
Abstract
Introduction to Radiation-Induced DNA Damage and Repair
IR-induced DNA damage includes single DNA strand breaks (SSBs), double DNA strand breaks (DSBs), and base modifications such as oxidation, alkylation, deamination, loss of base residues to produce apurinic or apyrmidinic sites (AP sites), all of which can indirectly lead to SSBs and/or DSBs. There are also crosslinks formed involving DNA–DNA and DNA–protein interactions (see Fig. 1).
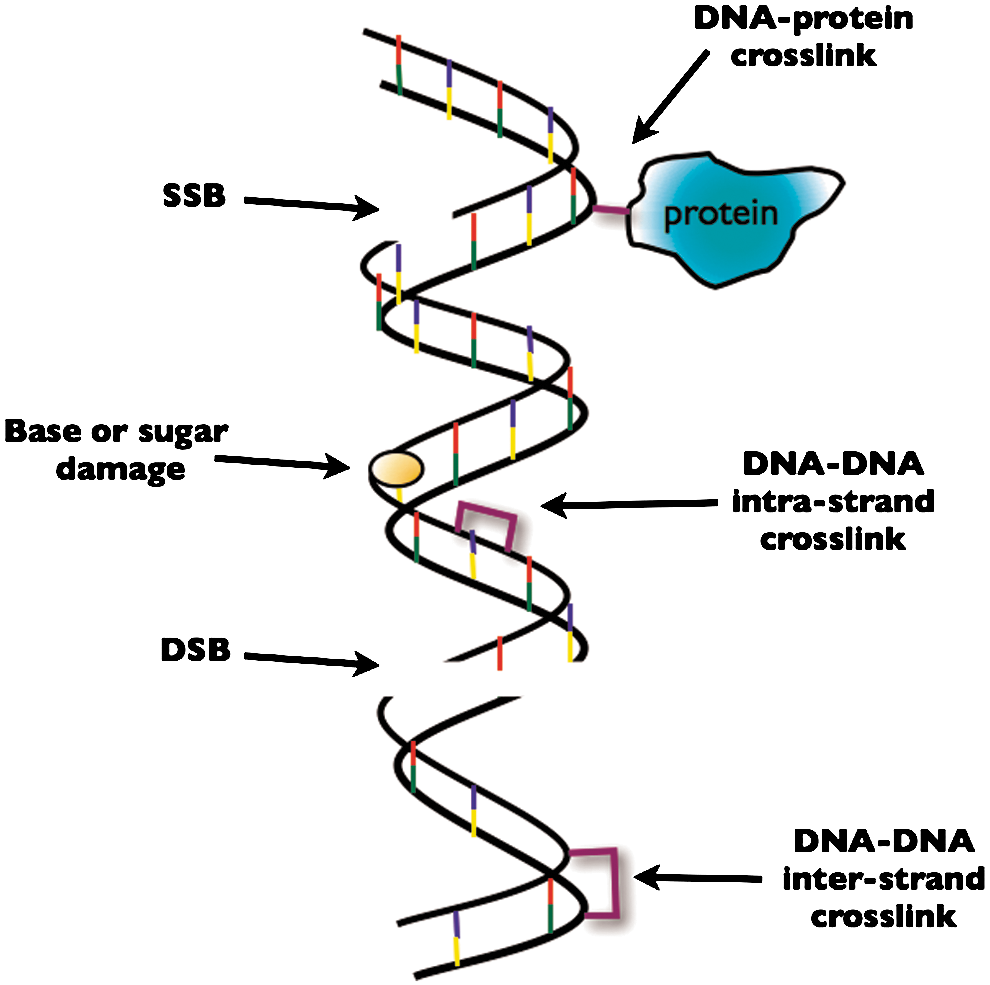
The most critical of these lesions in terms of lethality and mutation probability is the DSBs, which is considered to be a form of “complex DNA damage” also termed clustered damage or multiply damaged sites (132). Complex DNA damage is usually described as two or more lesions within one to two helical turns of the DNA arising from a single radiation track and distinct from endogeneous DNA damage (46). IR causes DNA lesions by direct interaction with DNA or indirectly via the generation of reactive species. In the case of X-rays, most DNA damage induced occurs via the latter method. Studies of DSB induction in mammalian cells exposed to X-rays suggest that ∼70% of the total DSBs result indirectly by interaction of reactive oxygen, such as hydroxyl radicals, that are generated by the ionization of other molecules, such as water, in close proximity to the DNA. In contrast, heavy ions have a higher probability of a particle traversal directly causing a DSB and therefore rely less on radical species for DNA damage induction.
This review will cover key aspects of the induction, sensing, and repair of DSBs caused by IR. Aside from the aspects of complex damage, most of the sections covered are of relevance to other agents that induce DSBs directly or indirectly.
Sensing DNA Double-Strand Breaks
In order for the DSB repair mechanisms (described in section “DNA Double Strand Break Repair Mechanisms”) to be successfully carried out, the DNA damage needs to be detected, cell cycle arrest must be induced, and the lesion can then be repaired. This process is known as the DNA damage response (DDR), which involves interplay between three distinct sets of proteins as proposed by Jackson (57). Damage is initially detected by sensor proteins, which recognize sites of damage within the DNA. These sensor proteins are recruited to damaged sites and amplify the damage signal to a set of proteins known as transducers, which function to relay the signal to downstream effector proteins (12) (see Fig. 2). It is the activated effector proteins, which then function to induce cell cycle arrest, DNA damage repair, or apoptosis (57).

Despite the method ultimately used to repair the damage [homologous recombination (HR) or nonhomologous end-joining (NHEJ)], DSBs are initially sensed by the Mre11-Rad50-Nbs1 (MRN) complex. This highly conserved complex also has a role in the processing and repair of DSBs (see section “DNA Double Strand Break Repair Mechanisms”) (123). Each of the proteins within this complex has different functions in DNA damage signaling. Mre-11 is a 70–90-kDa protein that possesses a DNA binding activity and appears to function to recruit other DNA repair proteins to the site of DNA damage. It also has a DNA endonuclease and exonuclease activity (25). Rad50 is a 150-kDa protein that directly interacts with Mre11 (59) and functions to partially unwind double-stranded DNA termini, enabling other proteins to access the site of damage and carry out repair functions. Finally, Nbs1 is a 65–85-kDa protein that binds to phosphorylated proteins via its N-terminus, thereby allowing the recruitment of various DNA damage repair and checkpoint control proteins to the site of DNA damage. Therefore, this complex functions both to detect damage and control checkpoint signaling in both HR and NHEJ repair pathways. Recent studies have indicated that MRN may be recruited to DNA DSBs by the single-stranded binding protein, human single-stranded DNA binding protein 1 (hSSB1), that has a high affinity for short regions of single-stranded DNA (ssDNA) at broken DSB ends (101). Recruitment of hSSB1 to IR-induced DSBs precedes MRN recruitment and, through a direct interaction with Nbs1, hSSB1 mediates MRN recruitment to DSB sites (102,103).
