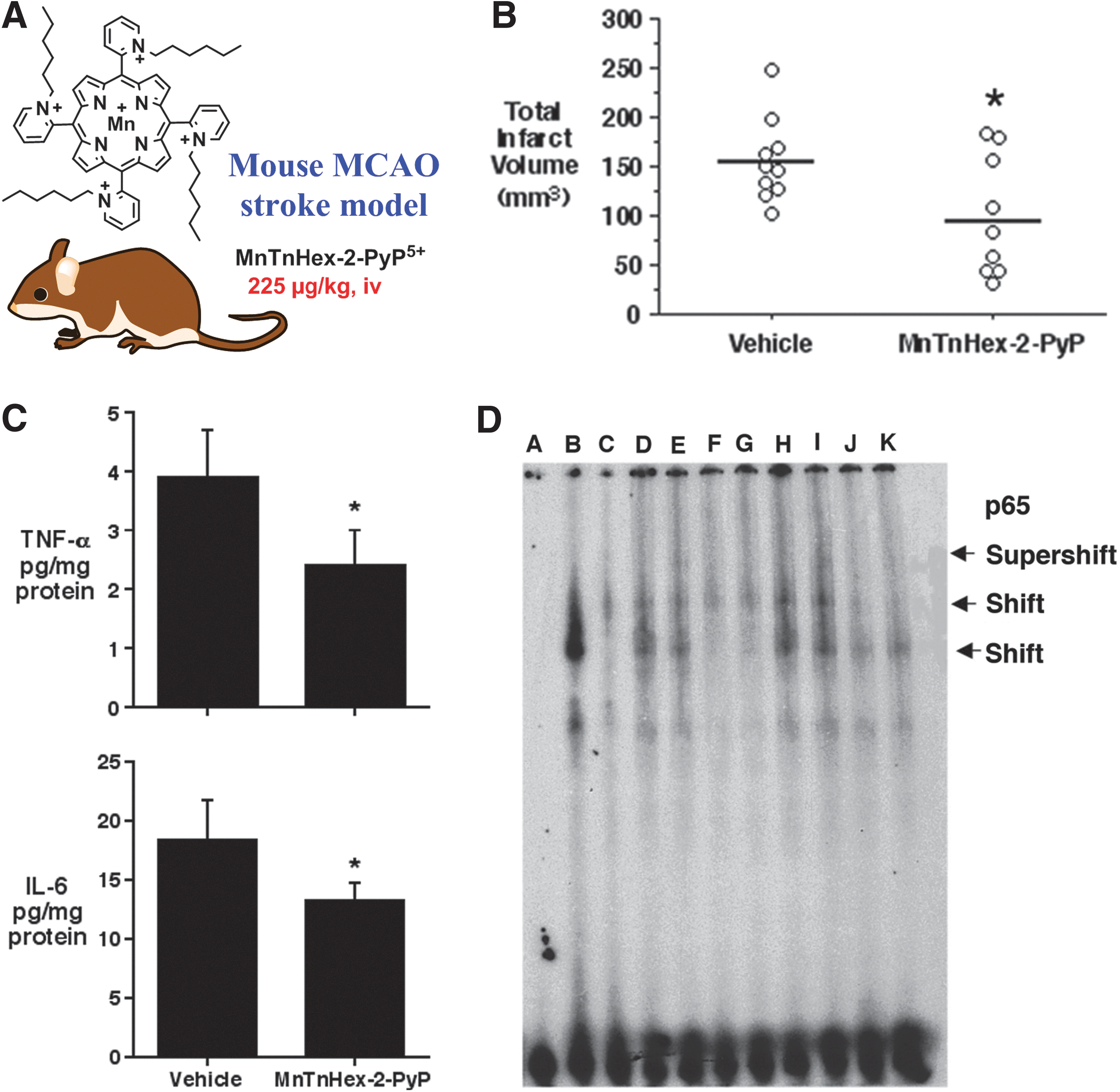
MnTDE-2-ImP5+ and MnTE-2-PyP5+ inhibited NF-κB activation in nuclear extracts of LPS-treated bone marrow-derived macrophages, and, therefore, suppressed pro-inflammatory cytokine production INF-γ and TNF-α. These results were recapitulated in human pancreatic cells cultured for 30 min in medium containing proteolytic enzymes and byproducts generated during cell isolation. The addition of 34 μM MnTDE-2-ImP5+ to isolated islet cells increased their survival and reduced levels of pro-inflammatory cytokines IL-6 and IL-8. Monocyte chemoattractant protein 1, MCAP-1 as well as PARP (poly(ADP-ribose)[polymerase]) were greatly suppressed and islets gained the capacity to normalize diabetic recipient mice (39, 40, 209). MnTE-2-PyP5+ also significantly delayed or prevented the diabetes altogether upon the treatment of young nonobese diabetic-severe combined immunodeficient mice with diabetogenic T-cell clone, BDC-2.5. All other aspects of NF-kB pathways were not affected, such as IKKα/β phposphorylation, and IKβ-α phosphorylation/degradation and p50/p65 nuclear translocation. Two additional experiments provided key support that MnP acted as a pro-oxidant, oxidizing cysteines whereby the antioxidant effects (listed earlier) were observed: (i) in a cell-free experiment (in the absence of GSH), MnP oxidized Cys-62 of p50 subunit of NF-κB, whereby disulfide formed prevented NF-κB activation (33, 278); (ii) in aqueous potassium phosphate-buffered solution, when only MnP and either cysteine or N-acetylcysteine or glutathione were present, the spectrophotometric evidence supported one-electron reduction of MnP (33). In vivo studies suggest that such NF-κB modification happens in the nucleus (33); data are supported by the biodistribution study in which MnTE-2-PyP5+ accumulated in the nucleus of macrophages at a 3-fold higher level than in the cytosol (33). Further on, the inactivation of NF-κB seems to play a major role in suppressing stroke injury with 2 hydrophilic and one lipophilic MnP. While in earlier studies hydrophilic MnPs were given intracerebroventricularly, most recently for the first time, the effect was demonstrated when lipophilic MnTnHex-2-PyP5+ was given subcutaneously for 6 days after initial iv bolus dose given at 30 minutes postreperfusion (Fig. 14) (238, 240). Again, similar to diabetes, the pro-oxidative action of MnP in stroke accounts for the antioxidative therapeutic outcome.
AIF—cardiomyocyte study
An elegant study of Miriyala et al. of a mouse model of doxorubicin-induced cardiac toxicity has shown the effect of mitochondrially located MnPs upon apoptosis inducing factor—mitochondrion-associated protein (AIFm2) (179). The AIFm2 is a p53 target gene and an AIF homologue. It appears to be a redox-responsive protein that resides in the mitochondria and plays a central role in caspase-independent cell death pathway (37, 153, 168, 194, 283, 295). The expression of AIFm2 is relatively low in tumor cells versus normal cells, suggesting the tumor-suppressive role of AIFm2 (178, 179, 296). If translocated into the nucleus, it serves as NADH-dependent oxidoreductase and is capable of nonsequence specific DNA binding, resulting in DNA fragmentation, that is, apoptosis. AIFm2 translocated into the nucleus after it formed the adduct with the product of lipid peroxidation, 4-hydroxynonenal. However, the three MnPs, MnTE-2-PyP5+, MnTnHex-2-PyP5+, and MnTnBuOE-2-PyP5+, which accumulate in mitochondria (see below under Bioavailability studies section), prevented the doxorubicin-induced mitochondrial lipid peroxidation, the 4-hydroxynonenal formation, and the formation of its adduct with AIFm2. Consequently, the translocation of AIFm2/4-hydroxynonenal adduct into the nucleus of H9C2 cardiomyocyte, as well as the initiation of the apoptosis was fully inhibited by MnPs (179). This appears to be a clear case of an antioxidative action of MnPs.
mTOR, c-Myc, and glucose-6-phosphate dehydrogenase
The studies of diabetogenic immune cells showed that MnTE-2-PyP5+ impedes diabetogenic autoimmune responses by restricting metabolic pathways for energy production (70). It suppresses aerobic glycolysis as demonstrated by reduced lactate production and Glut1 levels and inactivation of mTOR, c-Myc, and glucose-6-phosphate dehydrogenase (G6PD) (70). The mTOR, c-Myc, and G6PD are involved in signaling pathways that contribute to Wartburg effect (60, 111, 282). The c-Myc is widely described as an oncogene (282). The suppression of Nrf1 was also demonstrated (70).
Induction of adaptive response—up-regulation of endogenous antioxidative enzymes—rat kidney ischemia reperfusion model
Dorai's group substantiated the fact that MnP can cause adaptive response via a mild pro-oxidative stress (75). MnTnHex-2-PyP5+ was administered as a part of GMP treatment that contained specific renoprotective growth factors and mitochondria-protecting amino acids. It was given at a 24 h-time point before, at the time of ischemia (0 h) and at 24 h after 40–70 min of renal ischemia followed by 48 h of reperfusion. The up-regulation of antioxidant enzymes was demonstrated, suggesting that GMP treatment produced mild oxidative stress. In his subsequent work, Dorai introduced N-acetylcysteine into improved, I-GMP treatment. The aqueous solution chemistry shows that N-acteylcysteine readily couples with MnTnHex-2-PyP5+. In turn, I-GMP further enhanced oxidative stress via production of H2O2 (33, 52). Indeed, a much more robust up-regulation of endogenous antioxidative defenses was demonstrated: glutathione peroxidase, lactoperoxidase, inducible nitric oxide synthase, mitochondrial and extracellular SOD, thioredoxin reductase 1, and a set of peroxyredoxins (52). In addition, oxidative stress genes (HSP-70 and phospho-heat shock factor-1, pHSF-1) were up-regulated. Levels of independent tissue ischemia markers (galectin-3, lipacalin-2, and high mobility group B1 protein, HMGB1) were increased upon I/R, but were reduced upon the treatment with I-GMP (52). HSP-70 exerts a cytoprotective effect (91) and also functions in a “chaperokine-like” manner due to its capacity to transport peptides and present them to antibody-producing cells, boosting the innate immune response (305). A widely accepted marker of kidney function, creatinine was greatly increased by ischemia/reperfusion and was markedly suppressed by the I-GMP treatment. Had MnP acted as an SOD mimic, the up-regulation of two SOD enzymes should not have happened.
