
Abstract
Introduction
O
To control oxidative stress, delivery of exogenous antioxidants is a logical initiative. Studies have reported inconsistent effects of antioxidant administration, such as Vitamin E (79, 96) and butylated hydroxyanisole (34, 118), in autoimmunity. Therefore, synthetic catalytic antioxidant enzymes may be more useful in controlling inflammatory processes important in type 1 diabetes development. In particular, superoxide dismutase (SOD) mimics have a long half-life and can easily get into cells. Salen–manganese compounds and mesoporphyrins, two classes of SOD mimics, respectively, protected the beta-cells through inactivation of superoxide and nitric oxides (93) and in the case of Fe(III) tetrakis-2-(N-triethylene glycol monomethyl ether)-pyridyl porphyrin (FP15), peroxynitrite decomposition (122). Another group of mesoporphyrins, manganese porphyrins (MnPs), constructed with a manganese center, four quaternized cationic pyridyl nitrogens, and varying side chain alkyl groups imparting lipophilicity differences (Fig. 1), have also shown promise in ameliorating oxidative stress (10, 117). Similar to SODs, MnPs dismutate superoxide with a high rate constant, and react with other species (peroxynitrite, lipid peroxyl radicals, and hydrogen peroxide [H2O2]) with rate constants of different magnitudes (9, 28, 29, 36). They also display extreme stability, high bioavailability, and low toxicity, maximizing their therapeutic potential (11, 117, 128). The ability of the metalloporphyrins to reduce inflammation has been illustrated in models of ischemia/reperfusion injury (105, 140), radiation-induced injury (75, 76, 97), and stroke (111). Although these studies support the benefits of metalloporphyrins in alleviating oxidative stress in a variety of experimental models, this review will focus on the innate and adaptive immune response effects specifically associated with MnP use in autoimmune type 1 diabetes.

MnP-Mediated Effects on Innate Immune Cells
The innate immune system is responsible for providing the initial defense and regulation when the host encounters a foreign antigen (12). Innate immune cell activation also facilitates an efficient adaptive immune response, imparting specific immune protection. In the case of autoimmunity, dysregulated innate cell-mediated inflammation may be the first breach in a cascade of normally appropriate events that lead to a loss of tolerance. For example, nonobese diabetic (NOD) mice spontaneously develop type 1 diabetes, characterized by an early infiltration of innate antigen-presenting cells (APCs), such as dendritic cells (DCs) and macrophages, into the islets (104, 112). After initial damage to the beta-cells from an as-of-yet undefined trigger, ROS in the islets are enhanced, activating infiltrating APCs. APCs exacerbate oxidative stress and inflammatory conditions by activating redox-dependent transcription factors, such as nuclear factor kappa B (NF-κB), which produce proinflammatory cytokines, such as tumor necrosis factor-alpha (TNFα), interleukin (IL)-1β, and more ROS. Together, cytokines and ROS directly damage pancreatic beta-cells (5, 46, 53, 77, 78). In an individual genetically susceptible to type 1 diabetes, APCs prime autoreactive T-cells in the lymph nodes, which then home to the pancreas and cause even greater damage to the beta-cells. Additionally, DC and macrophage abnormalities have both been identified in type 1 diabetes patients (6, 12). Controlling the activation and early inflammatory events mediated by innate immune cells may be critical in hindering overt type 1 diabetes development.
The ability of MnPs to affect innate cell-mediated inflammation was first tested using peritoneal macrophages from NOD mice in vitro (99). After lipopolysaccharide (LPS) stimulation in the presence of an MnP, macrophages exhibit a reduced superoxide burst, likely resulting from decreased NOX activity, as these enzymes are key players in macrophage effector function (82, 87, 92). Further, IL-1β and TNFα secretion are also reduced, sparking interest in how MnP was regulating proinflammatory cytokine production. A study was conducted to characterize redox-dependent transcription factor activation in APCs after MnP treatment. Transcription factors, such as activator protein-1 (AP-1), specificity protein 1 (SP-1), and NF-κB, are all regulated by ROS. Redox reactions control subcellular localization and DNA-binding abilities of these transcription factors (71). AP-1 regulates several cell processes, including proliferation and differentiation (57). Upon MnP treatment, DNA binding of AP-1 is reduced, resulting in lowered tumorigenesis in a model of skin carcinogenesis (146). Similarly, SP-1 regulates cell growth and differentiation, and its DNA-binding efficiency is modulated by redox changes (2). MnP administration prevents DNA binding of SP-1 (131), implicating the potential for controlling innate immune responses as well as processes contributing to hypertension (59), aging (2), and cancer (30). In addition to AP-1 and SP-1, NF-κB DNA binding is also lowered after MnP treatment (131). NF-κB upregulates a plethora of genes involved in host defense, cell survival, and cytokine production, making it critical for an effective immune response. NF-κB is held within the cytoplasm by an inhibitory molecule IκB. Upon APC activation, NOX enzymes are activated, leading to the generation of secondary messengers such as H2O2. These secondary redox messengers cause degradation of IκB via phosphorylation/ubiquitinylation, and NF-κB is then translocated into the nucleus for transcriptional activation (85). MnP does not inhibit the translocation of NF-κB into the nucleus, but instead inhibits its DNA-binding abilities. Moreover, this report demonstrated the specificity of MnP administration for redox-dependent transcription factors; CCAAT-enhancer-binding protein, a transcription factor lacking a redox-sensitive amino acid necessary for DNA binding, is not affected by MnP treatment (131).