Following DNA damage recognition by the MRN complex, activation of a series of transducer and effector proteins occurs to form the DDR. Ataxia telangiectasia mutated (ATM), another primary sensor of DNA double-strand breaks, is recruited to sites of DNA damage by the MRN complex, inducing the autophosphorylation of ATM at ser 1981 and its activation. Deficiency of the ATM gene results in the neurodegenerative disorder AT (108). AT is characterized by neurodegeneration, high risk of cancer, and hypersensivity to IR, a discovery that lead to the proposal of its key role in detection and repair of DNA DSBs. In undamaged cells, ATM exists as inactive homodimers and multimers. Phosphorylation of ATM at ser 1981 allows dissociation of these into monomers with the kinase activity capable of downstream signaling (7). ATM can then phosphorylate the histone variant 2AX (H2AX) at ser 139 up to ∼1 Mb on either side of a DSB to form γH2AX, which forms foci at the sites of damage that are visible by fluorescent microscopy (105). H2AX can also be phosphorylated by the other phosphatidylinositol 3-kinase (PI3-K) like proteins ataxia telangiectasia and Rad3-related protein (ATR), which is primarily activated at stalled replication forks (130), and DNA protein kinase (DNA-PK) (90), which is required for NHEJ. Phosphorylation of ATM, ATR, and H2AX leads to an amplification of the DNA damage signal and the recruitment of a large number of DNA repair and checkpoint control proteins to the site of DNA damage, including the mediator of damage checkpoint protein 1 (MDC1) (114,115), the p53 binding protein (53BP1) (131), the breast cancer susceptibility protein 1 (BRCA1) (21,136), checkpoint kinase 1 (Chk1) (140), Chk2 (2,35), and p53 (18) (see Fig. 2). Also recruited are members of the Fanconi anemia (FA) family of proteins such as FANCD2 (118), which co-operate with BRCA1 and BRCA2 in the FA/BRCA DNA repair pathway. A core complex of 8 FA proteins is recruited to sites of DNA damage, leading to ubiquitation of FANCD2 and recruitment of BRCA2 and RAD51 (65). Depending on the proteins recruited, the cell can undergo a number of fates, including cell cycle arrest, DNA repair via HR or NHEJ, apoptosis if the damage cannot be repaired, or senescence (96).
Recruitment of the MRN complex, phosphorylation of ATM, and the subsequent phosphorylation of H2AX occur within minutes of DNA DSBs being induced. The localization of the MRN complex to sites of DNA damage has been shown to be upstream of ATM phosphorylation and localization, as silencing of MRN proteins results in impaired ATM signaling (19,122). However, the interactions between ATM and the MRN complex are not fully understood, as MRN seems to be required for the recruitment and activation of ATM, but ATM also appears to phosphorylate members of the MRN complex, allowing further downstream signaling (122). Recruitment of ATM may be through an interaction of its PI3-K domain with the C-terminal of Nbs1 (34). ATM is not only activated in response to IR-induced DNA DSBs. Conditions that alter the chromatin structure such as exposure to topoisomerases also initiate ATM autophosphorylation; however, under these conditions, many of the key DDR substrates of ATM [such as H2AX, MDC1, structural maintenance of chromosome 1 (SMC1)] are not phosphorylated indicating that amplification of the DNA damage signal cascade by ATM through its downstream effectors requires recruitment of ATM to DSB sites (68).
Some studies suggest that ATM may be targeted to sites of DSBs by the monoubiquitination of H2AX by the BMI1/RNF2/RING1 E3 ligase complex (135). H2AX is then further di- or polyubiquitinated by RNF8 thus allowing recruitment of the BRCA1/RAP80 complex, which is required for DNA repair. ATM also stabilizes hSSB1 by phosphorylation at thr 117 in response to IR (101).
DNA Double-Strand Break Repair Mechanisms
DNA DSBs consist of two SSBs within one helical turn on opposing strands of DNA. IR-induced DNA DSBs are not usually simple lesions, but rather contain multiple complex lesions and overhanging ends (or ragged ends) of DNA that cannot be ligated directly and therefore must be removed before repair can begin.
DSBs are primarily repaired by one of two pathways, HR or NHEJ (see Fig. 3), irrespective of their cause (see sections “Choice of Repair Pathway” and “DSB and Radiation Quality”). Although the reasons why one repair pathway may be chosen over the other are not fully understood, the choice is thought to depend on the phase of the cell cycle, the presence of an intact sister chromatid, cell type (110), chromatin complexity (45), and the complexity of damage induced (87) (see section “Choice of Repair Pathway”).
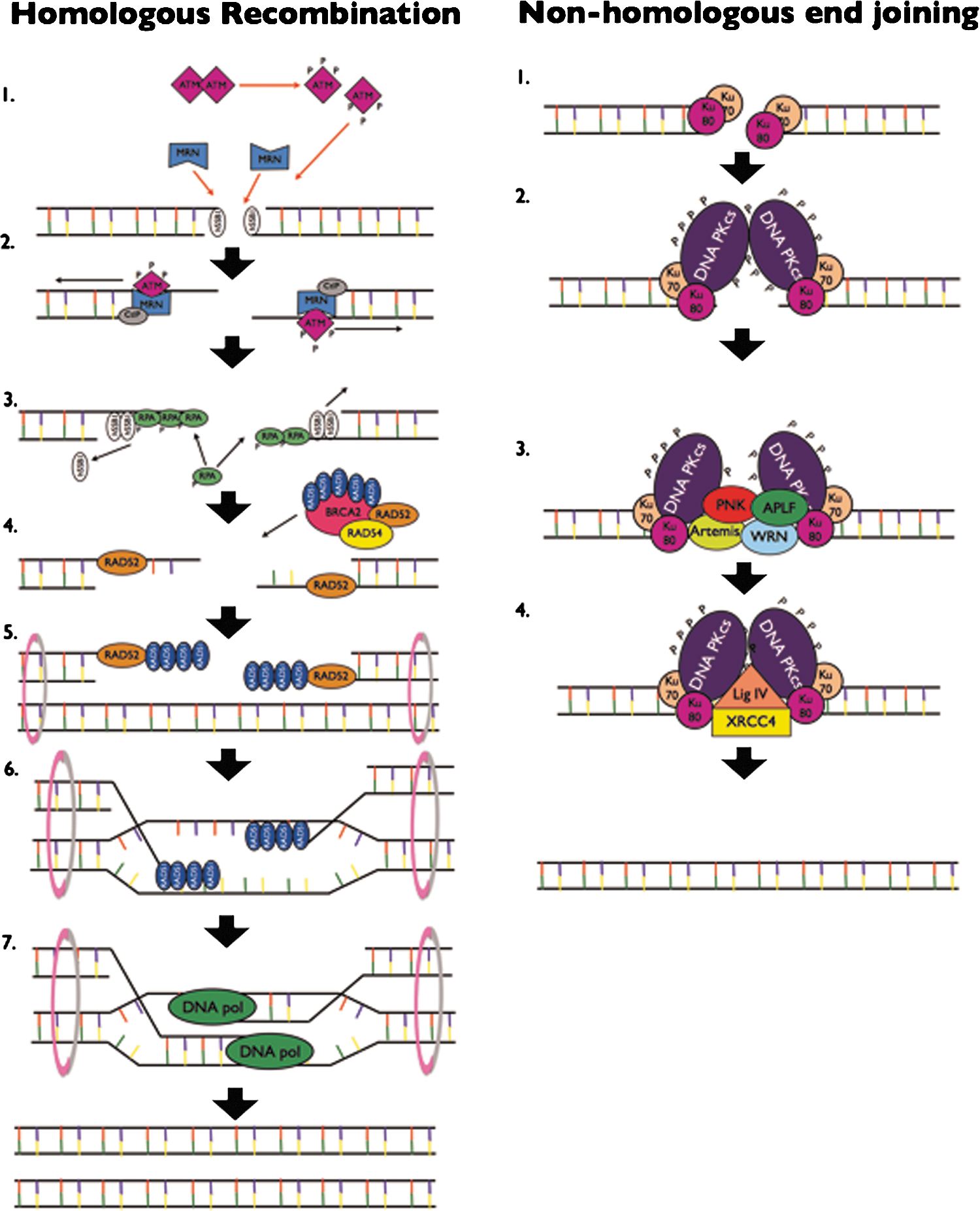
HR requires the presence of an intact sister chromatid as a template, and thus, this pathway is only available during late S and G2 phases (53). Therefore, during G1 (and early S-phase), when this option is not available, DNA DSBs are repaired by NHEJ. Although HR is considered to be a relatively error-free DNA repair method, even in the presence of a homologous chromatid, repair factors of each pathway compete for binding to DNA DSBs.
