
Abstract
Introduction
T

Since the extracellular environment is also highly oxidizing, the oxidative folding of proteins destined to the extracellular space is critical for their stability (76). In contrast, cell cytosol and nucleus are reducing environments due to the high levels of redox enzymes and reduced glutathione (GSH), and absence of enzymes generating reactive oxygen species (ROS) as part of normal metabolic function. Therefore, most cysteines in cytosolic and nuclear proteins are free (Fig. 1). Thanks to the low background of ROS, a specific signaling mechanism dependent upon ROS produced by membrane-bound enzymes, such as NADPH oxidases (NOX) and mitochondria systems, may operate in the cytoplasm (21).
These signaling ROS (73) mediate reversible oxidation–reduction of cysteine residues of cytoplasmic proteins that serve as transducing elements. The redox signaling based on cysteine modifications is implicated in many vital cell functions, including cell proliferation and survival.
Newly generated ROS are rapidly detoxified by the abundant cell antioxidant systems; however, an excess of ROS generated for increased mitochondrial oxidative metabolism or in response to xenobiotics, cytokines, and pathogen invasion, may overcome the antioxidant capacity of the cell and lead to oxidative stress (26). It is generally accepted that oxidative stress damage cell structures, including lipids and membranes, proteins, and nucleic acids and is implicated in various disease states, including cancer (Fig. 2).

Are ROS Causative Agents in Cancerogenesis and Tumor Progression? Failure of Antioxidants in Cancer Prevention and Therapy
Traditionally, ROS, mainly consisting of superoxide anion radical (O2.−), singlet oxygen, hydrogen peroxide (H2O2), and the highly reactive hydroxyl radical, have been simply viewed as a group of molecules harmful to cells, tissues, and organisms. In the late 1990s, however, it was proposed that ROS may act as signaling molecules (30). More recently, it became clear that ROS are essential component of the cell signaling pathways that directly interact with critical molecules to initiate a broad variety of cellular processes.
In spite of the acknowledgement of ROS as fundamental players in cell life, the excess of ROS, and the consequent oxidative stress, is commonly considered to cause cell damage and disease (102). In oncology, the most credited view reckons that high levels of intracellular ROS, induced by exogenous oxidizing conditions (ionizing radiations, xenobiotics) or endogenous distress, induce cancer (11, 33) and its progression by targeting DNA and proteins, increasing mutagenesis, converting protooncogenes into oncogenes, inactivating tumor suppressor genes, and eventually resulting in differentiation block, uncontrolled cell growth, and impaired cell apoptosis (102). Cancer development and progression is also tightly linked to inflammation (60). ROS produced by cells of the innate immunity would contribute to increase the risk for cancer initiation and help sustaining cancer progression, thereby strengthening the vicious circle inflammation, oxidative stress, and cancer (102).
Based on these considerations, much effort has been put in developing therapies directed at eliminating or normalizing ROS and ROS-related cell damage. Unexpectedly, in spite many cell culture and animal experiments showed apparent benefits of antioxidants in anticancer therapy, in human antioxidants fail or, worse, show evidence of unexpected harm, as demonstrated by several meta-analyses of randomized clinical trials (13, 31, 42, 44, 65, 70). These results weakened the ROS hypothesis of cancer and raised the alternative hypothesis that oxidative DNA damage is an epiphenomenon to an ongoing pathophysiological process, and elevated levels of ROS do not have a role in carcinogenesis (19).
Limits and Contraindications of the Available Experimental Systems to Study the Role of Redox State and Response in Cancer
The divergences between the expected results, based on the plethora of data obtained from basic and preclinical studies, and the actual results obtained from clinical trials with antioxidants in tumor prevention or therapy may indicate that the experimental systems used to study the role of redox in tumorigenesis and tumor progression are inadequate. The experimental systems most commonly used in this field are in vitro study on cultured cells, animal models, and analyses of samples from primary human tumors.
In vitro experiments have many limitations, including the difficulty to mimic tissue microenvironment, which is known to strongly affect tumor development and progression.