The inhibition of NF-κB binding to DNA was further investigated. Interestingly, MnP treatment oxidizes the NF-κB p50 subunit (51, 131), likely at cysteine 62, indicating oxidoreductase capabilities of the catalytic antioxidant when present in the nucleus. To delineate the oxidoreductase capabilities of MnP [specifically MnTE-2-PyP5+ (Mn(III) meso-tetrakis(N-ethylpyridinium-2-yl)porphyrin) for this and all upcoming figures] versus a commercially available SOD mimic, Mn(III) meso-tetrakis(4-carboxylatophenyl)porphyrin (also known as MnTCPP3− or MnTBAP3−), in modulating NF-κB activation, both were tested by electrophoretic mobility shift assay (EMSA) (Fig. 2). Unlike MnP, which has an overall cationic charge, MnTBAP3− has an overall anionic charge (8), which may affect its cysteine-binding capabilities. Indeed, MnTBAP3− was unable to inhibit the NF-κB p50 subunit binding to a consensus NF-κB-oligo, suggesting that the difference in charge determines oxidoreductase characteristics. These data are confirmed by a study from Batinic-Haberle et al., which clearly showed that anionic or neutral compounds do not prevent NF-κB p50 DNA binding (11). Another study by Jaramillo et al. depicted glutathionylation of NF-κB as a potential mechanism of MnP-mediated DNA-binding inhibition in lymphoma (51). Since the response to self-antigen elicits the same oxidative stress-dependent activation of proinflammatory cytokines necessary for maturing the immune response in nonautoimmune disorders, it is likely that MnP would facilitate glutathionylation of p65 NF-κB in diabetes as well.
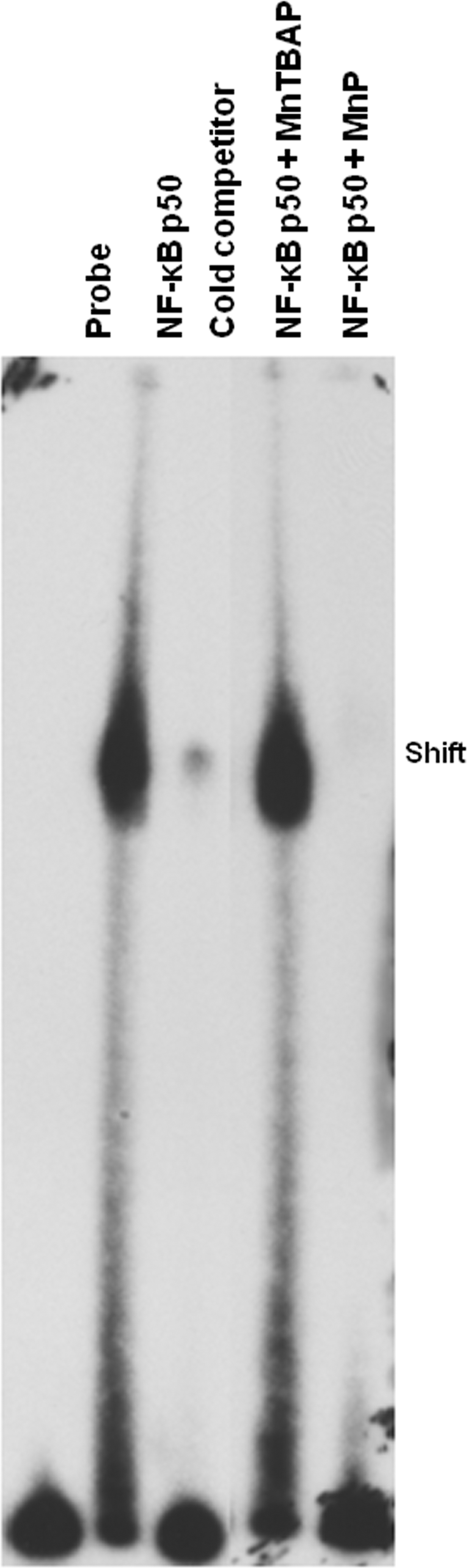
Reduced NF-κB activation after MnP administration thus provided a rationale for decreases in proinflammatory cytokines, such as IL-1β and TNFα, released from LPS-stimulated bone marrow-derived NOD macrophages (131). Alleviation of innate cell inflammation in early stages of islet insulitis may be beneficial in reducing both beta-cell damage and priming of the adaptive immune response. Recent evidence has demonstrated that stimulation of macrophages with poly(I:C), a viral dsRNA mimic, in the presence of MnP dampened type I interferon (IFN) production (110). Dampening type I IFN synthesis during the early stages of insulitis may have significant protective effects, as these cytokines can indirectly facilitate pancreatic beta-cell destruction. IL-1β and TNFα, on the other hand, directly destroy beta-cells through synergistic cytolytic mechanisms (18, 78, 94, 126, 134). Importantly, IL-1β and TNFα also serve as soluble signal mediators for instigation of a T-cell response. Soluble or third signal synthesis between APCs and T cells is critical for differentiation into specific T-helper subsets and for promoting effector functions (35, 73), as will be further discussed below. Therefore, MnP modulation of the innate immune system could be valuable in prevention and treatment of disease.
MnP-Mediated Effects on Lymphocytes
The adaptive response is primarily made up of T- and B-cells. Each of these cells produces a surface and/or soluble receptors that are specific for antigens presented by the innate immune cells. B-cells first produce a surface B-cell receptor and then later differentiate into antibody-secreting plasma cells, whereas T-cells only generate a surface T-cell receptor (TCR) (48).