Cancer studies
AIF—inflammatory breast cancer study
The effect of MnTnBuOE-2-PyP5+ on AIF, ERK, and p38(MAPK) kinases and X-linked inhibitor of apoptosis protein (XIAP) has been demonstrated in cellular inflammatory breast cancer study when MnP was co-administered with ascorbate (82). The MnP/ascorbate system enhances the oxidative stress via production of H2O2. In such pro-oxidative scenario, the GSH levels were reduced and the nuclear translocation of apoptosis-inducible factor AIF was enhanced, and so was the cellular apoptosis. With normal cells, such as cardiomyocyte, the mitochondrially localized MnP prevented the doxorubicin-based AIF-NHE adduct formation and nuclear translocation, and, in turn, inhibited the apoptosis (see earlier AIF cardiomyocytes). In an inflammatory breast cancer cell study (82), similar to a lymphoma study, the NF-kB was also suppressed (125, 126); in the former study, MnP/ascorbate, while in the latter, MnP/dexamethasone was the source of H2O2 (see below NF-kB—lymphoma).
HIF-1α–mouse breast cancer model
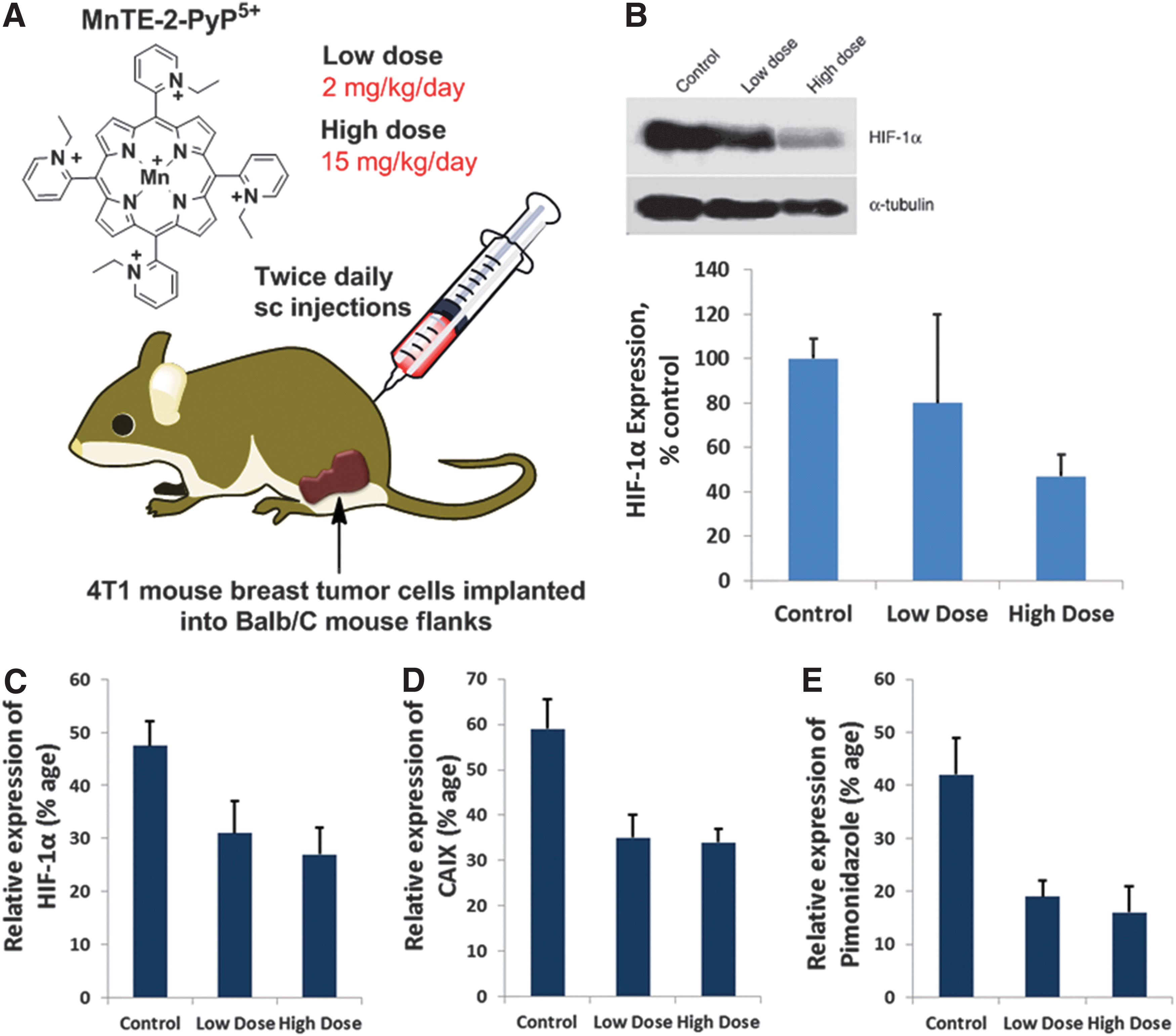
In a 4T1 breast cancer study, the suppression of hypoxia-related proteins was observed as well as the decrease in the levels of 8-oxo-2′-deoxyguanosine (8-OHdG), protein 3-nitrotyrosine, and NADPH oxidase. This could be a consequence of the anti-oxidative action of MnP. However, an MnP-driven oxidation of signaling proteins may be also operative; such an action would suppress secondary oxidative stress and, in turn, the up-regulation of various genes such as VEGF which would have otherwise contributed to the tumor growth delay (Fig. 15) (213). The data from Kim et al. (134) as well as the most recent data on the inhibition of NF-κB by Tome's group (125, 126) provide the basis for such reasoning (see below). NF-κB is known to control HIF-1α and NADPH oxidases (279). Kim et al. report indicates that a mild pro-oxidative event may cause the up-regulation of endogenous antioxidative defenses which action may result in antioxidative effects. If such scenario is operative, the effects could be wrongly assigned to the anti-oxidative action of MnP (134).
AP-1–a mouse skin carcinogenesis study
In skin carcinogenesis model, St. Clair's group has shown the inhibition of AP-1 by MnTE-2-PyP5+ and the reduction in the cell proliferation accompanied by the reduction in markers of oxidative stress (8-OHdG and protein carbonyls), all of which led to the remarkable suppression of skin papillomas (304). An antioxidative mechanism was proposed (304).
NF-κB—a lymphoma study
In a comprehensive study, Tome's group showed the pro-oxidative action of MnTE-2-PyP5+ on the enhancement of corticosteroid (dexamethasone)-induced lymphoma cell apoptosis (Fig. 16). The effect was ascribed to S-glutathionylation of p65 in cytosol, which prevented NF-κB DNA nuclear binding and, in turn, inhibited the up-regulation of anti-apoptotic genes (125, 126). Such modification required H2O2 and GSH. In turn, the cells were deprived of GSH—a major contributor to the physiological redox status. Tome's group also demonstrated that H2O2 was produced primarily in mitochondria by the action of dexamethasone (269)]. In addition, glucocorticoid treatment inactivates MnSOD. Were MnP a mimic of MnSOD, it would suppress (and not enhance) glucocorticoid-induced cell death.