HR repair of IR-induced DNA DSBs
The major event that determines the decision for repair by HR is DNA end resection. A crucial event therefore is binding and the activity of the MRN complex was described in section “Sensing DNA Double Strand Breaks.” The MRN complex binds to DNA at the site of a lesion, recruiting and activating ATM (1,7,28). The endonuclease activity of Mre11 then facilitates 5′–3′ resection to produce 3′ ssDNA ends (58,138). This endonuclease activity is regulated by an interaction with the CtBP-interacting protein in an ATM- and BRCA1-dependent manner (107).
Following the formation of long tails of 3′ ssDNA regions, hSSB1 molecules are initially bound, and then displaced by the replication protein A (RPA) to begin the formation of nucleoprotein filaments that will eventually invade the homologous DNA strand of the sister chromatid. A soft X-ray microbeam has been employed to show that binding of the hSSB1 protein to resected 3′ ssDNA ends not only occurs before RPA binding, but is required for the RPA focus formation, thus elucidating an early step in the HR pathway (102). RPA is activated in response to IR by extensive phosphorylation (66) and plays a crucial role in DNA damage checkpoint signaling, control of DNA replication, as well as each of the main DNA repair mechanisms (143).
The RAD52/BRCA2/RAD51/RAD54 complex is then recruited to the ssDNA by a BRCA1/PALB2 (partner and localizer of BRCA2) complex. This facilitates the replacement of RPA with RAD51, thus stabilizing the filament and catalyzing the invasion into the sister chromatid and subsequent Holliday junction formation (55,78). Additionally, direct binding of RAD52 to the ends of DSB lesions protects them from the exonuclease activity. These vital roles of RAD52 in HR may indicate that it plays an important part in the decision for DSB repair by HR versus NHEJ (51). RAD51 focus formation can be observed in cells after exposure to IR. This focus formation does not occur in cells deficient for RAD54, and HR in these cells is less efficient (32).
To facilitate invasion into the sister chromatid strand, the two chromatids are tethered together by the SMC proteins 1, 3, 5, and 6 (also called cohesins) (40). Following invasion to the sister chromatid, the two strands are aligned with homologous regions within the sister chromatid. DNA synthesis, catalyzed by DNA polymerase δ (DNA pol δ), occurs from the 3′ end to replace the sequence of DNA disrupted by the break. Once this DNA replication is complete, the Holliday junction is resolved by DNA resolvases through a complex process that remains unclear (56). Finally, the DNA ends are ligated (55).
NHEJ repair
In late S and G2 phase, HR competes with NHEJ and in G1 cells, the latter, a relatively more error-prone process, dominates. The key factors involved in NHEJ are the Ku70/Ku80 heterodimer (Ku), the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs), the nuclease Artemis, XRCC4, DNA ligase IV, and the XRCC4-like factor (XLF). As IR-induced DNA DSBs frequently contain overhangs and often have phosphate or phosphoglycolate groups bound to their 3′ ends, these require processing by additional factors such as the DNA pol μ and DNA pol λ, polynucleotide kinase (PNK)/phosphatase, and Werner's syndrome helicase (WRN) before ligation can take place (76). The processing of these unligatable ends may result in a base loss from either strand and is likely to be the reason that NHEJ is error prone. For a comprehensive discussion of the protein and DNA interactions involved in NHEJ see Refs. (76,94).
The first step in this pathway is the rapid recruitment of Ku to IR-induced DNA DSBs (127), which occurs within a few seconds of irradiation (67). Ku70 and Ku80 together form a DNA binding core that has a high affinity for DNA without sequence specificity. This may explain the almost instantaneous recruitment of this complex to break sites. Shortly after recruitment to break sites, Ku translocates along the DNA leaving the ends accessible for processing. Binding of Ku results in conformational changes in the C-terminal regions of both Ku70 and Ku80. This further acts to recruit the other key NHEJ players to sites of DSBs. Two DNA-PKcs molecules are recruited to each DNA DSB via an interaction with the C-terminal region of Ku80 (111). These two molecules tether the DNA ends together and that interaction stimulates their kinase activity. This interaction of two DNA-PKcs molecules tethering the extreme ends of the broken DNA is often referred to as the “synaptic complex” (27). DNA-PK is then phosphorylated at more than 16 sites via autophosphorylation and potentially by other kinases such as ATM and ATR. The protein kinase activity of DNA-PKcs that is switched on by its interaction with DNA and Ku is thought to be a necessary step in NHEJ repair. Autophosphorylation of the ser 2023–2056 PQR cluster and the thr 2609–2647 ABCDE cluster of DNA-PKcs is required for repair of IR-induced DNA DSBs (20). There is also crossover at this point between the NHEJ and HR pathways. Deficiency of DNA-PKcs results in high radio sensitivity (81). Cells in which DNA-PKcs were mutated so that key autophosphorylation events were inhibited had reduced capacity to undergo HR and were more radiosensitive (4). Therefore, the kinase function of DNA-PKcs has a critical role in repair of IR-induced DNA DSBs. That function is believed to be important for its release from DNA DSBs and laser microirradiation studies have shown that when DNA-PKcs lacked this function it was retained longer at the sites of DNA damage (120).