T-cells
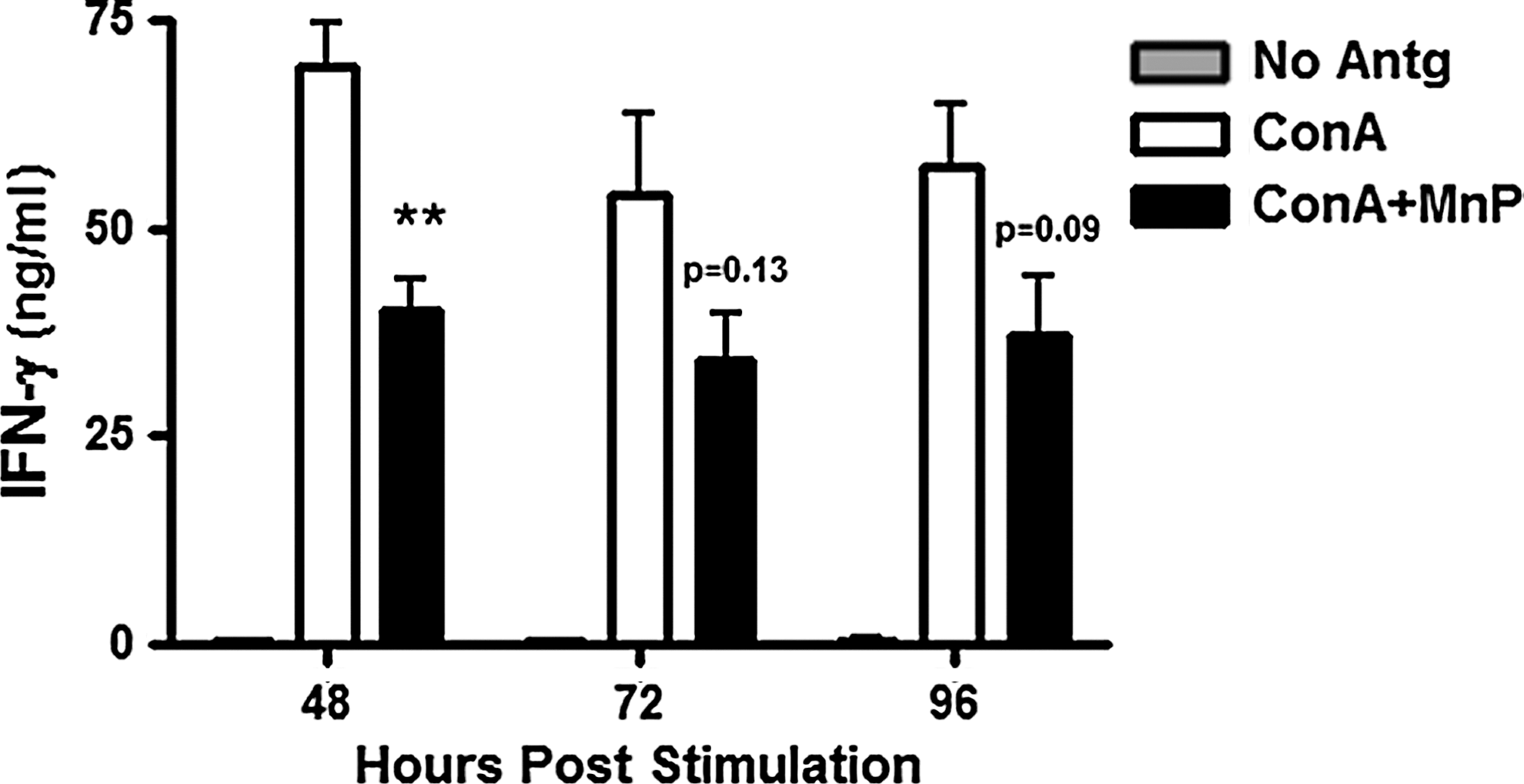
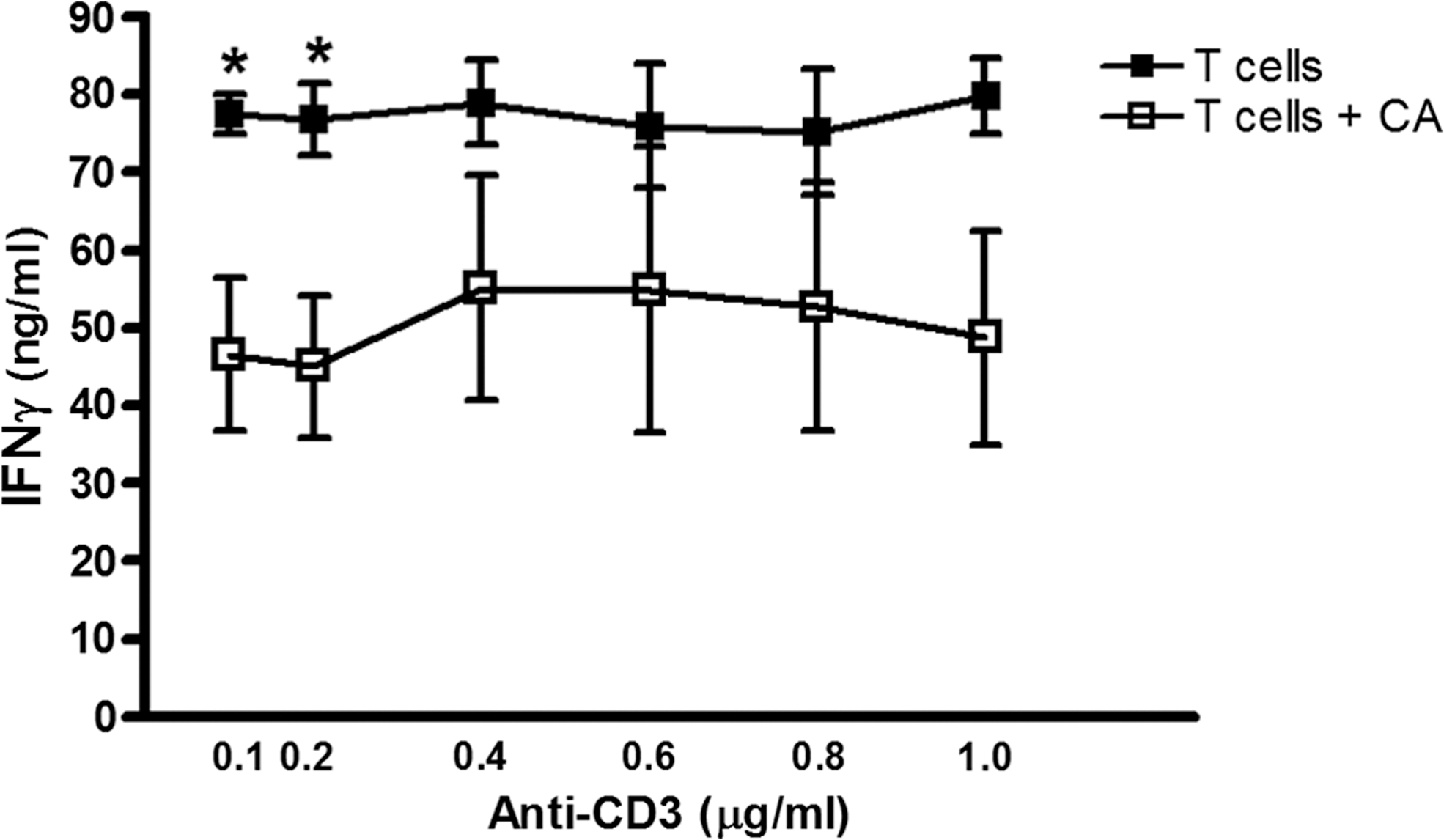
Autoreactive T-cells are the main effectors of beta-cell damage and apoptosis that eventually lead to the onset of type 1 diabetes. Interaction with APCs is necessary for T-cell activation and effector function. In type 1 diabetes, APC-derived soluble third signals drive T-helper (TH)1 immune responses (98, 127), fostering greater beta-cell damage. MnP inhibits APC-dependent autoreactive T-cell activation and proliferation, as depicted by ConA stimulation of diabetogenic T-cell clones cocultured with irradiated syngeneic NOD splenocytes (99). Likewise, ConA stimulation of NOD splenocytes alone plus MnP also displayed reduced IFN-γ production, but significantly only at 48 h post-stimulation (Fig. 3). MnP was only added once to cultures at time point 0 h; however, based on the reduced effects at 72 h and 96 h, repeat administrations may be necessary to sustain IFN-γ inhibition. In addition, MnP treatment of isolated T-cells stimulated with anti-CD3/CD28 does not afford the same IFN-γ reduction at high doses of polyclonal stimulation (Fig. 4) as is shown when APCs are present (Fig. 3). Our results and previous studies (99) thus indicate the necessity of APC antigen presentation for the MnP-mediated effect on the T-cell effector function.
