
Abstract
Introduction
Scope of the review
T
Overview of renal ROS and hypertension
ROS-generating and metabolizing systems
ROS are generated as a normal product of cellular metabolism (204). ROS, such as superoxide anion (O2
−•), hydrogen peroxide (H2O2), and hydroxyl anion (OH−•), are reactive byproducts of mitochondrial respiration or oxidases, including nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxireductase (XOR), and certain arachidonic acid oxygenases (316). ROS can be formed also by nitric oxide synthases (NOS) after depletion of the NOS substrate
Nitric oxide, ROS, endothelial dysfunction, and hypertension
Oxidative stress can be a cause, a consequence, or a potentiating factor for hypertension. Increased production of O2 −• in the vasculature impairs endothelium-derived relaxation factor/nitric oxide (EDRF/NO) and increases vascular smooth muscle cell (VSMC) contraction and proliferation, and attraction of inflammatory cells (75, 137, 161, 191, 294). Since the kidney contributes to the long-term control of blood pressure (BP) and this is, in part, dependent on NO (341) whose activity is regulated in the renal blood vessels and tubules by O2 −• (317), it is important to understand the renal mechanisms of generation and metabolism of ROS, their interaction with NO, and their relationship to hypertension.
Renal oxidative stress
An increased production of ROS in the kidney can initiate hypertension. For example, mice with the SOD-3 isoform knockout (−/−) had higher basal BP compared with wild-type (+/+), which was associated with increased production of O2 −• and inactivation of NO in the kidney (304). ROS also can accelerate the development of hypertension. For example, conscious SOD-3 (−/−) mice had an earlier and more rapid increase in BP with a slow-pressor infusion of angiotensin II (ANG II) than their wild-type (+/+) littermates, even though both achieved similar levels of BP after about 10 days (304). The lack of consistent findings of hypertension in SOD knockout models may relate to many factors. Some studies (38, 53, 89, 128, 243) reported tail-cuff measurements of BP, which lack precision. Others were limited to blood vessels or studied in animals under anesthesia (49, 53, 144). Those that used telemetry measurements of BP have reported no changes in SOD-1 (−/−) (28) or increased BP in SOD-3(−/−) (304) conscious mice. Importantly, these inconsistent conclusions cannot be explained simply by the different SOD isoforms, since, for example, SOD-3 (−/−) mice were reported to be hypertensive in one study (304), but normotensive in another (89). ROS promote or mediate hypertension initiated by many processes, such as activation of the renin-angiotensin-aldosterone system (RAAS) in rats with the two kidney, one clip (2K,1C) model of Goldblat renovascular hypertension (305). This was related to an activated neutrophil oxidase (NOX)-derived O2 −• production and a reduced metabolism of O2 −• by SOD (29, 304). Dietary salt loading increased renal and vascular ROS in many salt-sensitive models (24, 31, 136, 245, 315). This implies that oxidative stress contributes to both the renal and vascular mechanisms of both salt-independent and salt-sensitive hypertension (138, 179, 317).
Mechanisms of hypertension and renal ROS
Increased renal ROS production contribute to the development and progression of hypertension (2, 62, 159, 187, 245, 303, 304, 310) by increasing renal vasoconstriction (132), renin release (307), renal afferent nerve activity (31), contraction of afferent arterioles to increased renal perfusion pressure (myogenic response), or to ANG II (153), endothelin-1 (ET-1), and thromboxane prostanoid receptor (TP) activation (293). ROS also cause dysfunction of glomerular cells and proteinuria (73, 239, 246, 274, 330). Increased O2 −• in the kidney leads to vascular dysfunction and disrupts water and sodium (Na+) homeostasis (104, 193, 267, 317). Vascular O2 −• reacts with endothelium-derived NO and directly promotes vasoconstriction (167). The generation of O2 −• in specific nephron segments in response to endogenous vasoconstrictors such as to ANG II increases (264) or decreases (224) tubular Na+ reabsorption, depending on the nephron site studied.
Intrarenal expression of pro- and antioxidant systems and hypertension
NADPH oxidase
NADPH oxidase is the predominant source of renal O2 −•. NOX proteins are homologs of the phagocytic NADPH oxidase and are distributed widely in the renal vessels, glomeruli, and nephron segments, as summarized in Table 1 (27, 88, 170, 204, 254). NOX3 and NOX5 are expressed in the fetal, but not in the adult, kidneys (37). NOX1, NOX2, and NOX4 are expressed in the adult kidney. NOX4 is predominant in endothelial and tubular cells. All these NOX proteins require the chaperone protein p22phox and entail phosphorylation and/or upregulation of several cytoplasmic subunits (Rac1/2, p47phox, p67phox, p40phox, and Poldip2) for activity (88, 172). These subunits are demonstrated in most of the nephron segments, either by mRNA/protein expression or by physiologic experiments (Table 1).