Once DNA ends have been secured, they require processing to remove ragged ends. The specific enzymes that carry out this processing depend on the complexity of the breaks, whether gaps in the DNA need to be filled, and what groups are blocking the ends of the DNA. The exact timing of DNA end processing in NHEJ and the order of recruitment of many factors remain unclear, but the most likely key players in this stage of NHEJ are summarized next.
The nuclease Artemis has a 5′–3′ exonuclease activity and acquires the endonuclease activity in the presence of DNA-PKcs and adenosine triphosphate (ATP). The inactivation of the enzyme Artemis causes radiation-sensitive severe immunodeficiency indicating that it must have a role in repair of radiation-induced DNA damage. However, cells lacking Artemis still have some DSB repair suggesting that it is only required for the repair of a subset of breaks (see section “DSB and Radiation Quality” for more on this). One proposed mechanism of recruitment of Artemis during NHEJ is by an interaction with DNA-PKcs. Proper function of Artemis in NHEJ requires the activation of its endonuclease function. Exactly how this occurs remains unclear; however, in vitro Artemis has been shown to be a phosphorylation target of both DNA-PKcs and ATM. Artemis has a role in the processing of IR-induced DNA DSBs with DNA hairpin loops, 3′-phosphoglycolate groups, and functions in the repair of complex lesions in an ATM-dependent manner (76).
PNK is phosphorylated in response to IR although the kinase responsible is unknown (79). Knockdown of PNK results in increased IR sensitivity, most likely due to defective NHEJ DSB repair (62). PNK is involved in the removal of nonligatable ends at the termini of DSBs. Its DNA phosphatase and DNA kinase activities are key to this function. PNK may be recruited to DNA DSBs by an interaction with phosphorylated XRCC4. DNA pol μ and DNA pol λ are recruited to sites of IR-induced DSBs by their interaction with Ku (and possibly XRCC4-DNA ligase IV) and are required to fill gaps in the DNA created during the removal of ragged DNA ends. Their function in NHEJ is thought to be mainly through their respective BRCA1 C-terminus (BRCT) domains. DNA pol λ is phosphorylated in response to IR (79). Which DNA pol is required depends largely on whether a DNA template is required. While DNA pol λ normally requires a template to fill gaps, DNA pol μ can direct template-independent synthesis across a DSB with no terminal microhomology (86). Loss even of both these polymerases does not confer a significantly increased sensitivity to IR; therefore, it seems that they are required for the repair of only a small subset of IR-induced DSBs (92).
Aprataxin and PNK-like factor (APLF) also has both the exonuclease and endonuclease activity, is phosphorylated by ATM, interacts with Ku, and its downregulation results in defective DSB repair. These findings together suggest a role for APLF in NHEJ repair. There is also evidence that the Werner's syndrome protein (WRN) may have a role in NHEJ. WRN is a RecQ helicase that has a number of functions that would be useful in processing nonligatable DNA ends. These include 3′–5′ exonuclease, 3′–5′ DNA helicase, strand annealing, and DNA-dependent ATPase activities. Its exonuclease activity is stimulated by its interactions with Ku and the XRCC4-DNA ligase IV complexes. Finally, loss of the WRN activity in vivo is associated with cancer predisposition, premature aging, and genomic instability, thus suggesting a role in DNA damage repair.
To complete the repair of DNA DSBs, the processed ends must be ligated. In NHEJ, this is done by the XRCC4–DNA ligase IV complex. Although XRCC4 is required for NHEJ, it has no enzymatic function on its own, but instead acts as a scaffold bringing other factors to the sites of DSBs (61). XRCC4 has an α-helical region that interacts with a linker region between the two C-terminal BRCT domains of DNA ligase IV (48). This forms a very stable complex that stabilizes the activity of DNA ligase IV and via the interaction of XRCC4 with Ku brings it close to DNA ends requiring ligation (47). It has been proposed that in response to DNA DSBs, XRCC4 is phosphorylated by DNA-PKcs and that this stabilizes the complexes position at the break site and promotes ligation. Ligation, however, is not dependent on that phosphorylation. The nuclear localization of XRCC4 and its role in DSB repair is also dependent on its SUMO-ylation. Further to DNA end ligation by the XRCC4–DNA ligase IV complex, XLF is also required for a subset of DNA lesions where the processed ends are incompatible. The recruitment of XLF to DNA ends is independent of, but is stabilized by the XRCC4–DNA ligase IV complex.
Alternate NHEJ
An alternative end-joining pathway may be employed when some parts of the NHEJ pathway are missing or mutated. There is a crossover between this pathway and some components of HR and SSB repair (80) pathway are able to ligate broken DNA ends in the absence of DNA-PKcs, XRCC4, and DNA ligase IV. Instead, this pathway may employ PARP1, XRCC1, and DNA ligase III (128). In this case, PARP1 may bind the DNA DSBs and stimulate fusion. The XRCC1/ligase III complex then ligates the DNA ends. When Ku is present, it blocks the access of PARP1 to DSBs. Thus, alternative end-joining only occurs in NHEJ-deficient cells (129).
Choice of Repair Pathway
Following IR-induced DNA DSB induction, in cells in the G1 or S/G2 phase, there is competition for binding to the lesion by the Ku and the MRN complex. The outcome of that competition in S-phase dictates the DNA damage repair pathway. Binding of MRN and its activation and recruitment of ATM commits cells to repair by HR, whereas successful binding of Ku70-Ku80 takes repair down the NHEJ route. Localization of the MRN complex at DSBs is independent of the cell cycle phase (94). A further interaction that may dictate the choice of the repair pathway in late S-phase is that of 53BP1 and BRCA1. 53BP1 binds DNA near sites of DSBs and 53BP1 focus formation can be observed by fluorescence microscopy after IR exposure. It may have a role in inhibiting end resection by binding to dimethyl-histone H4 Lys20 and tethering DNA ends to facilitate ligation. The prevention of DNA end resection by 53BP1 indicates that 53BP1 binding may favor repair by NHEJ. Conversely, it is thought that BRCA1 can counteract the action of 53BP1, but the mechanism is unclear (13).
There is increasing evidence that the DSB repair rate and the choice of repair mechanism are dependent on chromatin complexity. DSBs that are repaired slowly may be in regions of heterochromatin that require prolonged chromatin relaxation for repair intermediates to access break sites. It has been shown experimentally that sustained activation and localization of ATM at these break sites is crucial for this and that ATM functions in this process to activate the KRAB-associated protein (KAP-1). pKAP-1 forms persistent foci that colocalize with γH2AX and these are thought to represent DSBs in heterochromatin. The same study also suggested that DSBs in regions of heterochromatin are repaired in G2 phase by an Artemis-dependent HR pathway. In contrast, most DSBs in euchromatic regions were repaired during G2 by NHEJ (45,142).