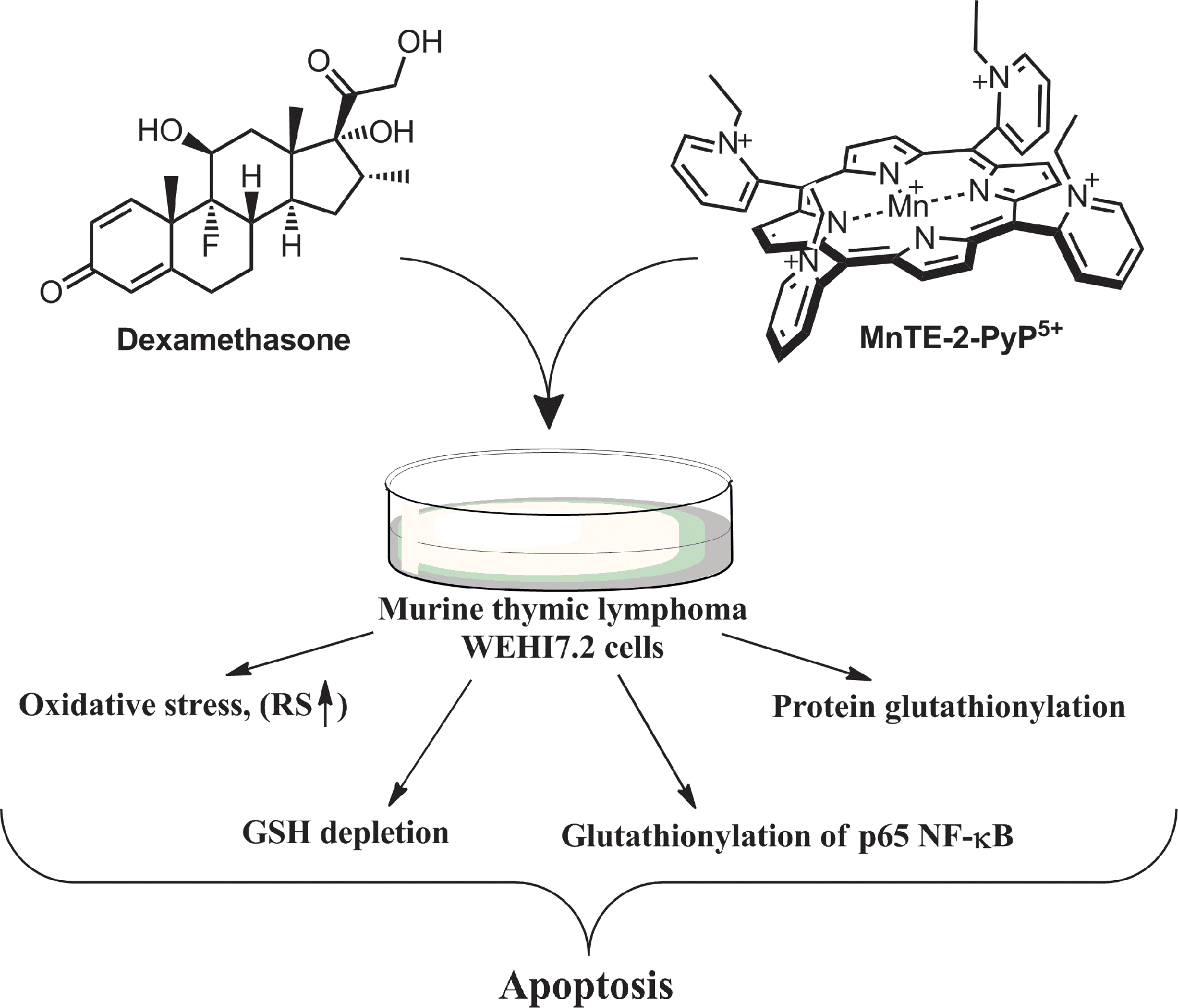
The aqueous-based studies have confirmed that MnP readily gets reduced one electronically by thiols (glutathione, cysteine, N-acteylcysteine, and dithionite), thereby producing thiyl radical. Thiyl radical production is a necessary step in S-glutathionylation. The glutathione peroxidase-like or cysteine oxidase-like activity of MnPs seems to be operative in S-glutathionylation. It is only natural that with similar thermodynamic features (reduction potentials), SOD enzymes and MnPs would undergo same reactions; however, the rates of such reactions would be limited by steric and electrostatic factors. While the low-molecular-weight MPs react readily with amino acids and proteins, the SOD enzymes have high specificity to small O2·− molecules, and will react with other large molecules that are orders of magnitude slower. For example, MnSOD reacts with ONOO− at log kred(ONOO−)=5, while MnPs reacts at log kred(ONOO−)=7.34 (28, 84, 86). Cu,ZnSOD was shown to have Px-like activity, and, most recently, Bonini's group showed that MnSOD has Px-like activity also (7). The cysteine oxidase activity of Cu,ZnSOD was reported to be log k ∼5 (294). Our preliminary studies in simple aqueous systems have demonstrated such thiol oxidase activity for cationic Mn(III) N-alkylpyridylporphyrins (272). Araujo-Chaves et al. have indicated GPx activity of MnTM-4-PyP5+ (9). The scheme in Figure 17 proposes the GPx-like and thiol oxidase activity of MnP. We have shown that MnP/ascorbate system kills cancer cells, while it may be nontoxic or mildly toxic to normal cells (Fig. 12) (220, 272, 299). Please see the anticancer mechanism elucidated by Tome's group originating from the impact of MnP/dexamethasone on cellular bioenergetics (124). In the presence of dexamethasone, the mitochondrially located MnP glutathionylates, and in turn, inactivates critical proteins in electron transport chain, complexes I and III of mitochondrial respiration. Tome's group suggested to further combine MnP/dexamethasone therapy with 2-deoxyglucose inhibitor of glycolysis to deprive cancer cells from both energy sources and to down-regulate the anti-apoptotic pathways (124). Their study has vast therapeutic implications. Somewhat similar studies were reported by Kalyanaraman's group, where breast cancer MiaPaCa-2 cells were co-treated with redox-active mitochondrially targeted nitroxide, Mito-carboxypropyl (Mito-CP), and 2-deoxy-D-glucose (2-DG), which resulted in suppression of energy resources (48).

NF-kB- brain tumor
In an MnP/radiation study of nude/nude Balb/c sc xenograft mouse study of pediatric D-341-MED medulloblastoma, the preliminary data show the effect on NF-κB pathways (see under Therapeutic effects section; cancer studies).
Will pro-oxidative action of MnP promote oxidation or prevent oxidation of biological targets?
Our data imply that, while the reactions of MnP are likely identical, the magnitude and the end results of such reactions may be profoundly different in normal versus cancer cells, as much as in one versus another cancer cell type. Based on our present understanding, the resulting effects of electron shuttling by MnPs may be pro-oxidative rather than antioxidative, and particularly so in cancer cells that are already under the conditions of increased oxidative stress (16). The modes of action, other than antioxidative, have also been speculated for Mn salens (229) and polyphenols, and discussed by Forman et al. for natural compunds commonly viewed as antioxidants (16, 107, 108, 260). It may, thus, be concluded that the redox environment of the cell, including the levels and activities of the endogenous antioxidative enzymes, are main factors determinining how the cell will respond to MnP that is, the outcome of the therapy.
Bioavailability Studies
Besides the compatibility of their redox properties with biological targets, bioavailability is the second critical property that determines the therapeutic potential of redox-active drugs. Bioavailability refers not only to the drug levels in organs, but also to their ability to reach the appropriate subcellular compartments. The impact of MnP bioavailability on its efficacy has been illustrated with SOD-deficient E. coli. The 10-fold lower kcat(O2·−) of a meta MnTE-3-PyP5+ porphyrin was fully compensated by its 10-fold higher lipophilicity relative to MnTE-2-PyP5+. Consequently, both compounds exerted identical efficacy in allowing SOD-deficient E. coli to grow aerobically (139). In E. coli and 4T1 mouse breast cancer cellular studies in the presence of ascorbate MnIIIP5+ was reduced which resulted in a loss of 1+ charge and increase in lipophilicity, and in turn bioavailability (251, 299).
In all bioavailability studies (reported below), chloride salts of MnPs were administered. In all oral bioavailability studies, the water solutions of MnPs were used, except with Drosophila melanogaster, where MnPs were dissolved in phosphate-buffered saline (130). The effect of counterion on the MnP bioavailability has not yet been explored.
PK studies
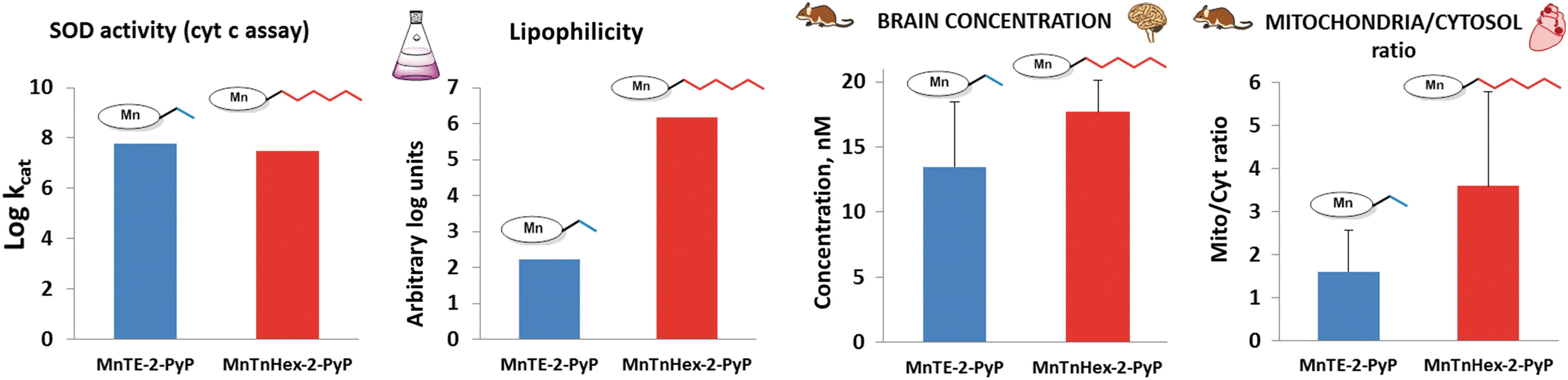
Comprehensive PKs of MnTE-2-PyP5+ and MnTnHex-2-PyP5+ via iv, ip, and oral routes of administration were reported. The LCMS/MS technique was employed to analyze MnP levels in plasma, organs, tumors, and subcellular fragments (288, 299). Figure 18 compares the key properties that affect the therapeutic potential of these MnPs. They exhibit similar redox potency described by the log kcat(O2·−), but are of different lipophilicities and, consequently, bioavailabilities that affect their mouse brain and mouse heart mitochondrial accumulation (Fig. 18) (288).