More important in the context of redox studies, cell culture experiments are often misleading in answering metabolic questions. In fact, redox status and metabolism are highly sensitive to culture conditions (39, 67, 75). Cultured cells are usually kept at an O2 tension of 21%, very distant from the 2%–4% O2 of normal tissues. Anti-oxidant and pro-oxidant enzymes, quiescent and substrate-limited at low O2 tension, are induced in hyperoxia (69). In addition, unlike the majority of normal and also neoplastic cells in living tissues, which are postmitotic, cultured cells are cycling and are enriched in NADPH to ensure macromolecule synthesis and hence proliferation (67). The net result of increased NADPH and hyperoxia is amplification of ROS production by NOX enzymes, and consequently, the induction of the antioxidant master regulator NF-E2-related factor 2 (Nrf2) (50) and of the adaptive response genes (69). Since antioxidants ensure faster growth, resistance to apoptosis and other benefits to immortalized cells, cells overexpressing antioxidant enzymes, are selected and predominate in most continuous cell lines. The abnormal redox state of cultured cells results in abnormal redox response to any trigger, often causing misinterpretation of the results (9, 75).
On the other hand, the study of the role of redox response in animal models and in primary human tumors is limited by the difficulty to directly measure ROS in vivo (14, 59).
Most ROS are highly reactive and short-lived and therefore hard to detect in complex biological matrices. A number of novel approaches based on genetically encoded sensor proteins for in vivo measurement of cell redox status have been developed (5, 24, 61, 63, 90). Although highly promising for the study of redox responses in vivo, a limit of these approaches in the field of cancer is that their use is restricted to transgenic animals or transfected cells. ROS can also be measured indirectly, following the formation of oxidative by products of lipids, proteins, or nucleic acids. Using this approach, several studies reported an increase in cancer patients of 8-oxo-2′deoxyguanosine, an established biomarker of oxidative stress (19). However, also many nononcologic pathological conditions exhibit elevated levels of oxidative DNA damage, with no increased incidence of carcinogenesis.
An additional constraint is represented by the high heterogeneity of tumor tissues, with the presence of (many) differentiated and (few) undifferentiated cells. To be relevant, the oxidative damage should occur at the level of undifferentiated, proliferating stem cells, but current analytical procedures for the detection of 8-oxo-2′deoxyguanosine do not allow discriminate whether the most important target cells are indeed more oxidized. We have recently shown that tissues staining with 2′,7′-dichlorofluorescein diacetate (H2DCFDA) is effective for detection of ROS in inflamed tissues such as lymph nodes and muscles (92, 94). However, we consistently fail to detect H2DCFDA staining in established tumor samples, both in the mouse 3-methylcolantrene (3-MCA)-induced sarcoma model and in human sarcoma 2 and carcinoma (not shown).
Defense Purposes of ROS
Thus, oxidative DNA damage may be an epiphenomenon to an ongoing pathophysiological process, and elevated levels may not have a role in carcinogenesis (19). But then, is there an alternative function for ROS produced in the tumor microenvironment? An appealing hypothesis has been recently proposed. According to Naviaux (67), ROS production is a stereotyped response to cellular injury or attack with protective purposes. ROS and the consequent oxidative changes in membrane lipids and proteins found in many chronic diseases are mediators of the oxidative shielding response that all eukaryotes adopt when placed in a hostile environment. In chronic diseases and cancer, oxidative shielding may defend cells by physically decreasing the cellular uptake, release, and exchange of any kind of potentially toxic substances from and with the environment. This response is very ancient: signs of ROS as a primitive defense mechanism against stress may be found as far as 1.5 billion years ago when eukariotic cell precursors in the precambrian sea underwent strong attacks by virus and intracellular bacteria that deprived them of metabolic resources (97). Only cells with effective defenses survived—that is, cells with internalized bacterial ancestors (mitochondria) that were able to sense and respond to the metabolic distress caused by the intracellular pathogens. Namely, the metabolic mismatch produced by the use of cellular resources by parasites diverts electron flow away from mitochondria in the cell, and mitochondrial oxygen consumption falls. Thanks to the presence of extracellular O2, cellular oxygen rises: under more oxidizing conditions, less electrons are available for carbon–carbon bond formation required to build intracellular pathogen scaffolds, but are instead attracted by intracellular oxygen to make ROS and reactive nitrogen species (RNS) and oxidize free thiols in cysteine and glutathione, a first example of oxidative shielding.