DSBs and Radiation Quality
Complex lesions
The type of DNA lesions that result from cellular exposure to IR is heterogeneous and, in general, the complexity of breaks strongly depends on the characteristics of the incident radiation. The term “radiation quality” is commonly used to discriminate the density of ionizations produced along the path of a charged particles track as this is correlated with the density of DNA lesions caused. Linear energy transfer (LET) is the amount of energy per unit distance that is transferred by a particle to the surrounding medium along its trajectory. This factor depends on the charge and kinetic energy of the particle and is normally reported in units of keV/μm. In first approximation, the LET is inversely proportional to the square of the particle kinetic energy, proportional to the square of the particle charge, and independent from its mass as indicated by the Bethe–Block formula (9);
v=velocity of the particle
c=speed of light
β=v/c
E=energy of the particle
x=distance travelled by the particle
Z=particle charge
e=charge of the electron
merest =mass of the electron
n=electron density of the target
I=mean excitation potential of the target
ɛ0=vacuum permittivity
Although LET is a quantity defined for charged particles, an LET value can also be calculated for X-rays leading to an average value of 2 keV/μm for 250 kVp X-rays. LET is therefore an indication of the ionization pattern (i.e., track structure) produced by a particle beam at a certain energy (141). The pattern of DNA damage caused by a particle traversal is directly related to the LET and track structure of the particle beam. As the primary charged particle traverses a cell, it causes ionizations itself that are capable of causing DNA DSBs due to their spatial and temporal proximity. These ionizations generate secondary delta electrons that have tracks away from the trajectory of the primary particle. The delta electrons are able to cause further ionizations. The result is a penumbra around the primary particle path, the width of which increases with increasing energy of the primary particle. As charged particles slow down (lose energy), the track length of the delta electrons also decreases so the pattern of ionizations has a higher density (and LET) near the end of the primary particles' path. This corresponds with the peak of energy deposition for charged particles known as the “Bragg peak.”
For charged particles, particularly heavy ions, the probability of causing complex, or clustered DNA damage increases as LET increases and therefore as a given particle slows down. Complex DNA damage caused by high LET radiation is believed to be more difficult for the cell to repair and therefore more lethal. An excess of small DNA fragments (<3 Mb) has been observed using pulsed field gel electrophoresis (PFGE) of DNA after irradiation of human fibroblasts with charged particles of different LET (80–300 keV/μm). This was not observed after low LET irradiation. These small fragments may be created as a result of intratrack ionizations (113).
A DSB is a type of clustered lesion as these breaks are usually formed from two DNA lesions on opposite strands within 10 bp of each other. However, complex DNA DSBs can also involve two or more breaks on each DNA strand within 10 bp (23). DSBs induced by high LET ions are ∼70% complex DSBs and 30% simple DSBs, compared to low LET particles such as X-rays that cause only 30% complex DSBs. Clustered lesions also include non-DSB damage and are most likely made up of closely opposed SSBs and abasic damage sites (49). These complex lesions, that potentially occur at four- to eightfold higher yields than prompt DSBs, are also less repairable than single lesions and can lead to a DSB during repair (30). Complex damages present a harder challenge for the cellular repair processes due to clustering of lesions and the loss of DNA fragments. Clustered DNA lesions are more likely to be misrepaired or unrepaired conferring an increased probability of chromosome aberrations, mutation, and cell death for high LET radiation (5).
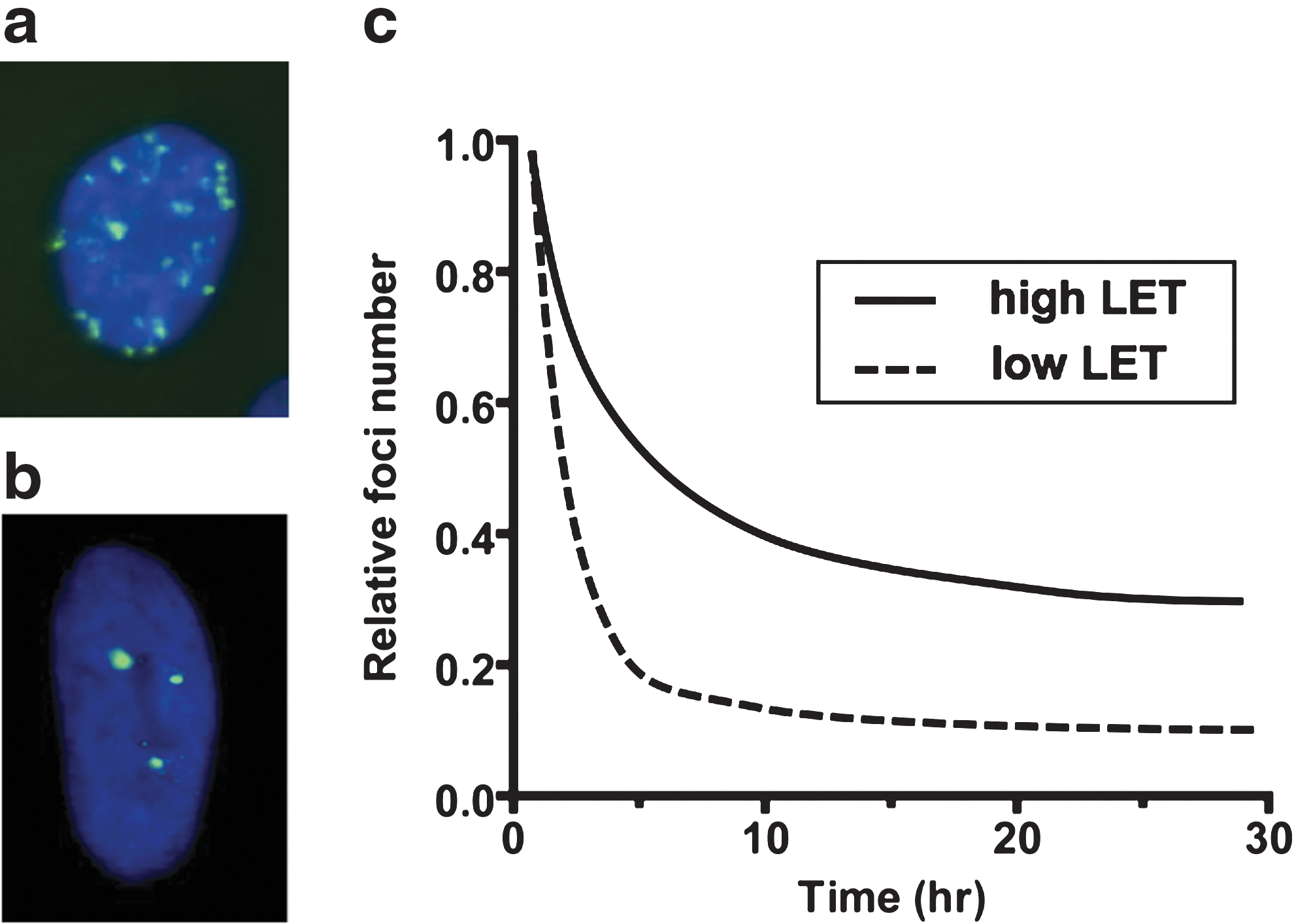
The main methods for studying induction and repair of DNA DSBs in radiobiology are PFGE and inspection of ionizing-radiation-induced foci (IRIF) (see Fig. 4a, b). Although historically the PFGE method has been used most frequently, IRIF-based assays provide higher sensitivity thus enabling their use at radiobiologically relevant doses (<2 Gy) and are a versatile platform for the study of DNA repair factor dynamics. The induction and resolution of IRIF has been employed for the investigation of the role of many factors involved in the sensing and repair of DNA DSBs, including, but not restricted to, pATM, 53BP1, BRCA1, γH2AX, MRN, RAD51, and DNA-PKcs (see Fig. 4c for a schematic representation of IRIF resolution over time post-IR exposure). With the advent of the green fluorescent protein and other live cell tags, it is now also possible to qualitatively and quantitatively monitor the DNA repair process in live cells and assess the protein interaction and turnover at the site of damage using techniques such as Forster resonance energy transfer and fluorescent recovery after photobleaching. The effectiveness of heavy charged particles for the induction of complex DNA DSBs has been shown using both methods (6,95,121). Imaging of IRIF has also shown that very early (<1 h) after irradiation with high LET radiation, foci are larger and more intensely fluorescing compared to those resulting from X-irradiation (22). IRIF localize in stripes through cell nuclei along heavy ion tracks and the colocalization of DNA repair factors involved in multiple repair processes suggests the highly complex nature of the lesions induced (85). These complex lesions may include multiple DSBs, SSBs, and abasic sites within close proximity that appear as a single larger and brighter focus, but are repaired slower than those observed following low LET exposures (23).