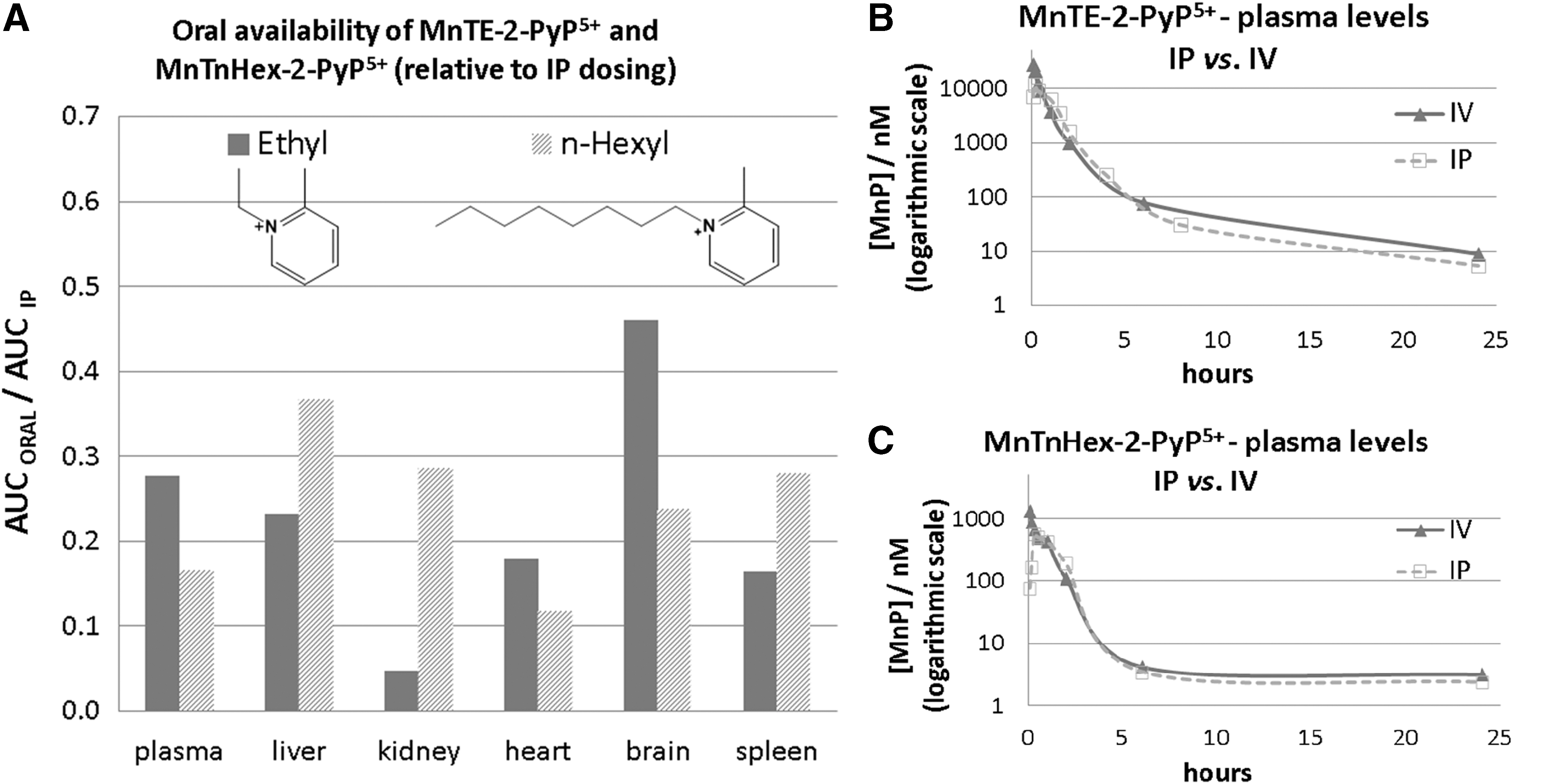
Regardless of high water solubility and pentacationic charge, these MnPs are orally available. The oral availability (based on plasma AUCORAL/AUCIV) is similar for all three compounds: 23% for MnTE-2-PyP5+, 21% for MnTnHex-2-PyP5+, and 22% for MnTnBuOE-2-PyP5+ (Fig. 19) (288). The oral availability was further substantiated in a model of galactosemia, Drosophila melanogaster, developed by Fridovich–Kiel's group (130). Galactosemia is a potentially lethal, autosomal recessive disorder that results from profound deficiency of galactose-P uridylyltransferase (GALT), the middle enzyme in the Leloir pathway of galactose metabolism. While newborns appear healthy, once exposed to milk that contains large amounts of galactose, they undergo devastating demise. At present, no cure exists. The survival of GALT-null mutant was extended for 30%–50% when Drosophila was fed with 10 μM MnTE-2-PyP5+ and 10 μM MnTnBuOE-2-PyP5+.

The availability of MnPs in organs when given orally, calculated as AUCORAL/AUCIP, ranges from 5% (kidney) to 46% (brain) for MnTE-2-PyP5+, and from 12% (heart) to 37% (liver) for MnTnHex-2-PyP5+ (Fig. 20). The AUC values for liver, heart, and spleen are higher for MnTnHex-2-PyP5+ than for MnTE-2-PyP5+ (and quite comparable for other organs) despite a fivefold lower dose, clearly demonstrating the better tissue penetration and tissue retention of a more lipophilic MnTnHex-2-PyP5+ (Fig. 20). We have also performed a dose-dependence study via an oral route (single dose administration) and showed that at higher doses, both MnPs exert toxicity in mice; they are well tolerated at ∼2 mg/kg for MnTnHex-2-PyP5+ and ∼10 mg/kg for MnTE-2-PyP5+ (288).
Mitochondrial availability
Factors that govern mitochondrial distribution relative to cytosol are cationic charge and lipophilicity (177). In a mouse study, both MnTE-2-PyP5+ and MnTnHex-2-PyP5+ prefer to accumulate in heart mitochondria relative to cytosol (5 days sc twice daily 2 mg/kg) (Fig. 18), which is in agreement with their beneficial effects demonstrated in animal models of mitochondrial disorders and ascribed to mimicking of MnSOD (177). The ratio of mitochondria/cytosol distribution for more lipophilic MnTnHex-2-PyP5+ is 3.6, while for hydrophilic MnTE-2-PyP5+, it is 1.6 (289) (Fig. 18). The accumulation of MnTnBuOE-2-PyP5+ in mitochondria relative to cytosol is similar to MnTnHex-2-PyP5+, likely due to their similar lipophilicity (Spasojevic et al., unpublished). Our data obtained by LCMS/MS agree with synchrotron-radiation-induced-X-ray emission studies of MnTE-2-PyP5+ and MnTnHex-2-PyP5+ (3).
Brain availability
The trend in mouse brain distribution (5 days sc twice daily 2 mg/kg) of 13 and 18 nM for MnTE-2-PyP5+ and MnTnHex-2-PyP5+ (MnTnHex-2-PyP5+/MnTE-2-PyP5+=1.4) is in good agreement with their mouse heart mitochondrial distribution (MnTnHex-2-PyP5+/MnTE-2-PyP5+=3.6/1.6=2.2) (Fig. 18) (288, 289). For the purpose of comparison, we averaged their organ oral availability (expressed as AUCoral/AUCip, where AUCip/AUCiv is 83–84% for both compounds) (288). The ratio of AUCoral/AUCip values for several major organs for MnTnHex-2-PyP5+ and MnTE-2-PyP5+ was calculated to be (33%)/(13%)=2.2. The similar ratios of these two MnPs in mouse heart mitochondria (1.4), brain (2.2), and all organs (2.2) strengthen the conclusion on the large impact of lipophilicity on their biodistribution.
Tumor availability
The fivefold higher levels of MnTnHex-2-PyP5+were found in a tumor (4T1 mouse breast cancer sc xenograft study) than in normal muscle extracted from the opposite leg (299) (Fig. 21B). MnPs bear potential as MR imaging agents (183). In agreement with a 4T1 mouse tumor study, the MRI mouse prostate tumor study shows that MnTE-2-PyP5+ accumulated almost exclusively in tumor and not in a surrounding tissue (Fig. 21A).
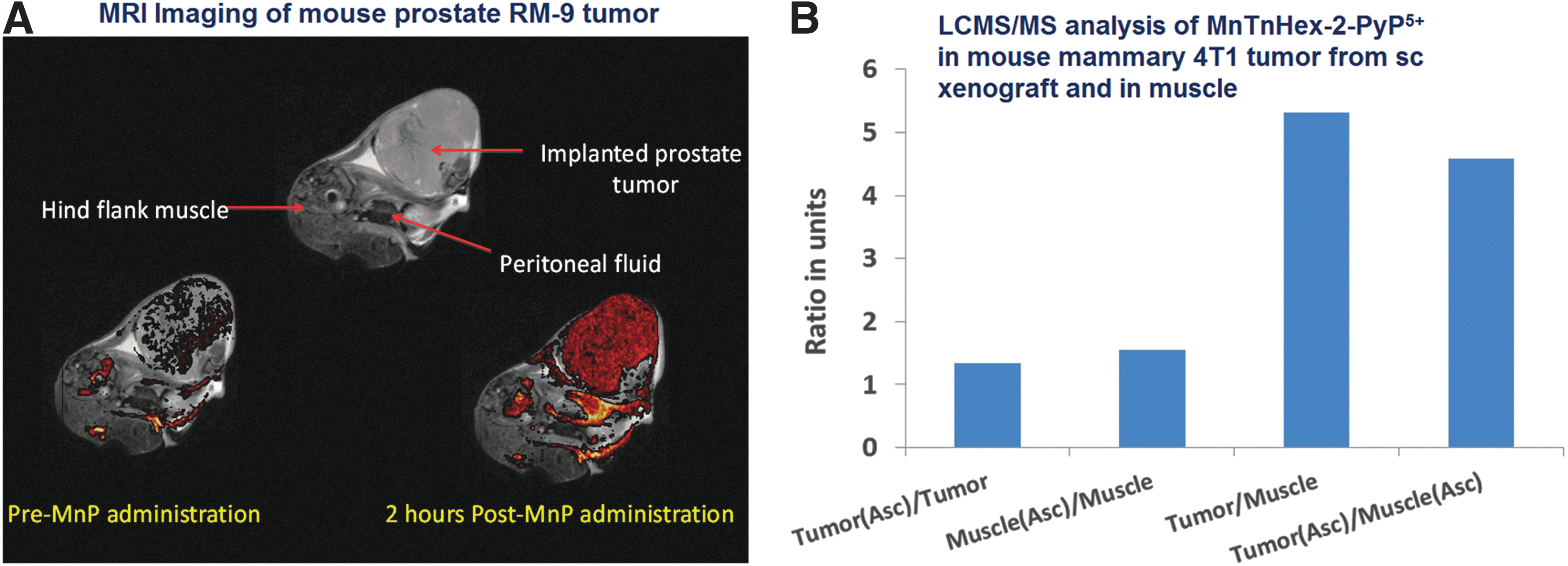
Comprehensive PK studies of redox-active compounds are rare (217). No reported PK data on Mn salen derivatives and Mn cyclic polyamines exist. The ip accumulation studies were performed with anionic Ga corrole; the drug reaches brain vessels but does not cross BBB (197). Due to the low total charge, the Fe corrole, derivatized with three meso pentaflurophenyl groups and two slufonatopyrrolic substituents, is orally efficacious; however, related PK studies are not available (105). Due to the dianionic charge, it does not cross BBB (105, 197).