T-cells are broken into two main classes: CD4+ and CD8+ T-cells. CD4+ T-cells consist of several helper subset populations, including TH1, TH2, and TH17 cells, and regulatory T (Treg) cells, whereas CD8+ T-cells are primarily cytotoxic. An imbalance between the TH1 and TH2 cells is thought to exist in type 1 diabetes. TH1 cells, which mainly secrete IFN-γ, TNFα, and IL-2, drive pathogenesis, while TH2 cells, which mainly produce IL-4, counteract the TH1-mediated proinflammatory disease state (32, 69, 101). Further, a loss of immunosuppressive Treg cells allows for enhanced TH1-cell responses and advancement of insulitis to overt diabetes (108, 125, 133). T-cells can promote apoptosis through direct lysis and proinflammatory cytokine production, which induces stress in the highly sensitive beta-cells (25, 49, 109, 116, 143). Moreover, T-cells can activate other immune cells, perpetuating the damage. In type 1 diabetes immunopathology, CD4+ TH1-cells migrate to the beta-cells and trigger activation and localization of other T-cells, such as CD8+ T-cells, and more APCs, resulting in the commencement of invasive insulitis and eventual break in self-tolerance. The leukocyte infiltrate into the islets can be visualized in diabetic animal models, mimicking what is thought to occur in humans (40). In an adoptive transfer model of type 1 diabetes using the diabetogenic Barbara Davis Center (BDC)-2.5 T cell clones into NOD.scid recipients, MnP treatment inhibits leukocyte infiltration into the islets (99). This result suggests the ability of MnP not only to protect islets from destruction, as the majority of mice remained diabetes-free, but also to hinder immune cell activation and subsequent localization to the pancreas.
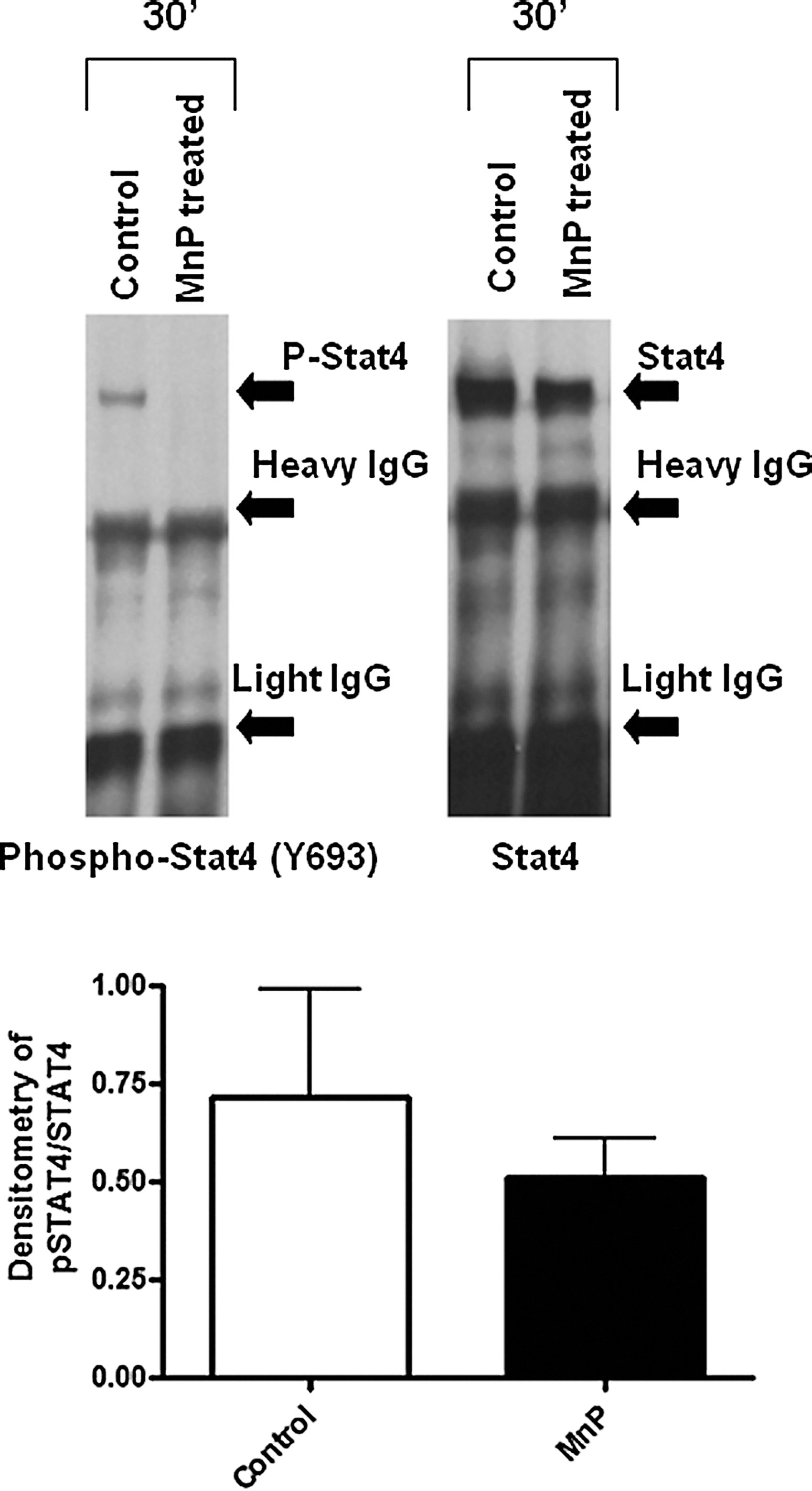
Naïve T-cells become activated by APCs through the major histocompatibility complex (MHC)–peptide interaction with the TCR, costimulation, and the aforementioned soluble third signal. In particular, differentiation of T-cells toward a TH1 lineage requires IL-12 secretion from APCs and signal transducer and activator of transcription (STAT)4 activation. IL-12 is primarily produced in response to Toll-like receptor ligation on APCs, part of the initial activation of innate immune cells (130). As a third signal, IL-12 triggers T-cell proliferation and IFN-γ secretion (61), characteristic of a TH1 T-helper cell lineage. Upon MnP treatment of murine splenocytes, the IL-12 p70 levels are significantly reduced (132). IL-12 propagates TH1-cell differentiation through STAT4 activation (42). STAT4 is a transcription factor essential for promoting IFN-γ generation (80). In type 1 diabetes, STAT4 plays a significant role in contributing to disease pathogenesis. NOD.STAT4 −/− mice display dramatic reductions in insulitis and diabetes onset in comparison to control NOD mice (141). Additionally, STAT4 polymorphisms have been characterized as a potential risk factor for type 1 diabetes development in humans (144). Upon immunization of NOD mice in the presence of MnP, activated (phosphorylated) STAT4 was decreased in the inguinal lymph nodes, despite an accumulation of T-cells in the lymph node (Fig. 5). Based on the reduction in immune responses demonstrated by MnP treatment, delayed migration of T-cells out of the lymph nodes is expected. Furthermore, in vitro stimulation of splenocytes demonstrated similar reductions in phosphorylated STAT4 (Fig. 6), indicating the ability of redox modulation to affect the transcription factor activation necessary for establishing the TH1 effector function.