Mechanism for repair of high LET complex DSBs
Repair kinetics studies have identified two specific components: a fast one that is responsible for the majority of the repair and which dominates the repair response in the immediate hours postirradiation, and a slower component that protracts for 24 h and longer. It has also been shown that the slow phase of DNA repair is longer for high LET-induced DNA DSBs than for low LET DSBs. This is most likely due to a higher frequency of complex clustered damage that requires additional processing before DNA ligation can occur and due to the presence of unrepairable lesions. Analysis of DNA rejoining by PFGE demonstrated an LET-dependent increase in rejoining time up to ∼200 keV/μm (106,133). Studies of IRIF resolution kinetics has provided further evidence for the complex nature of high LET particle-induced lesions. The disappearance of heavy ion-induced foci is slower compared to X-rays, and heavy ion (such as 1 GeV/nucleon iron ion) exposures induce a proportion of foci that persist even beyond 48 h after exposure (6,88,121). These persistent foci are generally believed to represent unrepaired DSBs; however, these have also been associated with IR-induced senescence and chromatin alterations (116,117).
Precisely how high LET DNA DSB processing differs from that of simple DSBs is yet to be elucidated. The kinase ATM and nuclease Artemis have been associated with the repair of complex DNA DSBs (73,100). Recent evidence suggests that complex clustered DNA damages that include DSBs with other DNA lesions close to them are processed in the slow phase of repair by a subpathway of NHEJ that is dependent on ATM, DNA-PKcs, and Ku (99). That study showed that simple DSBs (induced by ultrasoft X-rays) are repaired by an NHEJ pathway that does not require ATM or DNA-PKcs; in contrast, those factors were necessary for repair of more complex lesions (induced by multiphoton near-infrared microbeam). This is probably due to a necessity for extensive DSB end processing in these complex lesions. Finally, evidence suggests that slow kinetics of repair of high LET radiation lesions may be strongly influenced by chromatin complexity. Proposed methods for dealing with heterochromatic DSBs are described in section “DNA Double Strand Break Repair Mechanisms.”
DSBs and Replicative Stress
Oxidative stress-induced DSBs
DNA DSBs arise following direct damage to DNA by IR. However, other mechanisms involving oxidative stress can also lead indirectly to DSB induction (see Fig. 5). ROS can be produced in cells as a consequence of various cellular processes, including CO2 metabolism, stimulation of immune responses, or release of cytokines and chemokines from neighboring cells. ROS can have a number of different effects on carbohydrates, proteins, lipids, and DNA (70), including causing the oxidation of purines and pyrimidines, AP DNA sites and SSBs. The majority of this damage is repaired using base excision repair (BER) or nucleotide excision repair (NER). BER is important for the removal of nonbulky modifications and abasic sites and involves removal of a damaged base by a DNA glycosylase, incision at the remaining site, addition of an undamaged base by DNA pol and finally, religation of the DNA by a DNA ligase. NER is important in repair of bulky base modifications, which cause distortion of the DNA helix and requires the damage to be recognized by xerodermapigmentosum/cockayne syndrome proteins, DNA unwinding and incision, addition of an undamaged base and ligation. However, it is estimated that ∼1% of these single-strand lesions are converted into DSBs (118), particularly at replication forks during DNA replication. Recognition of these DSBs differ from DSBs induced by direct IR in that ATR appears to play an important role, as well as ATM. They are then consequently repaired by NHEJ or HR.

Hypoxia
Relative oxygen concentrations within cells have also been reported to influence both levels of DNA damage induced and how this damage is repaired. Hypoxia within tumors has been associated with increased radioresistance and decreased disease-free survival (126). DNA repair pathways, including HR and NHEJ, have been shown to be less effective in hypoxic conditions (89). This is thought to be due to the alteration of expression of DNA DSB-associated genes [reviewed by Bristow and Hill (14)]. Cellular responses to hypoxia are mediated primarily by the hypoxia inducible factor family of transcription factors. DNA DSB repair proteins downregulated in response to hypoxia include BRCA1 (10), RAD51, RAD52, and BRCA2 (82). The result of this is that DNA DSB repair by HR is compromised in hypoxic conditions, potentially leading to an increase in biological effects, including translocations or chromosomal deletions.
Oxidative stress may also positively or negatively impact on DNA damage recognition and repair. ATM has been shown to be activated by oxidation in the absence of either a DSB or MRN recruitment indicating that it may be a key mediator in the cellular response to oxidative stress (50), while DNA-PKcs, which stabilizes pP53, has been shown to be negatively regulated by Artemis during oxidative stress, and therefore preventing G1 arrest and apoptosis (139). Further, repair of DSBs by NHEJ under conditions of oxidative stress is impeded due to an inability of Ku to bind to the lesion. This may be due to an oxidation-mediated conformational change of Ku (8).