Toxicity
The subcutaneous route of administration appears to be the least toxic for MnPs while affording the highest body exposure (AUC) (288). The single TD50 (the dose toxic to 50% of mice) determined for such a route is 91.5 mg/kg for MnTE-2-PyP5+ and 12.5 mg/kg for MnTnHex-2-PyP5+ (210). Given the up to 120-fold higher efficacy of MnTnHex-2-PyP5+ versus MnTE-2-PyP5+ in animal studies, the therapeutic window for the former is superior to the latter (28, 210). A single maximal tolerated dose via oral route was found to be ∼10 mg for MnTE-2-PyP5+, and ∼2 mg/kg MnTnHex-2-PyP5+ (288).
The first lead drug candidate of Mn(III) N-substituted pyridylporphyrins was MnTE-2-PyP5+ (AEOL10113, BMX-010) and has been GMP scaled up and Drug Master File filed. A full GLP nonclinical assessment was conducted in order to evaluate the safety of MnTE-2-PyP5+ (92). The safety evaluation included in vitro genotoxicity studies (Bacterial Reverse Mutation Assay, mammalian chromosome aberration test using Chinese hamster ovary cells), hemolysis, flocculation, safety pharmacology (in vitro hERG, respiratory and CNS in mice, and cardiovascular in monkeys), venous irritation in rabbits, and iv toxicity in mice (single dose, five-day, and 28-day) and monkeys (single dose, 5-day, and 14-day). MnTE-2-PyP5+ was not genotoxic or hemolytic, did not cause flocculation or elicit adverse pharmacologic effects on respiration, the central nervous system, or the cardiovascular system. The intended iv clinical solution did not cause venous irritation in rabbits. The NOAEL [No observable (toxic) effect level)] in mice after 28 days of dosing was 10 mg/kg. The NOAEL in monkeys after 14 days of iv dosing was 5 mg/kg. Based on the results of these studies, a conservative safe initial starting clinical dose of 5.0 mg (0.083 mg/kg in a 60 kg adult) is proposed for human trials. Due to patient life issues, the use of MnTE-2-PyP5+ as a transplantation aid was not pursued past the preclinical stages (92).
The 2nd lead in the class of Mn(III) N-substituted pyridylporphyrins, MnTnBuOE-2-PyP5+ is also GMP scaled up. The safety evaluation on mice and monkeys is in progress.
The lead compound in the class of N,N′-di-ortho substituted imidazolylporphyrins, MnTDE-2-ImP5+ was in phase I clinical Trials on ALS (198) and was well tolerated. The limited PKs has been reported (174). It is currently in development as a radioprotector by Aeolus Pharmaceuticals and supported in large part by Federal funds.
Therapeutic Effects
Introduction
Among the most striking therapeutic effects reported thus far is the effect of MnTE-2-PyP5+ in a rat spinal cord injury model, where 1 mg/kg/day given subcutaneously for a week after T10 injury (starting 30 min after T10 crush) resulted in a remarkable near-full recovery of rat limb function. The data are discussed with regard to inhibition of NF-κB activation (204). In a rat stroke model, the efficacy of MnTnHex-2-PyP5+ is at least in part due to the inhibition of NF-κB (238)]. It seems that in many models of diseases the activation of the master transcription factor, NF-κB, which controls inflammatory and immune pathways, is implicated. A very similar effect on rat spinal cord injury was observed with estrogen and is supposedly orchestrated via glucocorticoid receptor (67, 110, 129, 185, 243). A significant association between testosterone level and severity of SCI has been reported (79, 83). Since males greatly outnumber females in serious spinal cord injuries, it is unlikely that estrogen will be accepted as a treatment. The prevalence of testosterone deficiency was significantly greater in participants with complete motor dysfunction compared with those with less severe injuries (79, 83). Significant reduction in neuropathic pain and complete inhibition of chronic morphine tolerance were exerted by MnTE-2-PyP5+ and MnTnHex-2-PyP5+ (24, 76). Remarkable data were also obtained in a prostate radioprotection study, where the effect on complete reversal of erectile dysfunction and prevention of testes shrinkage was observed (100, 164, 165). Near-full elimination of papillomas was reported in a mouse skin carcinogenesis model when 5 ng MnTE-2-PyP5+ was applied on skin for 14 weeks at 4 days per week (304). All existing studies of cancer, radiation, and radiosensitization are listed in Tables 2 and 3; while possible mechanisms are discussed under reactivity toward signaling proteins section and next. The effects of MPs on central nervous system injuries are summarized by Warner et al., and their effects on inflammation and autoimmunity by Delmastro-Greenwood et al. in this Forum (208a). Next, we show some of the data related to the radioprotective and radiosensitizing effects of MnPs. We have chosen to include other redox-active drugs also, though some may be neither true SOD mimics (catalysts of O2·− dismutation) nor O2·− scavengers. The reason is that the observed effects are frequently similar to the effects of true SOD mimics. The most recent example is the study reported at 2012 Annual Meeting of the Society of Free Radical Biology Medicine from Kalyanaraman's group on the effect of nitroxide Mito-CP on cancer cell bioenergetics (48). The results resemble those observed with MnPs by Tome's group (124). Finally, we discuss in detail only those cancer studies that have not been covered in our earlier reviews (26 –28, 208a, 236, 270). The anticancer research is becoming increasingly important and prevalent due to (i) the increase in aging population, and, thus, the number of cancer patients; and (ii) the lack of anticancer drugs, which is at least in part due to the ability of cancer cells to mutate and become resistant to therapy. Thus far, we have tested them in animal models of lymphoma, leukemia, skin, brain, breast, prostate, and head and neck cancer studies.
Data relate to Mn porphyrins as monotherapy and to their combination with other agents and radiation. In addition, data for some of the other redox-active compounds most frequently studied, natural and synthetic, are listed.
Mito-CP, Mito-carboxypropyl; m, multiple dosing.
multiplicity; multiple (m), single (s).
Radioprotection studies
Pulmonary studies
In a rat pulmonary radioprotection study, 6 mg/kg MnTE-2-PyP5+ normalized breathing rate frequencies, as well as suppressed HIF-1α pathways and its gene VEGF when the two-week-long sc administration started at different time points between 2 h and 8 weeks after radiation. MnTnHex-2-PyP5+ was radioprotective also and at as low a dose as 0.05 mg/kg/day; the sc dosing started at 2 h after radiation and lasted for 2 weeks (95, 96). A significant decrease in HIF-1α, TGF-β, and VEGF A, as well as an overall reduction in lung damage (histopathology), was observed in animals in which MnP treatment started at the time of fully developed lung injury 8 weeks post-IR (96).
In another mouse pulmonary radioprotection study, the forty-four genes associated with metabolism, cell growth, apoptosis, inflammation, oxidative stress, and extracellular matrix synthesis were up-regulated throughout 6 months postradiation (122). The messenger RNA expression of adrenomedulin (Adm), 1-acylglycerol-3-phosphate O-acyltransferase 2 (Agpat2), N-acetyltransferase ARD 1 homolog (Ard1), connective tissue growth factor (CTGF), Enolase 1, α noneuron (Eno1), HIF-2α, guanine nucleotide binding protein, α11 (Gna11), protein kinase, AMP-activated, α catalytic subunit (Prkaa1), sorbitol dehydrogenase (Sdh1), and tubulin α3 (Tub α 3) were elevated in irradiated animals relative to control. The expression of DNA damage repair (Dr1), fatty acid-binding protein 4 (Fabp4), formin-binding protein 3 (Fnbp3), and solute carrier family 2, member 1 (Slc2a1) was elevated only at 6 month postradiation. The animals were treated with imidazolium derivative, MnTDE-2-ImP5+ (AEOL10150) (Fig. 1) starting at 24 h after 15 Gy whole thorax radiation with sc loading dose of 40 mg/kg, followed by 20 mg/kg (sc) every other day for 4 weeks. The impact of MnTDE-2-ImP5+ was evaluated at 6 weeks after radiation. The elevated expression of 31 of these genes was attenuated in animals treated with MnTDE-2-ImP5+, suggesting that expression of a number of hypoxia-associated genes is regulated by early development of oxidative stress after radiation. Such genes are: IL-1 β, TGF- β1, PPAR-α (peroxisome proliferator-activated receptor α), HIF-2β, carbonic anhydrase 12 (Car12), neuroblastoma myc-related oncogene (Nmyc1), cell division cycle 42 homolog (Cdc42), denactin 2 (Dctn2), death-associated kinase 3 (dapk3), matrix metalloproteinase 14 (MMP14), CTGF, Sdh1, and so on (122). Data substantiated the earlier observation (96) that MnP radioprotects even when its injections started at 24 h after injury.