The reduction of TH1 responsiveness is corroborated by several other MnP studies. Redox modulation significantly decreases proliferation, IFN-γ, and TNFα secretion from stimulated splenocytes in vitro and upon antigen-specific recall assay of immunized NOD and BALB/c mice (132). Moreover, diabetogenic BDC-2.5.TCR. transgenic (Tg) splenocytes similarly show a reduction in TH1 activation, as measured by diminished CD69+ expression, proliferation, and IFN-γ secretion after in vitro stimulation plus MnP treatment (31). Diabetogenic TH1 hyporesponsiveness was further characterized in a study signifying the importance of ROS for activating redox-dependent metalloproteases. As depicted by fluorescein-5-maleimide staining of reduced thiol-containing membrane proteins (Fig. 7), MnP administration inhibited overall membrane protein oxidation, providing more evidence for its oxidoreductase activity [in the nucleus, MnP displays oxidizing properties (131)]. The membrane protein tumor necrosis alpha-converting enzyme (TACE) is an enzyme responsible for cleaving immunological ligands from transmembrane proteins such as pro-TNFα (13), epidermal growth factor receptor (EGFR) (39), Notch (86), and lymphocyte activation gene 3 (LAG-3) (67). TACE is activated via cysteine oxidation, releasing its inhibitory domain from its active subunit and subsequently localizing it to the plasma membrane (88). Both TNFα and LAG-3 are important for regulating TH1 responses. TNFα, as stated above, can serve as the third soluble signal within the immunological synapse between APCs and naïve T-cells. Moreover, TNFα is produced from TH1 cells as an effector cytokine (19, 44). In a previous study, MnP treatment reduced TNFα secretion from nondiabetogenic splenocytes (132). Using diabetogenic BDC-2.5.TCR.Tg splenocytes, stimulation in the presence of MnP also decreased TNFα production (Fig. 8). LAG-3, on the other hand, serves as a negative regulator of T-cell activation. Its early expression on T-cells inhibits CD4 binding to MHC class II (47), regulating T-cell clonal expansion (138, 139). TACE cleaves LAG-3 from the cell surface, allowing for progression of T-cells to effector function (67). In the presence of MnP, TACE activity is reduced, leading to decreased LAG-3 cleavage and blockade of diabetogenic CD4+ T-cell progression to effector function (31). Moreover, MnP administration inhibits diabetes transfer of BDC-2.5.TCR.Tg splenocytes into NOD.scid mice in correlation with decreased serum-soluble LAG-3 levels, indicating lower autoreactive T-cell activation (31). MnP antioxidants, therefore, not only block innate cell functions, but can also hinder diabetogenic TH1-cell activation and effector function, resulting in delayed spontaneous diabetes in the NOD mouse (31).