Bystander effects
DNA DSBs can also result following signaling from neighboring cells that have been damaged by radiation. The radiation-induced bystander effect is defined as the ability of an untreated cell to respond to signals from a neighboring irradiated cell (96). Bystander effects have been observed in a wide range of cell lines and in vivo (96). The damage can arise in two ways, either through intercellular communication via gap junctions, or via the release of soluble factors, which can travel to distant cells. The soluble factors released are dependent on the cell type, which has been irradiated, but have been reported to include chemokines/cytokines (29,91,109), death ligands (74), ROS (75), nitric oxide (75,109), growth factors (29), and Ca2+ (75). Bystander effects are reported to be the result of increased ROS generation in nonirradiated cells as a consequence of signaling from irradiated cells, most likely due to increases in intracellular Ca2+ and subsequent mitochondrial depolarization and ROS release (75). Endpoints for which bystander effects have been observed include DNA damage, mutations, terminal differentiation, and apoptosis [reviewed by Prise and O'Sullivan (96)]. DNA damage occurring in cells following bystander signaling differ from the responses of directly irradiated cells, with damage being detected at later time points. Bystander-induced DNA damage is also primarily detected by ATR, with ATM found to act downstream of ATR in bystander cells (see Fig. 5) (16,96).
Targeting DSB Repair in Cancer Therapy
The aim of radiation therapy is to induce DNA damage in the form of DSBs, which ultimately leads to cell death within the tumor. However, resistance is a major mechanism limiting the effectiveness of radiotherapy as a cancer treatment. One way in which tumor cells can develop resistance to radiation treatment is by improving the effectiveness of DNA repair pathways, thereby enabling the cells to repair the damage and allowing the cells to survive and replicate. Targeting these repair pathways may therefore be a good therapeutic option to try to improve the effectiveness of radiation treatment.
Expression of DNA damage repair proteins in cancer
AT is a condition resulting from mutation of the ATM gene. It is characterized by a predisposition to cancer and very high cellular sensitivity to radiation treatment. As explained in sections “Sensing DNA Double Strand Breaks” and “DNA Double Strand Break Repair Mechanisms,” ATM plays a role early in the DDR network, being one of the first proteins to be phosphorylated following DNA damage detection. It is therefore easy to see why loss of function of ATM can lead to an increased predisposition to cancer development, as loss of ATM has a major impact on the cells ability to repair damage efficiently. Mutations in ATM lead to a much higher risk of development of breast cancer (98,119) and leukemia (77), while also reducing time to recurrence. Mutation of ATM has also been reported to lead to an increased sensitivity to radiotherapy treatment (17), indicating that this protein may be important in radiation response.
Altered expression of other proteins in DNA repair pathways have been shown to both increase risks of developing cancer and influence the effectiveness of treatments designed to induce DNA damage. For example, overexpression of members of the FA family of proteins, which function to co-operate with BRCA1 and BRCA2 in DNA damage repair by HR, leads to a higher risk of developing various types of cancer, including breast cancer (31,41,97,112,124), leukemia (33,54), and pancreatic cancer (24,52,60). In addition, low expression of FA proteins have been shown to predict a response, with low FANCD2 expression associated with an increased response of esophageal cancer patients to treatment with a combination of chemotherapy and radiotherapy (3). It is also widely known that mutations of the DNA damage repair proteins BRCA1 and BRCA2 significantly predispose carriers to development of breast and ovarian cancer, with up to 85% of these tumors associated with changes in expression levels of BRCA1 or BRCA2 (39). BRCA status may also be important in patient outcome, with BRCA2 mutations reported to correlate with improved survival and chemotherapy response in ovarian cancer patients (137). PARP, which is important for detecting single-strand DNA breaks, plays a key role in the response of HR defective cells to DNA damage, as unrepaired SSBs can be converted to DSBs, which the cells cannot effectively repair. PARP is overexpressed in breast cancer compared to normal breast tissue, and its overexpression has also been correlated with high grade, tumor size, and worse survival (44). These studies highlight the importance of DNA repair pathways in cancer treatment outcomes and the need to target these pathways to improve response rates.
Therapeutic inhibition of DNA repair pathways
Various inhibitors have been developed to target DNA repair proteins. A dual PI3-K/mTOR inhibitor, NVP-BEZ235, has been reported to potently inhibit both ATM and DNA-PKc, thereby inhibiting both HR and NHEJ pathways. This inhibitor has shown promising results, showing significant radiosensitization in a panel of cell lines and in tumor xenograft models (85). In addition, this inhibitor is currently in Phase 1/2 clinical trials for the treatment of solid tumors. Inhibition of ATM using different inhibitors has also shown to significantly increase sensitivity to radiation treatment [reviewed by Kuroda (71)]. Interestingly, the inhibition of ATR function in hypoxic cells, which show higher levels of radiation resistance, using the specific inhibitor VE-821 has been shown to sensitize these cells to radiation treatment, indicating that this approach may be important in a therapeutic setting for the treatment of tumors with hypoxic regions (93).
Chk1/Chk2 inhibitors have also been tested in combination with DNA damaging agents. The inhibitor AZD7762 has been used in pancreatic cancer cells both in vitro and in tumor xenograft models and has shown to be effective at inducing cell death and tumor regression both as a single agent and in combination with gemcitabine and radiation (84). Mitchell et al. have also shown that AZD7762 can sensitize a panel of human tumor cell lines to radiation treatment, and in addition, can sensitize HT29 xenograft models to radiation with minimal side effects from treatment with the inhibitor both alone and in combination with radiation (83), indicating the potential of this inhibitor as a clinical agent. Other Chk inhibitors have also been shown to increase radiosensitivity of tumor cells, such as XL-844 (104) and PF-477736 (37), which has shown potential both as a single agent and in combination with DNA damaging agents (11).
Synthetic lethality
Many types of cancer arise from cells that have developed mutations in DNA DSB repair pathways. Alterations in these pathways allow cells to accumulate damage and other mutations, but still retain their ability to divide. However, cells possess numerous DNA repair pathways, which can be activated following DNA damage. If one of these pathways is inactivated in tumor cells due to mutations of key proteins within the pathway, the tumor cells can become more dependent on other repair pathways to repair replication-associated and other types of DNA damage. This opens up important therapeutic options, as targeting the intact pathways should prevent the tumor cells from repairing any DNA damage incurred and allow the cells to undergo apoptosis. This concept has been named synthetic lethality (see Fig. 6).