Presumed by physiologic experiments (28).
+, present; ±, weak; N/A, no data available.
mRNA detected in preglomerular vascular smooth muscle cells of WKY rats (170).
AA, afferent arteriole; EA, efferent arteriole; GLOM, glomerulus; Podo, podocytes; MC, mesangial cells; PT, proximal tubule; TAL, thick ascending limb; JGA/MD, juxtaglomerular apparatus/macula densa; DT, distal tubule; CCD, cortical collecting duct; MCD, medullary collecting duct; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, neutrophil oxidase; WKY, Wistar Kyoto.
Specific NADPH oxidase subunits in the kidney are implicated in the development of hypertension in animal models. Thus, p67phox was required for salt-sensitive hypertension (62), p22phox and p47phox for ANG II-induced hypertension (103, 153, 191, 207) and for genetic hypertension in spontaneously hypertensive rats (SHR) (29, 265), and Poldip2 for hypertension in models of chronic kidney disease (CKD) (150).
Studies using gene-deleted (−/−) mice and pharmacological inhibitors implicate NADPH oxidase-dependent ROS generation in the kidney cortex in increased renal vascular resistance (RVR), enhanced mitochondrial respiration, and decreased efficiency of O2 usage for Na+ transport (TNa/QO2) in models of hypertension (301) and CKD (150). NADPH oxidase-dependent ROS generation in the renal medulla is limited by the availability of O2 (34). Medullary ROS enhance tubular Na+ reabsorption and diminish pressure natriuresis (209). Indeed, there are increased ROS and decreased NO, activities in the renal outer medulla in many hypertensive and salt-sensitive models (41, 209). An increased expression of p67phox in the renal outer medulla of the Dahl salt-sensitive (DS) rat mediates increased NADPH oxidase activity, salt-sensitive hypertension, and renal injury (62).
Activation of NADPH oxidase by p47phox in the renal afferent arteriole leads to endothelial dysfunction, the development of an endothelium-dependent constrictor factor (EDCF) response, enhanced contractility to ANG II, ET-1, and TP agonists, and enhanced myogenic responses to increased renal perfusion pressure (2, 129, 153, 193).
NADPH oxidase-derived O2 −• has nephron-specific effects on tubular Na+ transport and fluid reabsorption (217, 224, 264). ROS decrease Na+ reabsorption in the proximal tubule (PT) (224), but increase Na+ reabsorption in the loop of Henle (LH) and in distal segments of the nephron (177, 264, 272). Activation of specific isoforms of NOS has the opposite effects at these nephron segments.
Enhanced production of O2 −• by NADPH oxidase in the macula densa (MD) diminishes NO signaling and increases tubuloglomerular feedback (TGF) responses (312). These likely contribute to enhanced preglomerular vasoconstriction and reduced glomerular filtration rate (GFR) that would restrict the rapid elimination of a salt load and predispose to salt sensitivity (70, 165, 207, 312).
Superoxide dismutases
Increased ROS in the kidney results not only from overproduction by prooxidants enzymes but also from decreased metabolism by antioxidant enzymes (187, 245, 304). The ability of an intact antioxidant system to reduce or remove ROS is a key step in the limitation of tissue injury. Indeed, antioxidant enzymes are heavily expressed in the kidney where they restrict the levels of ROS and limit hypertensive tissue injury (29, 75, 304). The major scavenging system for removal of O2 −• is the SOD family of enzymes that catalyzes the dismutation of O2 −• to H2O2. H2O2, more stable than O2 −•, induces a vasorelaxation that has been ascribed to the endothelium-derived hyperpolarizing factor (72, 166), upregulated eNOS (72), and to blunted myogenic responses (150). H2O2 contributes to the ANG II-dependent hypertrophic and remodeling responses in blood vessels (75).
Three SOD isoforms are detected in the normal kidney: the copper–zinc containing SOD (SOD-1), located predominately in the cytoplasm; the manganese-containing SOD (SOD-2) in the mitochondria; and the extracellular SOD (SOD-3) in the extracellular space, where it is anchored by a heparin-binding domain that is required for its full activity (67, 208, 210, 215, 278, 279, 342).