Thus, cellular defense against infection, a task of the modern innate immunity still largely based on ROS production, was a crucial function of ancient mitochondria. Much later specialized enzyme systems, such as the NOX system, with the task of producing ROS evolved in eukariotes out of the mitochondria (37, 54). Yet, any stressful event induces ROS generation by mitochondria. Moreover, new evidence suggests that ROS derived from mitochondria act as signal-transducing molecules that may provoke the upregulation of inflammatory cytokine subsets (66, 103).
Overexpression of Antioxidants in Tumorigenesis and Tumor Progression
As discussed above, the theory of oxidative shielding claims that ROS and oxidative changes in cancer (like in other chronic disorders) are the symptoms of disease and not the cause (67). In fact, in a first phase ROS, at lower levels, would protect the cell both as signaling molecules and by forming the oxidative shielding; in a second phase, when the local harmful environmental conditions threaten to spread to neighboring cells and put at risk the survival of host. In preneoplastic lesions, ROS-mediated death of stressed cells, in which high ROS production has possibly caused dangerous mutations, prevent cancer formation (Fig. 3A). Following this approach, we can reason that a strong activation of cell antioxidant systems in stressed cells, aimed at restoring the redox homeostasis after ROS production, would lower the intracellular ROS levels, and consequently allow survival of mutated cells and cancer development (Fig. 3B). Indeed, constitutive activation of Nrf2 via somatic mutations has been implicated in carcinogenesis (36, 68, 79); more recently, oncogene-directed increased Nrf2 activity with activation of cellular detoxification programs was shown to contribute to tumorigenesis (22), further questioning the actual role of ROS as cancer-promoting agents.

Antioxidant upregulation is likely to play a role also in tumor progression and drug resistance. We have previously reported that, in human, primary non-small-cell lung cancers display a highly reduced redox phenotype due to overexpression of antioxidants in both neoplastic cells and infiltrating inflammatory cells. In vitro and in vivo approaches with tumor cell lines expressing different levels of antioxidants have unraveled a correlation between redox phenotype and tumor malignancy, the higher antioxidant phenotype correlating with the higher aggressiveness (10).
An additional effect of the upregulation of ROS-adaptive response is protecting tumor cells against anticancer agents (Fig. 3C). We reported that the same dose of the pro-oxidant agent arsenic trioxide was able to kill tumor cell lines featured by a more reduced phenotype but induced upregulation of antioxidant systems and increased proliferation rate on cell lines displaying a less reduced redox state (10). In fact, a common mechanism through which ionizing radiations and chemotherapeutic agents kill neoplastic cells is the induction of oxidative stress (52, 87). However, this approach often fails, likely due the strong overexpression of antioxidant enzymes by neoplastic cells, which results in resistance to pharmacologically sustainable drug doses (45, 72, 85, 95, 100). Tumors most often comprise heterogeneous neoplastic cells that conceivably display different redox states (Fig. 3). Therefore, even in tumors with a less reduced phenotype, in which most cells are sensitive to oxidizing therapeutic agents, a cell fraction displaying a strong upregulation of antioxidant systems, pre-existent or induced by the pro-oxidant treatment, may survive (45, 80). The selection of resistant cells with a highly reduced redox phenotype represents an additional constraint, due to the aforementioned correlation between antioxidant phenotype and tumor aggressiveness. More importantly, it has been clearly demonstrated that both normal and cancer stem cells and early progenitors contain lower levels of ROS and higher levels of antioxidants than their more mature progeny (22, 41, 82, 88). These differences in the redox state are critical for maintaining stem cell function. As a consequence, cancer stem cells, as well as normal tissue stem cells, develop less DNA damage upon exposure to oxidizing agents compared to nontumorigenic cancer cells and are the cell population most resistant to oxidizing treatments (23).