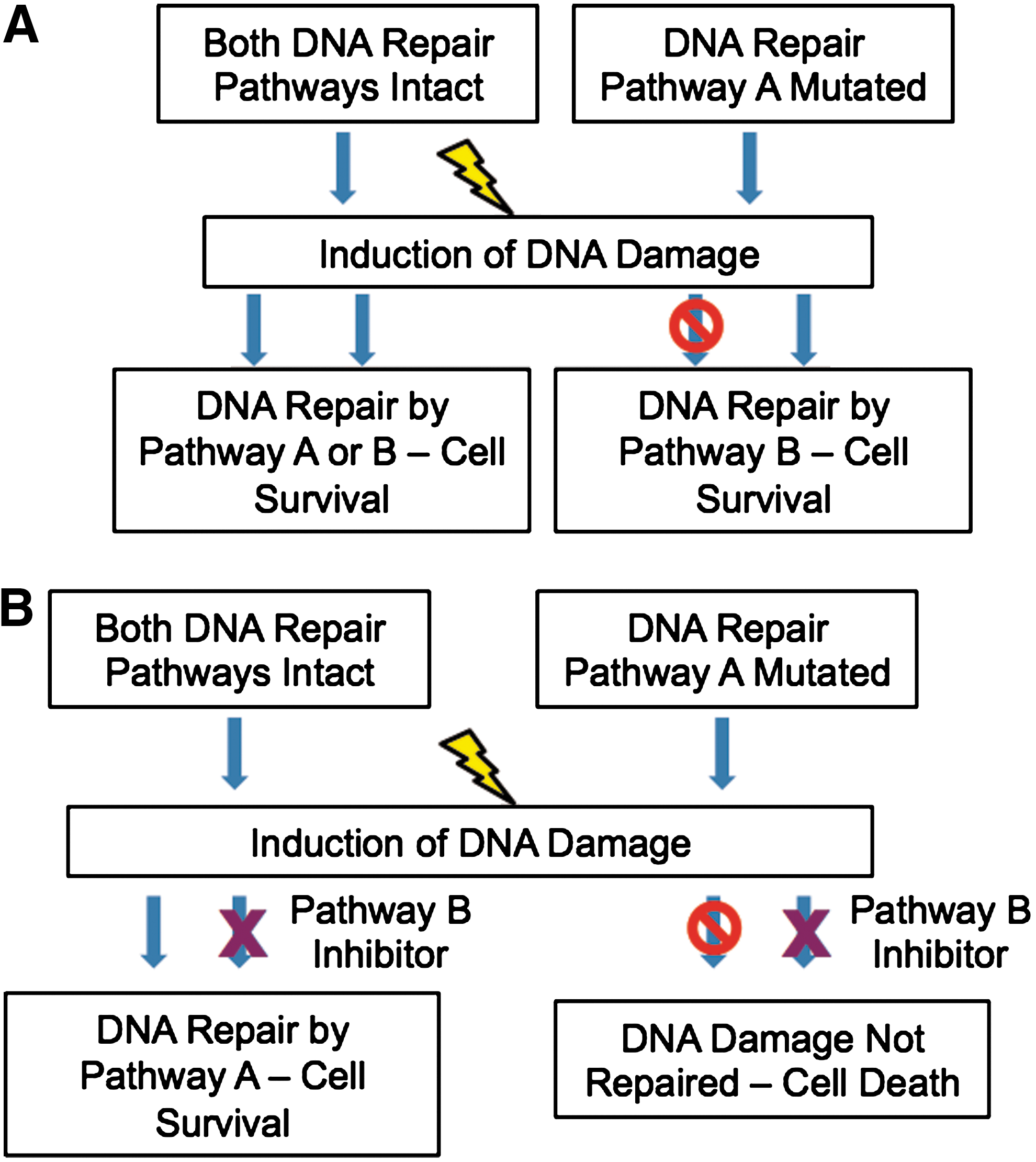
Synthetic lethal interactions can be seen between a number of proteins, most notably in the use of PARP inhibitors in a BRCA mutant setting (36). Cells that have developed a mutation in BRCA1 or BRCA2 have defective HR function. Inhibition of PARP results in an accumulation of SSBs, which can be converted into DNA DSBs at replication forks. These DSBs cannot be repaired in HR-deficient cells, resulting in the cells undergoing apoptosis. PARP inhibitors may be very useful in the treatment of patients with tumors possessing BRCA mutations, as tumor cells have developed mutations in BRCA1 or BRCA2, while the surrounding normal tissue will not have these mutations. This means that the inhibitors should specifically target the tumor cells, while sparing the surrounding normal cells (36), which have an intact HR pathway to repair the damage.
Inhibition of PARP has been shown to sensitize cells to radiation treatment (125). Importantly, the use of PARP inhibitors or PARP-targeted siRNA as a single agent has been shown to result in a significant reduction in cell survival of BRCA2 mutant cells (15,36). In addition, xenograft models developed from patient tumor samples have shown that PARP inhibitors induce significant apoptosis and reduce proliferation of BRCA2 mutant tumors, both as a single agent and in combination with DNA damaging agents, while BRCA2 wild-type tumors show greatly reduced responses to PARP inhibition (69). These studies and others have indicated the importance of PARP inhibitors as a treatment strategy, leading to the testing of PARP inhibitors in clinical trials. In a Phase I clinical trial, the PARP inhibitor olaparib was shown to exhibit an antitumor activity in patients with BRCA1 or BRCA2 mutations, while causing few adverse side effects (38). This inhibitor was also tested in Phase II clinical trials. Gelmon et al. reported that 41% of BRCA1 or BRCA2 mutant ovarian cancer patients showed a response following treatment with olaparib, compared to 24% of patients without mutations (43). However, another study reported that there was no statistically significant improvement in progression-free survival in ovarian cancer patients with BRCA1 or BRCA2 mutations treated with the inhibitor (63). Astra Zeneca have withdrawn olaparib from Phase III trials after analysis of the data indicated that there was no improvement on overall survival following treatment of BRCA1 and BRCA2 mutant ovarian cancer patients. However, it is still being investigated in other disease settings, such as BRCA1/2 defective breast cancer. There are also other PARP inhibitors in various phases of clinical trials, including iniparib (which was the first PARP inhibitor to be tested clinically), rucaparib, and veliparib.
PARP inhibitors also show synthetic lethality when combined with ATM mutations (134), as well as mutations in MRE11 and NBS1 (26). Synthetic lethal interactions are also observed between mutations of the FA family members and ATM inhibitors (64).
Summary
DNA double-strand breaks are critical lesions for cells, which if not repaired can lead to a high probability of mutations and cell death. Taking IRs as an example of an effective inducer of DNA DSBs, lesion complexity, where additional damages are associated with a DSB due to the radiation track structure, is also important causing additional challenges for repair pathways. DSBs can also be produced indirectly via oxidative stress and the consequences of replication fork stalling, typically seen for IR-induced bystander responses. Cells have evolved several options for sensing and repairing DSBs with the molecular mechanisms being defined. The restricted options of repair pathways available to tumor cells, when specific mutations in key repair enzymes are present, allow selective targeting of these particularly via synthetic lethality approaches. Our expanding knowledge of DSB induction, using IR as a paradigm, is providing new opportunities to target these in cancer therapy.
Footnotes
Acknowledgments
The authors are grateful to Cancer Research UK (C1513/A7047), Medical Research Council (G1100014), Engineering and Physical Sciences Research Council (EP/1005714/1), Breast Cancer Campaign (2009MayPR03), and the European Commission (EPIRADBIO project No. 269553) for funding their work.