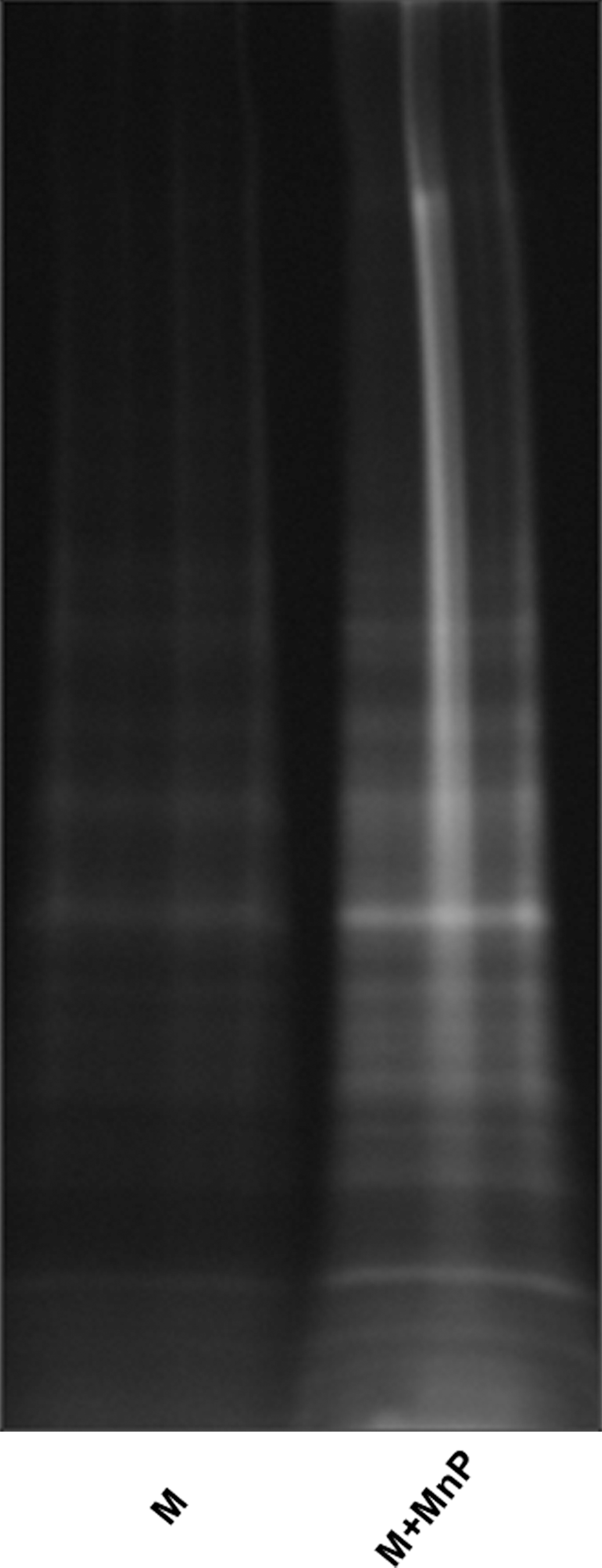
Synergy between CD4+ and CD8+ T-cells is necessary for transfer of diabetes in mice (23). While CD4+ T-cells secrete damaging proinflammatory cytokines and instigate activation of CD8+ T-cells and macrophages, CD8+ T-cells directly lyse beta-cells through Fas-dependent mechanisms and perforin release (3, 56, 135, 136). In addition to the effects on TH1 lymphocytes, MnP also modulates CD8+ T-cell effector function. MnP treatment of cytotoxic T-lymphocytes (CTLs) confers a reduced cytotoxic capacity (116). Moreover, CD8+T-cell proliferation and effector function (IFN-γ and TNFα) are also decreased after MnP administration. Finally, cytolytic mechanisms, such as perforin and granzyme B, are inhibited upon MnP treatment, indicating overall CTL hyporesponsiveness. These results highlight the potential control of both CD4+ and CD8+ T-cell populations with MnP treatment, likely contributing to reduced autoreactivity and diabetes onset.
B-cells
During infection, B cell-mediated defenses are indispensable, providing the basis for immunization strategies against seasonal flu and other pertinent diseases (45, 72, 74). However, in type 1 diabetes, the contribution of B-cells is relatively unclear, although autoantibody production serves as a biomarker of disease progression (16, 123). B-cells have been characterized as another APC population that supports T-cell activation (113), and along with DCs and macrophages, B-cells have also been detected in the infiltrate of the pancreatic islets that leads to insulitis (16). Therefore, the MnP effects on innate immune cells would likely hold true for B-cell activation, although this has yet to be elucidated in diabetes.
Contrastingly, a prostate cancer study conducted in mice demonstrated elevated B-cell numbers after treatment, potentially indicating an enhancement of antitumor immunity (75) and suggesting the possibility of heightened immune responses after MnP administration in cancer studies. Unlike type 1 diabetes, cancer requires greater immune responses. This boost in immunity, however, may not be a direct effect on lymphocytes, but instead an end result of tumor targeting. MnP administration reduces hypoxia inducible factor-1α (HIF-1α) activation and vascular endothelial growth factor and EGFR secretion, allowing for successful irradiation of localized tumor cells (83, 100). In addition, combination treatment of tumors with MnP and dexamethasone or ascorbate instigated the production of H2O2, effectively killing lymphoma (51) and breast cancer cells (142), respectively. During the irradiation process, however, bystander immune cells may be protected, as MnP is known to maintain nonmalignant cell viability (14, 116). These studies thus highlight the oxidoreductase activities of MnPs and further the applicability for modulating immunological responses in not only autoimmunity but also cancer.