Increased Redox Potential at the Tumor Site
Upregulation of antioxidant genes in tumor cells also affects the extracellular redox state through delivery of redox-active enzymes and nonprotein thiols (Figs. 2 and 3). The externalization of intracellular redox enzymes and free cysteine or GSH is not restricted to tumor cells, but it is also observed in normal cells, including immune cells, under stress conditions (77). For instance, protein disulfide isomerase (PDI), normally confined in the ER lumen, is secreted by activated platelet and reduce disulfide bonds in integrin, promoting thrombus formation (15). Thioredoxin (TRX) and free cysteine are abundantly released by dendritic cells during antigen presentation and mediate T lymphocyte activation (1).
Among tumors, TRX is actively secreted by many different neoplastic cells (74, 84). Likewise, PDI is externalized and expressed at the cell surface of glioma cells (34) (Figs. 2 and 3). Efflux of GSH by cancer cells occurs through the inducible multidrug resistance protein 1 (MRP1) transporter (17, 101), which also confers drug resistance and protect cells from toxic insults. Free cysteine is also abundantly released by tumor cells (3, 10, 57, 91) through the cystine/cysteine redox cycle (Fig. 4). This cycle is basically composed by system xc−; the redox couple cystine/cysteine; and intracellular oxido-reductases including TRX. System xc− is an oxidative stress-inducible, cystine/glutamate antiporter that imports oxidized cystine releasing equimolar glutamate into the extracellular space (18). The functional component of the system is the xCT protein, responsible for transport activity (18, 74). Once internalized, cystine is enzymatically reduced to cysteine.
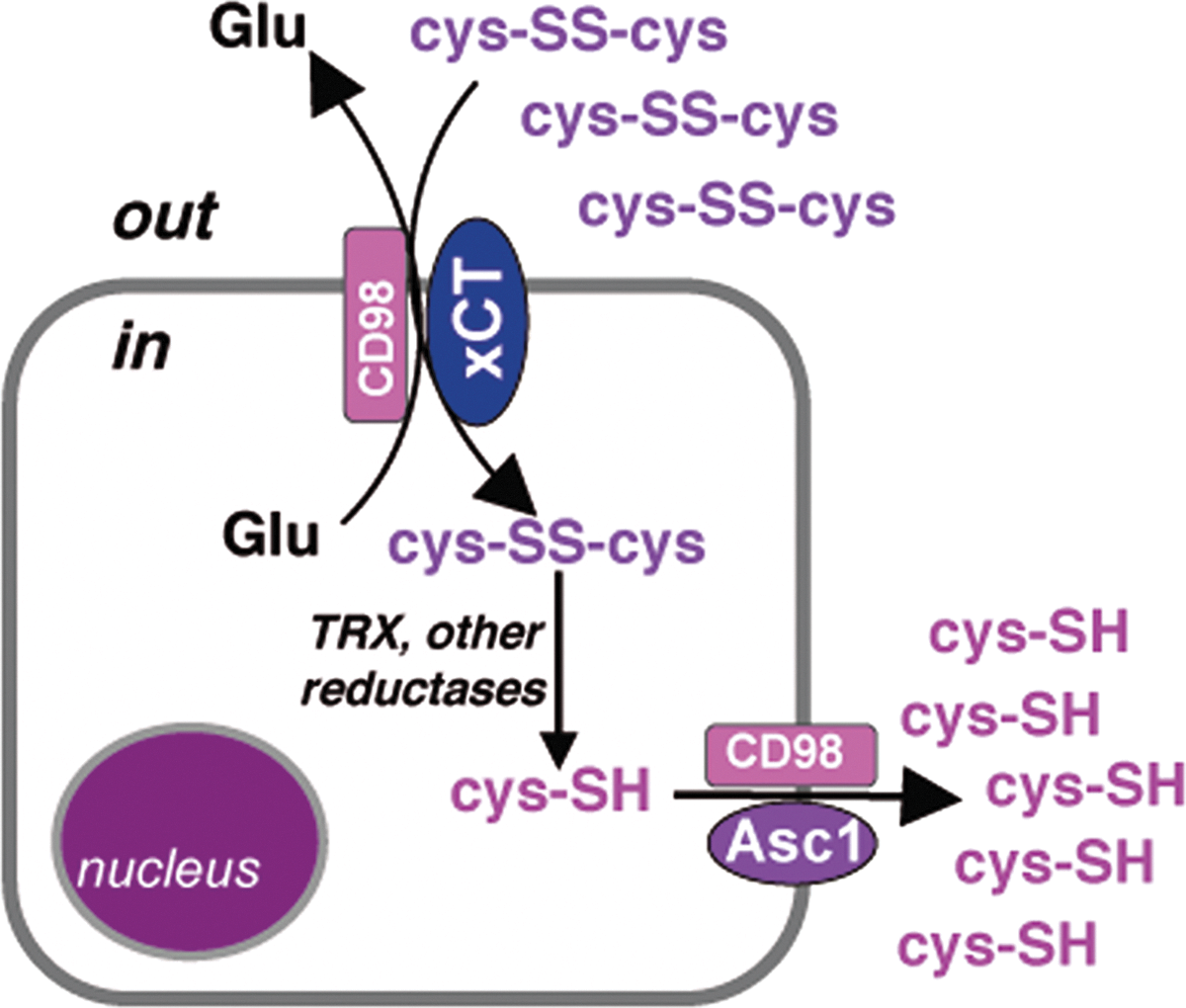
Internalized cysteine was long thought to be merely used for protein and GSH synthesis. However, increasing evidence indicates that some cysteine is actively released extracellularly via the neutral amino acid transport systems, expressed by most cell types (Fig. 4). The activity of this cycle results in replacement of oxidized cystine, the predominant extracellular form of the amino acid in physiologic conditions, with reduced cysteine (75). System xc− is strongly induced in tumors: In vitro evaluations of cysteine release by tumor