The impact of MnTDE-2-ImP5+ on apoptotic pathways as a consequence of mouse pulmonary irradiation was also explored (Fig. 22) (302). The involvement of oxidative stress was verified via observed up-regulation of NOX4 and 8-OHdG, which were increased at 6 weeks after radiation. The apoptosis was observed primarily in type I and II pneumocytes and endothelium. Apoptosis correlated well with increased phosphoinositide 3-phosphatase (PTEN) and transforming growth factor TGF-β1 (excreted by many cells, including macrophages and involved in apoptosis via Sma- and Mad-related proteins), SMAD [SMAD proteins are homologs of both the Drosophila protein, mothers against decapentaplegic (MAD), and the Caenorhabditis elegans protein SMA (from gene sma for small body size)] and death-associated protein 6 (DAXX transcriptional activities), as well as inhibition of downstream PI3K/AK signaling, and increased p53 and Bax protein levels. MnTDE-2-ImP5+ suppressed oxidative stress also and, thus, the pro-apoptotic signaling, which consequently reduced the number of apoptotic cells (Fig. 22) (302).
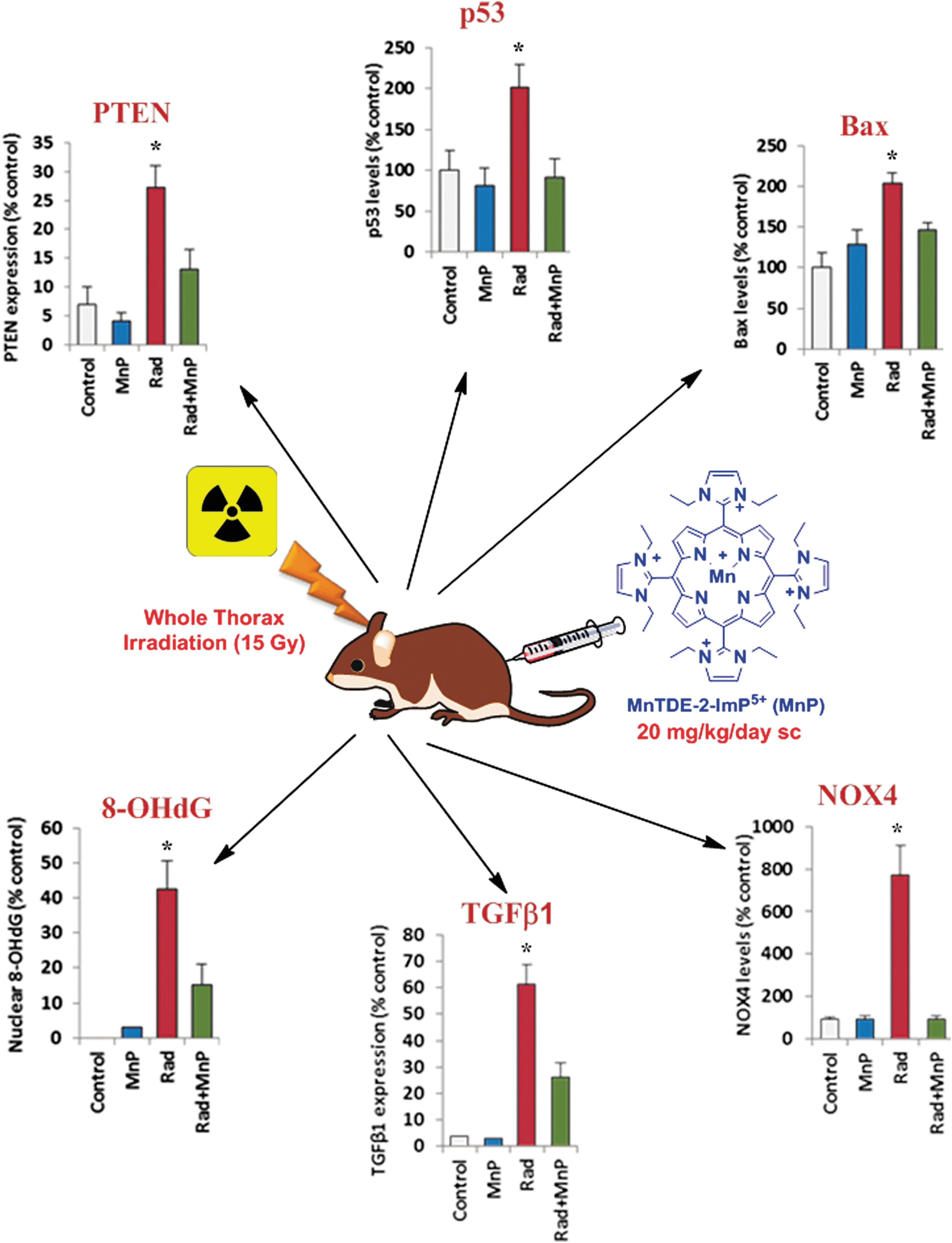
One study of pulonary radioprotection of nonhuman primates has been completed, and the other is in progress. Both studies administer low doses of MnTnHex-2-PyP5+. The treatment with MnP (sc twice daily at 0.05 mg/kg) delayed onset of radiation-induced lung lesions, prevented elevation of respiratory rate, and reduced lung weight, inflammation, edema, and epithelial hyperplasia (50).
Head and neck cancer studies
Significant radioprotection of mouse salivary glands were observed in a C57BL/6 study with MnTnBuOE-2-PyP5+ injected sc twice daily at 1.5 mg/kg for the duration of the study, starting one week before radiation (13). The study is a part of head and neck cancer studies (Ashcraft et al. in preparation). In preliminary studies, no protection of cancer was observed (Park et al., unpublished).
Hematopoietic studies
MnTE-2-PyP5+ protected bone marrow-derived hematopoietic stem cells (HSCs) against sublethal 6.5 Gy total body radiation (TBI) (152, 205). Mice were treated with 6 mg/kg MnTE-2-PyP5+ sc at 6 h after radiation and then, every day for 30 days. After treatment, the irradiated mice showed a significant recovery in the frequency and number of HSCs and a dramatic improvement in HSC and function of hematopoietic progenitor cells, HPC. The clonogenic function of HSCs from irradiated mice after MnP treatment was comparable to that of HSCs from normal controls, suggesting that MnP treatment inhibited the induction of HSC senescence by TBI. The effects were attributed to the inhibition of senescence p16 pathways, which is supported by the finding that MnP treatment also reduced the expression of p16Ink4a (p16) mRNA in HSCs induced by TBI and improved the long-term and multilineage engraftment of irradiated HSCs after transplantation (205). The authors reported that the reactive species produced by NADPH oxidases played a critical role.
Protection of erectile dysfunction during prostate radiation
The role of oxidative stress in prostate radiation-caused erectile dysfunction was demonstrated by Koontz group in a rat model (Fig. 23D–F) (136). Irradiated animals (eight to nine per group) received prostate-confined radiation in a single 20 Gy fraction. Radiation caused a significant decrease in intracavernous pressure and increased expression of NADPH oxidase isoaform 4 (NOX4), DNA damage (8-OHdG), and lipid peroxidation (measured by 4-hydroxynonenal) in prostate tissue and corpora cavernosa accompanied by a trend toward an increase in endogenous antioxidant defense, nuclear factor-erythroid-derived 2-like 2 (Nrf-2) (136).