Beta-Cell Transplantation for Type 1 Diabetes
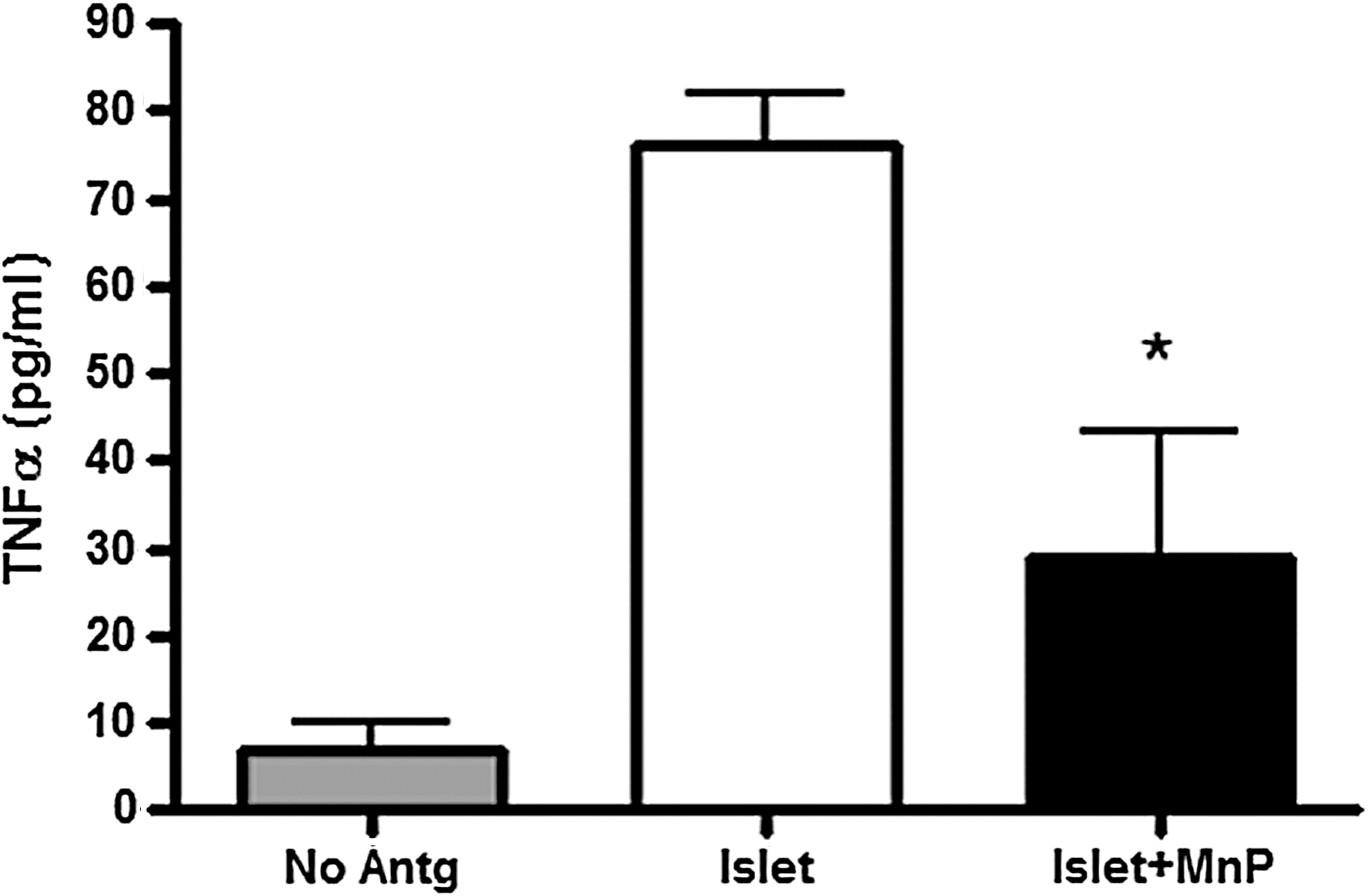
In cases of severe hyperglycemia and insufficient glucose control, islet cell transplantation is an option for type 1 diabetes patients. Islet cell transplantation, like every transplant, requires strict regulation of immune cells to protect against rejection. Immunosuppression therapies are consequently administered to allograft recipients. Long-term immunosuppression can be detrimental to the patient, resulting in secondary complications such as infections and cancer (33, 37, 119, 120, 124). Further, particular antirejection medications can themselves be toxic to the allograft, as abundantly displayed in islet transplantation (7, 54, 89, 145). Protection of an islet allograft without the harmful side effects of immunosuppression is, therefore, highly desirable. After allograft transplantation, if no immunosuppression is given, rejection is mediated by T-cells via direct and indirect pathways (43, 137). Recipient T-cells can either directly recognize donor alloantigens in the context of donor APCs or can respond indirectly against processed alloantigens presented by self-MHC. CTLs play a major role in mediating allograft rejection, through cytotoxic mechanisms such as granule release, FasL upregulation, and TNFα secretion (4, 137). In addition to the studies depicting reduced CD8+ T-cell effector responses after MnP administration (116), redox modulation was also tested in allorecognition experiments. In primary mixed leukocyte reaction (MLR) studies, MnP treatment blocks effector cytokine production and splenocyte proliferation (116). Together, these results suggest that MnP administration inhibits transplant rejection by impeding CTL responses.
MnP treatment was also tested in numerous studies for its ability to protect islets during isolation and after transplantation. In an early study by Bottino et al., MnP better preserved human beta-cell mass in culture for 7 days in comparison to the control conditions (14). Further, MnP treatment allows a suboptimal level of islet cells to stabilize euglycemia, unlike control-treated islets. These studies were followed by a more in-depth characterization of the islet cell health after MnP treatment. Similar to the studies in bone marrow-derived macrophages (131), islet cells treated with MnP display reduced NF-κB DNA binding (15). Total islet cell loss is also reduced after MnP administration, correlating with decreased caspase-dependent activation of apoptotic pathways, increased insulin production, and reduced IL-6/monocyte chemoattractant protein-1 generation after 60 h in culture (15). Islet function was most recently tested after MnP treatment in syngeneic, allogeneic, and xenogeneic transplantation models. In each of these instances, redox modulation enhances glucose sensing and graft survival in comparison to controls (115). Notably, in all of the studies mentioned, MnP maintains cell and islet viability (14, 15, 115, 116). Collectively, MnP affords graft protection, making it an effective agent for islet transplantation and encouraging the possibility of porcine islet xenotransplantation in humans, considering the donor shortage of human islets (50, 107). Additionally, the autoreactive immune cell hyporesponsiveness caused by MnP administration acts in conjunction with islet preservation to inhibit autoimmune responses and potentially allow for the reversal of type 1 diabetes. With success in islet transplantation, future studies will investigate the use of MnP in other transplant models.