cells revealed that the levels of extracellular cysteine may reach surprising amplitudes (10, 90). Therefore, the cystine/cysteine redox cycle affects also the redox state of the microenvironment, with important pathophysiological consequences (see below). Not only do cancer cells actively modify the redox microenvironment, but also infiltrating inflammatory cells contribute to change the redox state. As introduced above, cells of the innate immunity (i.e., macrophages, dendritic cells) increase their store of antioxidants upon activation and, like cancer cells, overexpress the xc− system and TRX, resulting in extracellular release of reduced cysteine (1, 86). Furthermore, also inflammatory cells secrete redox enzymes, including TRX (1) and the gamma-interferon-inducible lysosomal thiol reductase (GILT, Figs. 1 and 2) (51).
Impact of the Extracellular Redox State on Tumor Microenvironment
Effects on surface molecules and cytokines
As stated above, most cysteines in secretory and membrane proteins are linked in disulfide bonds formed into the ER lumen (Fig. 1). This oxidative folding provides stability to extracellular proteins. However, recent evidence indicates that, like cysteines in cytoplasmic proteins, also cysteines in extracellular proteins may serve signaling tasks (46). It has been shown that some disulfide bonds in membrane proteins are exposed at the protein surface, while others, structurally important, are hidden by the protein folding.
Proteomic/mass spectrometry studies allowed identify nonstructural disulfide bonds that are redox-labile in around 30 membrane proteins of leukocytes, belonging to several classes of proteins (integrins, receptors, transporters) (62). Reducing labile disulfides often results in a change in protein structure and function. Thus, the simultaneous presence of labile disulfide bonds and redox enzymes extracellularly offer a mechanism for regulating protein function through redox events. For example, a labile disulfide bond was identified in CD132, the common gamma chain of several cytokine receptors. This receptor is susceptible to reduction by the redox enzymes that can be present extracellularly at the tumor/inflammation site, namely, TRX, PDI, and GILT (34, 51, 74). Remarkably, CD132 reduction prevents interleukin (IL)-2 signaling (62).