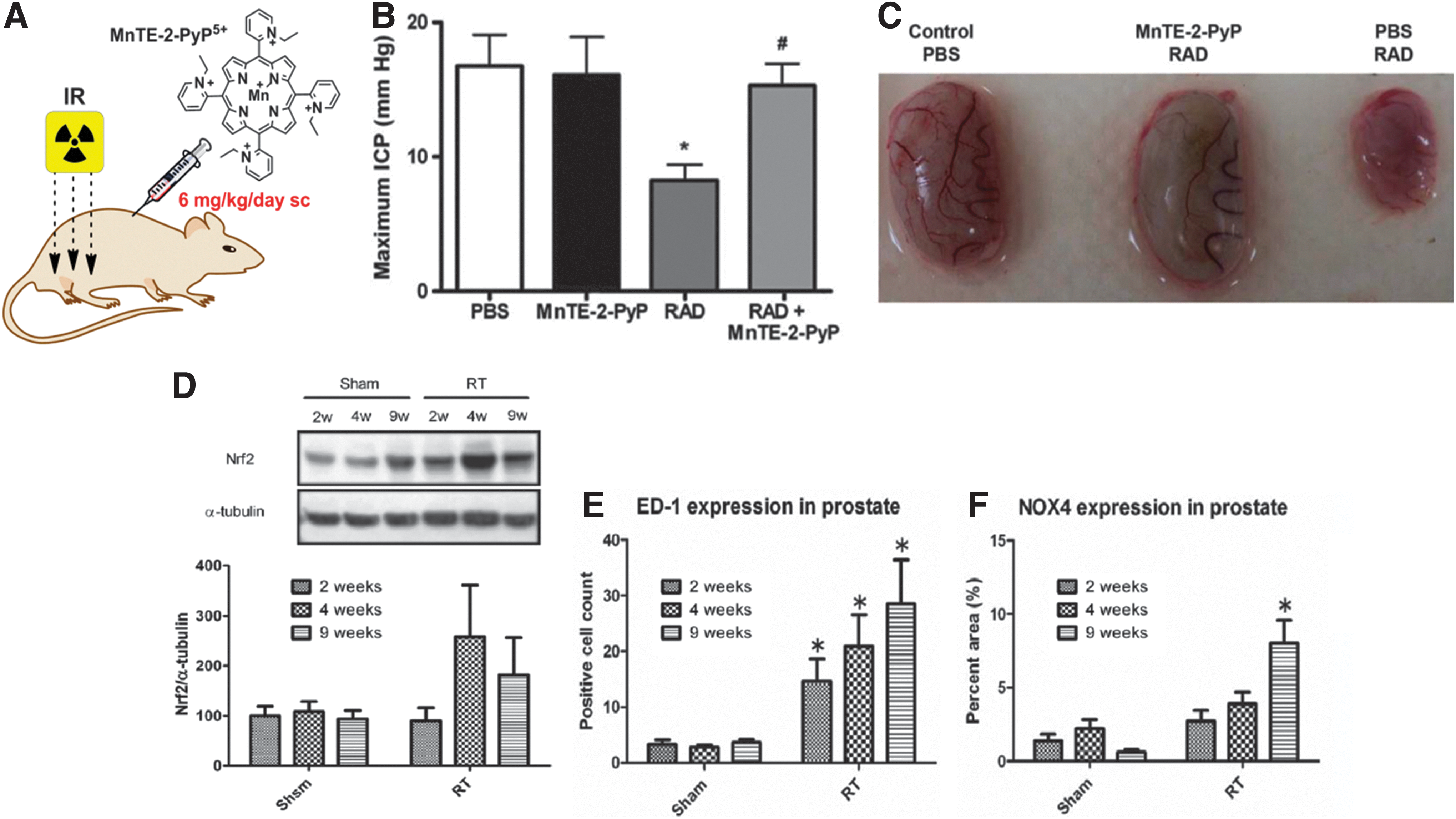
The MnTE-2-PyP5+ fully reversed the erectile dysfunction as a consequence of prostate tumor radiation (Fig. 23) (192). Mice were irradiated for five sequential days at 7.5 Gy/day in a lower pelvic region, which mimics the irradiation scheme of prostate tumor patients. MnP was injected at 5 mg/kg 24 h before starting with radiation and then, 2.5 mg/kg every other day for the next two weeks and 5 mg/kg once a week for 12 weeks postradiation. MnP reduced DNA damage as seen by the decrease in 8-OHdG levels in prostate epithelial cells. It also prevented the ∼60% of shrinkage of rat testes due to radiation, and prevented damage of penile tissue. Intracavernous pressure in was obtained after cavernous nerve stimulation as a measurement of erectile function 12 weeks postirradiation. A significant decrease in erectile function was demonstrated as compared with the nonirradiated mice and was greatly attenuated with MnP. The reduction in tissue damage and hair loss by MnP was demonstrated as well.
Is Mn pro-oxidant or antioxidant in radioprotection?
While thus far we have understood the radioprotective effects as resulting from scavenging reactive species, rising number of data argue that perhaps mild pro-oxidative action of MnP may predominate. The oxidation of NF-κB (which controls HIF-1α, and its genes) by MnP would prevent excessive inflammation, secondary oxidative stress and result in prevention of extensive oxidative damage of biological targets (208, 209, 236 –240, 278). Data on rat kidney ischemia/reperfusion indicate that MnP can also up-regulate endogenous antioxidative defenses (52, 75). Similar findings have been argued by Huang's group (134) and Forman (89). The action of another class of SOD mimics, cyclic polyamines, is discussed in terms of activation of Nrf-2 via oxidation of cysteine of Keap (176). The same is proposed based on the actions of curcumin, resveratrol, diallyl sulfide from garlic by Forman et al. (89) . Studies are needed to further substantiate such mechanisms, including the effect of MnP on the activation of the primary cellular defense against the cytotoxic effects of oxidative stress, Nrf-2. Nrf-2 translocates to the nucleus upon oxidative stress and binds there to antioxidant response elements, increasing transcription of those genes (159).
Cancer studies
In several different animal models, MnPs protected normal tissue again radiation; importantly, no protection of tumor tissue was demonstrated. Moreover, MnPs proved to be radio- and chemosensitizers (21, 22). MnTE-2-PyP5+ also enhanced hyperthermia in a B16F10 mouse melanoma study (182).
Prostate cancer
A study on the prostate cells evaluated the antitumor potential of MnTE-2-PyP5+. MnP was used in a dose-dependent fashion (1–30 μM). It significantly inhibited the growth of three prostate cancer cells: PC3 (98.2%±0.9%), Du145 (94.3%±0.7%), and LnCAP (100%±0%). MnTE-2-PyP5+ further reduced the growth of tumor cells when co-administered with radiation (5 Gy), but did not inhibit the proliferation of the prostate tumor cells. However, MnP significantly inhibited the invasiveness of PC3 cells on matrigel (67%±3.1%) and significantly inhibited Du145 cells (56.7%±5.6%) ability to migrate across a filter. MnTE-2-PyP5+ reduced NF-κB activity, which may inhibit tumor colony formation. It further reduced the phosphorylation of the focal adhesion kinase (involved in breaking down the extracellular matrix to allow cells to migrate), and VEGF-A in hypoxic prostate cells. VEGF-A is another protein of VEGF family that is involved in the growth migration of tumor cells (287).
Breast cancer
Significant radiosensitization via HIF-1α suppression and, in turn, vastly reduced vascular density exhibited by MnTE-2-PyP5+ was demonstrated in a 4T1 mouse breast cancer sc xenograft study (180). The MnTE-2-PyP5+exerted an anticancer effect as a single agent at a high (but not at low dose of 2×1 mg/kg/day) dose of 2×7.5 mg/kg/day for the duration of the study. Strong suppression of oxidative stress and angiogenesis (reduced NADPH oxidase expression, HIF-1α, VEGFR, macrophage infiltration, and protein nitration) was demonstrated (Fig. 15) (213).
Brain tumor—MnP-driven radiosensitization
Recently, a brain tumor study was conducted on sc xenografts of glioblastoma multiforme D-245 MG (eight mice per group). Similarly, lipophilic and redox-active drugs, MnTnHex-2-PyP5+ (shown in Fig. 2) and MnTnBuOE-2-PyP5+ exert similar radio- and temozolomide-sensitizing effects (21, 22). Tumor growth was delayed for ∼12 days as a result of MnP impact on radiation and temozolomide and in both of these treatments combined (Fig. 24). The significance in tumor growth delay with MnP/radiation therapy of pediatric medulloblastoma D-341 MED cell line (in a sc xenograft Balb/c nude/nude mouse was not reached under single conditions tested. However, the metastatic pathways (ctss, catepsin L, becn1, and beclin1) were largely down-regulated, as were antiapoptotic and NF-κB pathways (Nfkb1, Bcl211, and Bcl2) and PI3kinase and mTOR (Rsp6kb1). The protein translation changes were also implicated (EIF5b and Rsp6kb1)(22). The data are in agreement with the data on NF-κB inhibition by MnP obtained with a number of other models (see also under Reactivity towards signaling proteins section).
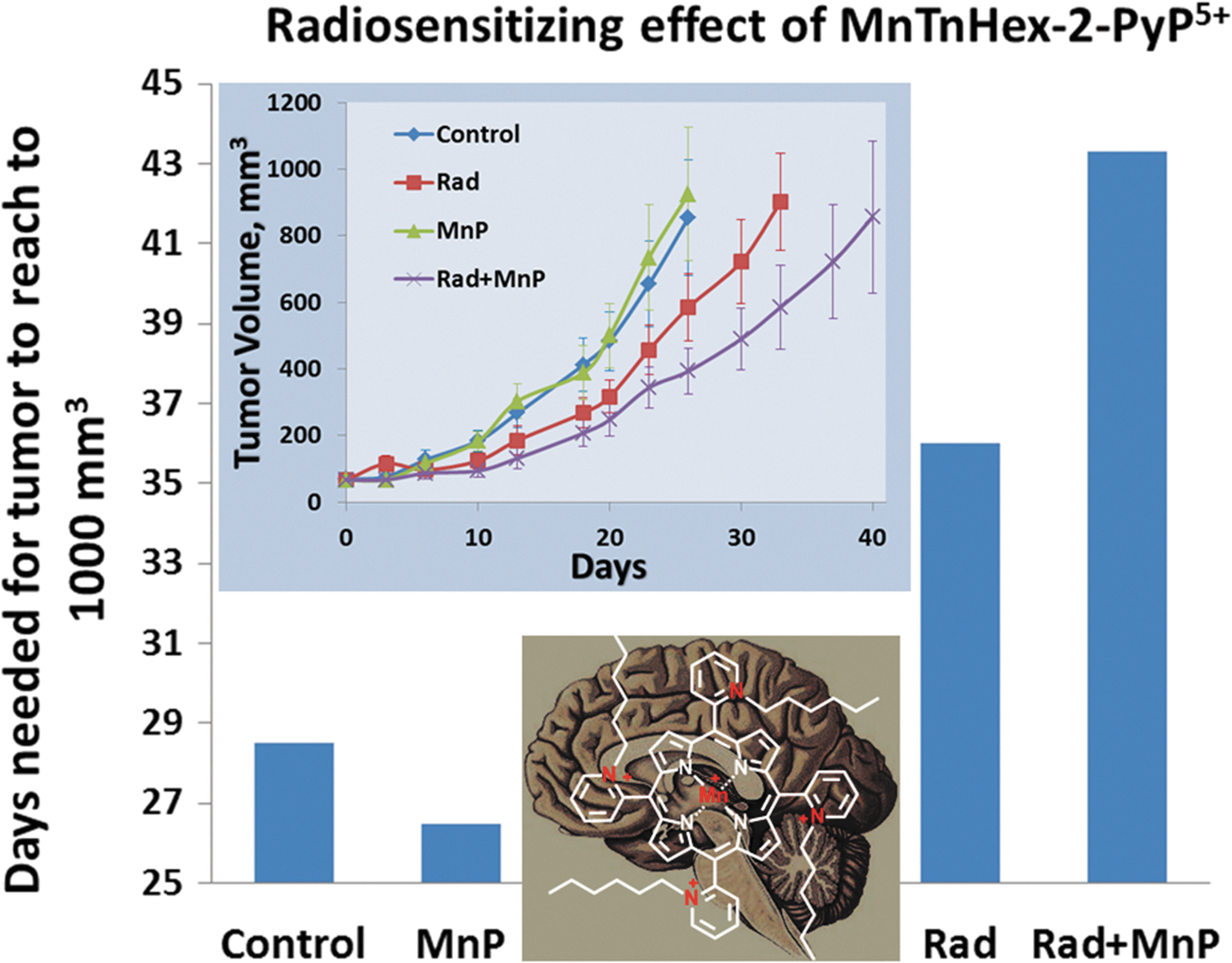
Lymphoma
As discussed under Reactivity toward signaling proteins section, MnTE-2-PyP5+ enhanced corticosteroid (dexamethasone) therapy in a lymphoma cellular study via NF-κB pathways (Fig. 11) (125). The most recent study indicates that MnP additionally inactivates complexes I and III of mitochondrial electron transport chain, suppressing largely mitochondrial ATP production (123). MnP also suppresses glycolysis in its own right and when combined with glycolysis inhibitor 2-deoxyglucose (124); in turn, energy production of the cell seems to be greatly suppressed by the action of MnP (123).