Global Immunosuppression Potential
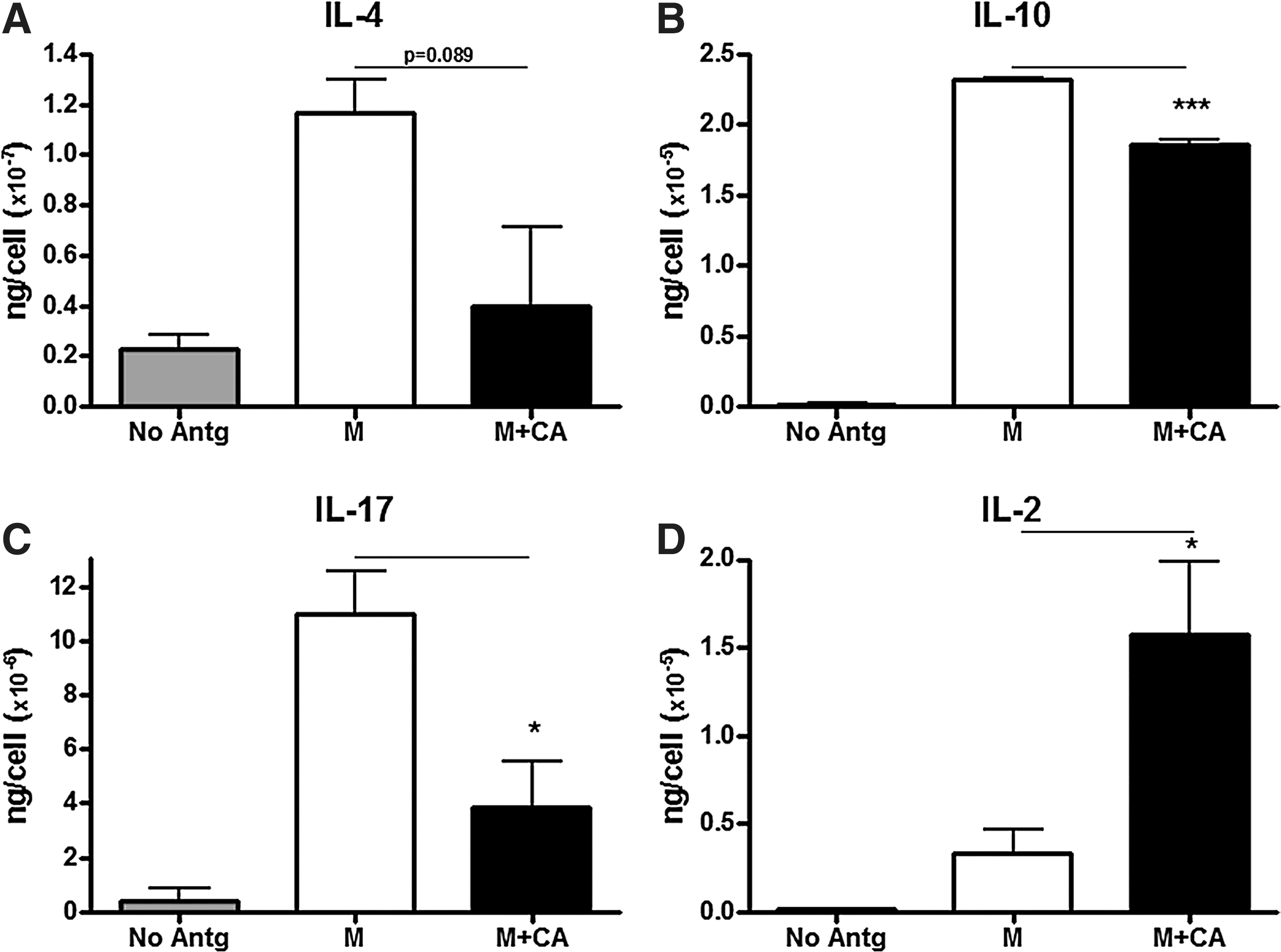
Because MnP decreases TH1 T-cell function, the compensation of and skewing toward a more TH2 subset was questioned. TH2 cells are known to be protective in autoimmune type 1 diabetes (38, 69), secreting IL-4, IL-5, and IL-13 to counteract damaging IFN-γ and TNFα, as mentioned previously. Moreover, Treg production of IL-10 would also mediate suppression of TH1 and TH2 cytokines (90). Interestingly, MnP treatment decreased IL-10 and IL-4 production (Fig. 9A, B). Our results are corroborated by a study by Jungsuwadee et al. displaying reduced TH2 activation and function in an ova-induced asthma model after MnP administration (55). IL-17 secretion was also significantly decreased after MnP treatment in both diabetogenic splenocyte stimulation (Fig. 9C) and in a primary MLR (116). In contrast, IL-2 production was actually enhanced after MnP treatment, suggesting a lack of cytokine utilization (Fig. 9D). This result correlates with the reduced proliferation (31, 131) and increased T-cell viability (116) seen upon MnP treatment. The T-helper cytokine data suggest that MnP globally suppresses T-cell responses instead of skewing from one lineage to another. The induction of a regulatory population is also unlikely, as ceasing treatment results in a loss of diabetes protection in the NOD mouse model (31). Therefore, further studies will need to be conducted to determine the mechanism behind overall T-cell hyporesponsiveness.
Additionally, as is the case with all immunosuppressive agents, MnP may impact pathogen clearance. Chronic administration of MnP for transplant maintenance, for example, may lead to infection susceptibility. Appropriate experiments will need to be completed to determine the effects on infection resolve. Nonetheless, MnP inhibits immune responses and maintains the islet beta-cell health, making it a more cytoprotective immunomodulatory agent in comparison to other clinically used drugs (i.e., rapamycin) for preventing and/or reversing autoimmune type 1 diabetes.
Concluding Remarks
MnP oxidoreductants are ROS-scavenging agents that can protect against a number of inflammatory pathologies (55, 59, 75, 76, 97, 105, 111). In autoimmune type 1 diabetes, innate and adaptive immune cells play critical roles in destroying pancreatic beta-cells. MnP usage reduced macrophage and T-cell effector responses, leading to overall protection against diabetes onset (31, 99) (Fig. 10). Moreover, MnP affords better islet transplantation, through blockade of CTL responses and enhanced islet preservation (14, 15, 115, 116). In regard to its oxidoreductase activity, a combination of MnP prooxidative functions in the nucleus and antioxidative mechanisms to scavenge ROS both attribute to pancreatic beta-cell protection. Lastly, MnP does seem to impart global immunosuppressive effects on T-cell responses; however, these mechanisms will have to be further elucidated for understanding the impact on infection clearance. Overall, redox modulation via MnP treatment can serve as an immunomodulatory and cytoprotective strategy for alleviating autoreactivity and islet transplant rejection, suggesting better outcomes for preventing and/or reversing type 1 diabetes. A better understanding of the MnP oxidoreductase function and its immunosuppressive mechanism may allow for its use in other chronic inflammatory conditions as well as organ transplantation beyond islets.

Footnotes
Acknowledgment
The authors thank Meghan Marré (Children's Hospital of Pittsburgh of UPMC) for critical reading.
Author Disclosure Statement
JDP is a consultant for BioMimetix Pharmaceutical, Inc.