In addition, secreted proteins have labile disulfides. Among cytokines, active IL-4 is endowed with a number of intramolecular disulfides. Upon reduction of these labile disulfide bonds, IL-4 is inactivated, because its binding to the IL-4R receptor is inhibited (20). β-defensin 1, a key effector molecule of innate immunity, is characterized by three intramolecular disulfide bridges. While the bioactivity of the oxidized form is limited, after reduction of disulfide bridges it becomes a potent antimicrobial peptide (78). In addition, the bioactivity of transforming growth factor (TGF)-β is redox regulated. However, unlike in IL-1, two free cysteine residues are required for the biological activity: when the two residues form a disulfide, TGF-β bioactivity is lost (6).
Other redox changes may modulate the signaling activity of extracellular molecules. Indeed, TRX and PDI have been shown to form complexes with tumor necrosis factor receptors on B lymphocytes from chronic lymphocytic leukemia through the SH groups of their cysteine residues (83). These modifications result in improved survival of the neoplastic B cells. The high incidence of labile disulfide bonds in membrane proteins, together with the evidence that changes in redox potential and secretion of disulfide-altering enzymes occur in both inflammatory and tumor microenvironment, points to a redox-regulator mechanism that can alter membrane protein activity (62). In normal inflammation, the increase of extracellular antioxidants is limited and temporary, and may serve to control the dangerous hyperactivity of proinflammatory cytokines. Then, the redox state rapidly returns to the equilibrium. Differently, in the tumor microenvironment the excess of anti-oxidant production by both neoplastic and immune cells may result in dysregulation of the correct outcome of the inflammatory response.
Dysregulation of inflammation and modifications of the redox state of exofacial thiols of membrane proteins on neoplastic cells, resulting in altered signaling, may cooperate in promoting tumor progression. In support to this hypothesis, the anti-tumor compound parthenolide, endowed with nucleophilic properties, has been found to exert its cytotoxic activity on lymphoma and melanoma cells by attenuating the levels of exofacial-free thiols (53, 81).
Effects on damage-associated molecular pattern molecules
Important components of the connection between inflammation and cancer are damage-associated molecular pattern molecules (DAMPs) (76). DAMPs are protein (e.g., HMGB1) or nonprotein (e.g., ATP, uric acid, lipids) molecules confined in healthy conditions within cells but delivered outside upon cell damage. The levels of released DAMPs depend on the abundance of intracellular DAMPs and are often higher and enduring in neoplastic tissues than in normal tissues (Fig. 5). DAMPs play many functions in tumors. They mediate endothelial cell activation, angiogenesis, and stem cell migration and contribute to recruit and activate innate immune cells to release cytokines that stimulate tumor growth and progression (58). In keeping with their primary cytoplasmic or nuclear localization, DAMPs in principle require reducing conditions to maintain their correct protein folding, and undergo oxidation in the oxidizing extracellular milieu (76). The amino acid residue most susceptible to oxidation is cysteine (cys-SH), at the level of its sulfur atom. In most proteins, the first oxidation product of cys-SH by H2O2 is cysteine sulfenic acid (cys-SOH) (20) (Fig. 6). Sulfenic acids are quite unstable and readily oxidized to sulfinic (cys-SO2H) or to disulfide (cys-SS-cys). The latter results from condensation of sulfenic acid with free sulfhydryl with formation of a disulfide bond. Sulfinic acid is a relatively stable intermediate, and is reducible to sulfenic acid by the ATP-dependent sulfinic acid reductase, sulfiredoxin (SRX)-8 (12). However, it can be oxidized to sulfonic acid (cys-SO3H), the most highly oxidized and irreversible thiol. Other oxidative modifications of cysteine include S-nitrosylation and S-thiolation (Fig. 6). The former involves the covalent incorporation of a nitric oxide moiety into thiol groups. This post-translational modification affects the function of a wide array of cell proteins, ranging from ion channels to nuclear regulatory proteins. The excess of S-nitrosylation perturbs the oxidative/nitrosative balance generating nitrosative stress with important consequences on the antioxidant systems. Recently, emerging data indicate the probability of crosstalk between signaling/regulation mechanisms involving protein S-nitrosylation and protein S-glutathionylation (49, 64). S-thiolation is the modification of cysteine residues with GSH (or cysteine). S-thiolation prevents the irreversible oxidation of cysteine residues during oxidative stress and is crucial for protection of thiol-containing proteins. Since as discussed above, both GSH and cysteine molecules can be released, especially in the tumor microenvironment (3, 10, 17, 91, 101), one can envisage that glutathionylation and cysteinylation are not restricted to inside cells but may occur also extracellularly. In most cases, oxidation of a cytoplasmic protein results in loss of the correct folding and inactivation. Consequently, some DAMPs have exploited mechanisms that delay oxidation. The lectin galectin-1 binds sugar moieties on the cell surface or extracellular matrix immediately following secretion (8). In this way, it hides its six cysteine-reduced residues preventing their oxidation. However, oxidation not always leads to inactivation. Rather, several cases of DAMPs whose function is modulated, or switched by various degrees of oxidation have been reported (Fig. 7). Here we will depict some examples.