Head and neck cancer
Preliminary data indicate the radiosensitizing effect of MnP, MnTnBuOE-2-PyP5+ in head and neck mouse sc xenograft study, which along with its ability to protect salivary glands and oral mucosis indicates its high therapeutic potential (13) (Ashcraft et al., in preparation).
Skin cancer
The anticancer effects of MnP as a single anticancer agent were previously reported in skin carcinogenesis model (304). See also under Reactivity towards signaling proteins section; Cancer studies. Please see Holley et al. contribution in this Forum, which suggests that the pharmacological intervention by an MnSOD mimic, MnTnBuOE-2-PyP5+, may play an important role in prevention of aberrant cell signaling that may contribute to carcinogenesis (117).
As discussed under Reactivity towards cellular reactants, the MnP is a promising anticancer agent in combination with ascorbate. Such a system, very similar to radiation therapy, is a source of cytotoxic H2O2 that MnP employs to oxidize a variety of biological targets (Tovmasyan et al., unpublished). Under certain conditions, such as in pediatric tumors, radiation should be avoided and could be potentially replaced with MnP/ascorbate. Related studies are still at cellular stage, and exploration of optimal ratio MnP/ascorbate is required as well as a thorough insight into the redox status of the each targeted cancer cell line. Another cycling system employing ascorbate is ascorbate/menadione combination, which is already in clinics for the treatment of prostate cancer (262, 284).
Is MnP acting as an oxidant or antioxidant in cancer?
Present lymphoma data clearly suggest that in the presence of H2O2 (produced synergistically by dexamethasone and MnP) and GSH, the MnP glutathionylates p65 of NF-κB and, thus, suppresses anti-apoptotic pathways (125, 127). These data, along with the data showing (i) that MnP/dexamethasone inactivates complexes I and III of mitochondrial respiration and, in turn, ATP production (124, 125), (ii) reduces cytosolic ATP production in glycolysis (124), and (iii) suppresses aerobic glycolysis via suppression of pathways involved in Wartburg effect (70), offer substantial proof of high anticancer pro-oxidative potential of MnP in an appropriate redox environment of excessive oxidative stress. Suppression of NF-κB pathway, when MnP was given along with radiation, was seen in a nude/nude Balb/c mouse sc xenograft study of D341-MED pediatric medulloblastoma. Similar to lymphoma, the radiation was the source of H2O2 in MnP/radiation studies (16, 231). In cellular studies, the MnP/ascorbate system was the source of cytotoxic H2O2 (25, 82, 299). Cancer cells produce abnormal levels of reducing equivalents that are necessary for proliferation and for maintenance of appropriate (cell-type dependent) levels of RS to prevent cell apoptosis and cell death but induce genomic instability (16). During cancer progression, its oxidative status increases and imparts the neighboring and distant normal tissue (16). Thus, even with no source of H2O2 added, cancer is under increased levels of H2O2 relative to normal tissue, supporting the likelihood of pro-oxidative action of MnP. Finally, the magnitude of the anticancer effect would be also highly dependent on the type of cancer cell, that is, its redox environment, which may differ largely from cell to cell.
Concluding Remarks
Being a true and very efficacious SOD mimic does not in itself mean that a drug will eventually be an excellent therapeutic. Bioavailability and low toxicity are other critical parameters that will determine its in vivo efficacy and therapeutic potential. However, the animal efficacy studies of cationic Mn and FePs, with E1/2 ∼+300 mV versus NHE, undoubtedly, show that those compounds of high SOD-like activity have the ability to readily shuttle electrons between their metal sites and a variety of biological targets (reactive species, proteins, lipid, nucleic acids, etc.), which, in turn, normalize cellular metabolism and, thus, have high prospects to become therapeutics. The high accumulation of cationic MPs in mitochondria and transport across BBB further contributes to their remarkable efficacy. Any efficacious SOD mimic is also an efficacious scavenger of peroxynitrite as well as of many other RS (such as hypochlorite, lipid radicals, ·NO, CO3·−, etc.). However, even compounds, which are not true and potent SOD mimics, and are not even very biocompatible redox-active drugs, scavenge a wide variety of highly oxidizing species (ONOO−, ClO−, lipid radicals, etc.), and if localized at critical cellular sites, may still be effective modulators of cellular redox status. The sum of bioavailability, redox compatibility with biological targets, and toxicity of MnPs, as well as the cellular redox environment would determine their final therapeutic potential.
Based on all said, it is more correct to describe SOD mimics as modulators of cellular redox environment (cellular signaling pathways or redoxome), or promoters/catalysts of processes already in place—rather than antioxidants. Years ago, we had assigned the therapeutic effects observed entirely to the MnP-driven removal of reactive species (in particular O2·− in accord with H2O2 removal) and, thus, to their antioxidant actions. Rising evidence, however, points to the prevailing pro-oxidative mechanism of action, even in normal cells, with final antioxidative therapeutic effects. Such effects may originate from (i) the inhibition of pro-inflammatory, antiapoptotic NF-κB-based pathways via oxidation of thiols of NF-κB subunits; and (ii) adaptive responses to MnP-driven increase in oxidative stress, with subsequent up-regulation of endogenous antioxidative defenses. In cancer in particular, the pro-oxidative action, via NF-kB pathway, may predominate, leading to cancer cell death; further studies in different models and on different cancer cell types are needed to gain thorough insights into the biological pathways of MnPs and other redox-active compounds.
The prediction, the anti-(normal tissue) or pro-oxidative (cancer) therapeutic outcome of MnP, would have higher chances to be correct if it is based on the knowledge of the expression and the activities of enzymes that control the cellular redox environment of a cell type of interest (redoxome, or, as Forman et al. (89) put it “nucleophilic–electron-rich- tone” of the cell): catalases, SODs, glutathione peroxidases, glutathione reductases, glutathione transferases, peroxyredoxins, thioredoxins, and so on. The redoxome may differ largely from one cancer cell to another, which would greatly affect the magnitude of the therapeutic effect. While a drug may reach clinics without full knowledge of its whereabouts, the mechanistic studies are critical for understanding the biology of diseases, which, in turn, would favorably affect drug development. While the tools are often available to track the pathways modified by the action of SOD mimics, the identification of the sources of modification is more challenging. Given the complexity of cells and the rich chemistry of SOD mimics, we are still far from comprehending the in vivo whereabouts of SOD mimics.
Footnotes
Acknowledgments
The authors are grateful to numerous researchers who have worked for years on in vitro and in vivo models of oxidative stress and help them revise their development strategies and keep them motivated. They are particularly thankful to those who made them believe that scientists of the highest integrity, kindness, and humbleness still exist; to mention the few who cross their pathways: Daret St. Clair, Margaret Tome, Jon Piganelli, Huaxin Sheng, David S. Warner, Judith Fridovich-Keihl, and Kam Leong. They are thankful to Garry Buettner for the useful information on the therapeutic potential of ascorbate in just completed Phase I Clinical Trials. As always, they are lucky to just step down the corridor to discuss many topics and confirm the correctness of their thoughts with Irwin Fridovich. They appreciate the financial support from Duke University's CTSA grant 1 UL 1 RR024128-01 from NCRR/NIH (AT, IBH, IS), W.H. Coulter Translational Partners Grant Program (IBH, IS, AT), and NIH/NCI Duke Comprehensive Cancer Center Core Grant (5-P30-CA14236-29) (IS), NIH U19AI067798 (ZV, IBH, IS, AT), and IBH general research funds (AT).
Author Disclosure Statement
Both Drs. Batinic-Haberle and Spasojevic are consultants for BioMimetix Pharmaceutical, Inc. Duke University, and Drs. Batinic-Haberle and Spasojevic also have patent rights and have licensed technology to BioMimetix Pharmaceutical, Inc. related to this technology.