S100 proteins
S100 proteins are a large group of calcium-binding proteins with an intracellular role in migration and cytoskeletal metabolism. After release into the extracellular space due to cell damage, they become danger signals with functions in innate immunity (28). Many different functions have been described for these proteins, possibly deriving from the different redox state. In fact, under reduced conditions, S100 proteins are chemotactic, but, when oxidized, they lose this property. Interestingly, reversible redox modifications of S100, such as S-nitrosylation and S-glutathionylation, trigger antiinflammatory activities that may affect the microcirculation (64). In contrast, other modifications, such as oxidation of Met residues to Met sulfones and Cys-Lys sulfinamide formation, are irreversible, and excessive accumulation of these, as found in human atheroma and in brain extracts from patients with Alzheimer disease, may contribute to the pathogenesis of chronic inflammatory conditions (55). In addition, oxidative-crosslinking or oligomer formation in S100 has been found to strengthen the proinflammatory action of S100A4 mediated by the receptor for advanced glycation end products (RAGE) (35).
Galectin-1
Like S100 proteins, galectin-1, a cytosolic member of the beta-galactoside-binding lectin family that also plays DAMP function, exists in both reduced and oxidized states. Galectin-1 displays carbohydrate-binding activity when it is in the reduced state, but loses this activity upon oxidation. Conversely, oxidized galectin-1 stimulates axonal regeneration by suppressing inflammatory responses as well as by stimulating the release of axonal regeneration-promoting factors in macrophages (27, 38, 40).
HMGB1
In addition, the activities of HMGB1, a nuclear protein that signals tissue damage when released into the extracellular medium, are redox-dependent. In a first study, terminal oxidation of HMGB1 due to high ROS levels produced during apoptosis was found to inhibit the DAMP activity of HMGB1, redirecting its ability to activate acquired immune response toward tolerance (48). Later, the ability of HMGB1 to induce proinflammatory cytokine production by innate immune cells was shown to depend on the redox state of its three cysteine residues: C23 and C45 must form a disulfide bond, and C106, which is unpaired, must be in the thiol state (98, 99). Either terminal oxidation of these cysteines to sulfonates (CySO3 −) or their complete reduction to thiols (CySH) abrogates the cytokinestimulating activity. More recently, it has been demonstrated that the reduced form of HMGB1 can recruit motile cells. Therefore, the role of extracellular HMGB1 as a chemoattractant or as a proinflammatory mediator relies on its redox state. When the three cysteine residues are reduced, then HMGB1 is a chemokine, whereas the presence of a C23-C45 disulfide bond inhibits the chemoattractant function of HMGB1 and makes it a proinflammatory cytokine (93). Further oxidation of C106 to sulfonates by ROS abrogates both activities. The different forms of HMGB1 are thought to bind different receptors: RAGE the reduced form, Toll-like receptor 4 the one with disulfide (93).
Combined Therapies May Interrupt the Vicious Circle Orchestrated by Redox Signaling at the Tumor Site
The data reviewed above point to a couple of peculiarities of the tumor microenvironment: (i) abundance of DAMPs; (ii) abundance of antioxidants. As discussed, both features may promote tumor progression. DAMPs mediate angiogenesis and stem cell migration, and recruit innate immune cells (58). DAMPs can also induce autocrine growth of tumor cells: in the case of malignant mesotelioma, motility, survival, and anchorage-independent growth are sustained by HMGB1 secreted by the neoplastic cells (47). In turn, a highly reduced redox state promotes tumorigenesis (22) and tumor progression (10) and contributes to drug resistance (57). In addition, the excess of DAMPs and the reduced redox state may collude in a vicious circle that favors tumor progression. In fact, due to the high level of antioxidants in the extracellular tumor microenvironment, DAMPs can be maintained in a reduced redox state longer (Fig. 7), so that the possibly damaging effects exerted by the reduced DAMPs are not downmodulated by oxidation. As an example, in the case of mesotelioma, HMGB1 has been proposed to bind and activate RAGE (47). Since only the reduced form of HMGB1 binds RAGE (93), HMGB1 is likely released by cancer cells in a reduced state. One can assume that the maintenance of the reducing folding by the antioxidant environment would then perpetrate RAGE stimulation. HMGB1 is overexpressed by many tumors (Fig. 5); therefore, a similar process can take place in many tumors, when neoplastic cells undergo spontaneous or drug-induced cell necrosis (91). On the other hand, many DAMPs recruit innate immune cells that contribute to increase the extracellular redox potential by releasing free cysteine and redox enzymes (1, 43, 86). These observations strengthen the rational for the employment of anti-inflammatory and redox-directed drugs in oncology, possibly in combination with chemotherapeutic or tumor-specific biologic drugs. Among anti-inflammatory compounds, epidemiological studies revealed that long-term use of aspirin and other nonsteroidal anti-inflammatory drugs reduces cancer risk (4, 7, 32); cyclo-oxigenase (COX) inhibitors were found to exert antineoplastic activity in some cancers (96). Among redox active compounds, we have observed that the simultaneous treatment with inhibitors of the antioxidant response enhanced the sensitivity to a pro-oxidant drug (arsenic trioxide) in melanoma cells. While inhibition of GSH production was highly effective, cystine/cysteine cycle blockers also prevented the increase of the extracellular redox potential, an added value in view of the detrimental effects of the extracellular redox state on tumor progression and drug resistance, discussed above (90). Along this line, sulfasalazine (SASP), a potent inhibitor of xCT-mediated cystine uptake, has been reported as an effective antitumor drug in a variety of cancers (16, 25, 56).
Interestingly, we have recently shown the synergy of the combined treatment with the anti-inflammatory compound Ibuprofen and SASP in the tumor model of 3-MCA-induced mouse sarcoma. This pharmacologic association strongly delayed tumor growth, reduced tumor size, and increased survival of treated mice (2). The two drugs target not only tumor cells but also tumor-associated macrophages that were dramatically decreased in the tumor infiltrate of mice subjected to the combined treatment (2).
Conclusions and Perspectives
Over the last years, increasing evidence has demonstrated that not only ROS production but also the antioxidant systems they elicit have profound impacts on pathologic processes implicated in the neoplastic disease, including inflammation, tumor progression, and drug resistance. An emerging point is that the antioxidant response, in both neoplastic cells and inflammatory cells, through enhanced activity of the cystine/cysteine redox cycle goes beyond the cell border and increases the extracellular redox potential, generating a reduced microenvironment. In turn, the reduced extracellular redox state may drive thiol modifications in proteins of the microenvironment, both normal (membrane associated and secreted) and pathologic (DAMPs). Changes in redox folding may alter the activity of membrane and secretory proteins, as well as of DAMPs, with promoting effects on tumor development and progression. In addition, upregulation of antioxidants increase chemoresistance. A better understanding of the redox-related events in the tumor microenvironment has tremendous application potential in the development of rational combinations of molecular-targeted redox-active drugs with chemotherapeutics.
Finally, the encouraging results obtained using antiinflammatory compounds and redox-directed drugs in association support the use of novel combined therapies aimed at restoring the redox balance and turn off inflammation as a novel approach in the treatment of cancer.
Footnotes
Acknowledgments
We gratefully acknowledge the members of our laboratory for helpful discussion and suggestions, Caterina Pellecchia for secretarial assistance, and Compagnia San Paolo (FINA 333/11) for funding our research.