Studies have reported the distribution of the different SOD isoforms along the nephron using immunohistochemical or in situ hybridization techniques (67, 208, 210, 215, 278, 279, 342). These have yielded somewhat different conclusions depending on the species studied. (67, 208). A high level of SOD-3 immunoreactivity in the mouse is observed on the cortical and juxtamedullary PT and in VSMCs of renal blood vessels, whereas medullary areas are only weakly reactive, and glomeruli are not stained (66, 215). Although rats have a diminished tissue SOD-3 expression, it is demonstrated clearly in the rat kidney (29, 303). SOD-1 is expressed in the kidney of rats and dogs (278, 279). Its immunoreactivity in the dog kidney is prominent in the cortical thick ascending limb (cTAL) while, in the rat kidney, it is confined to the PT. SOD-1 represents ∼60% to 80% of total SOD activity in the kidneys of several species (181), but only about 30% of total SOD activity in the vasculature, where it preserves endothelial NO activity (28). Wild-type mice and transgenic mice expressing the human SOD-2 isoform have high levels of SOD-2 immunoreactivity in the mitochondria of all tubular cells. However, only transgenic mice have prominent SOD-2 immunoreactivity in endothelial cells (ECs), VSMCs, and interstitial and glomerular cells (210).
Generation and metabolism of O2 −• in hypertension
A growing body of evidence supports the hypothesis that both overproduction of O2 −•, notably by NADPH oxidase and mitochondria, and reduced O2 −• metabolism by SOD and other antioxidant enzymes can initiate or potentiate the development of hypertension. Two weeks of salt loading or 2 weeks of prolonged ANG II infusion both increase renal oxidative stress by increasing O2 −• generation via NADPH oxidase and decreasing O2 −• metabolism via SOD (29, 138, 317). Since salt loading reduces renin release and ANG II, several hypotheses can be proposed to explain the mechanisms involved in O2 −• production induced by ANG II or high salt. Some studies reported selective changes in the expression of NADPH oxidase components or SOD isoforms in the kidney after salt loading or ANG II infusion. Salt loading increases the expression of NOX2, NOX4, p22phox, p47phox, and p67phox and decreased SOD-1 and SOD-2 (15, 32, 71, 138, 187), whereas ANG II acting via AT-1 receptors (AT1R) increases the renal expression of NOX2, NOX4, p22phox, p47phox, p67phox, Rac1, and SOD-2 and decreased expression of SOD-3 (29, 30, 62, 70, 90, 93, 129, 138, 153, 187, 207, 312). Other studies reported increased mitochondrial generation of H2O2 by the medullary thick ascending limb (mTAL) cells in response to increased tubular flow that secondarily increased NADPH oxidase activity (213). A paradoxical activation of the intrarenal RAAS observed in DS rats fed high-salt diet (142) could represent another mechanism of NADPH oxidase activation in the kidney in salt-sensitive hypertension. Salt loading increased ET-1 production in the collecting duct (CD) of normal rats and mice (173). However, DS rats had lower medullary levels of ET-1 and endothelin receptor type-B (ETB) receptor in response to a salt load than Dahl salt-resistant (DR) rats (269). Since ET-1 negatively regulates Na+ reabsorption in the CD, the increased Na+ transport at this site provides another potential pathway for salt-dependent renal oxidative stress. The activation of parallel pathways may explain the very severe hypertension and renal damage caused by an infusion of ANG II into rats fed a high-salt diet.
As reviewed comprehensively (319), short- or long-term administration of polyethylene glycol covalently linked to SOD (PEG-SOD) to increase cellular uptake or SOD-mimetic compounds such as the redox-cycling nitroxide tempol decreases oxidative stress, improves vascular and renal function, and lowers the BP in conscious or anesthetized rats and/or mice in genetic, salt-sensitive or -resistant, ANG II-dependent or -independent models of hypertension (28, 134, 245, 251, 319).
Welch et al. (304) reported decreased SOD activity and decreased SOD-3 expression in the kidney cortex of mice after 12 days of ANG II infusion at a slow-pressor rate. In contrast, Fukai and Harrison (75) reported increased SOD-3 expression in blood vessels from mice infused with ANG II at pressor rates. ANG II infusion in the rat decreased the renal cortical mRNA for SOD-3, yet increased SOD-2 (29). Renal expression of SOD-1 and SOD-2 was unchanged in SHR, but SOD-3 was significantly decreased (2). The expression of SOD-1 and SOD-2 was reduced in the renal medulla of DS rats compared with DR rats (187). SOD-2 is inactivated rapidly by nanomolar concentrations of peroxynitrite (ONOO−). Guo et al. (95) reported that the nitrotyrosine content of SOD-2 in the kidneys of rats increased 13-fold after 2 weeks of infusion of ANG II at a pressor rate, and that renal SOD-2 activity was decreased by 50%, despite unchanged protein expression. This was ascribed to post-transcriptional modification by tyrosine nitration, which decreases the SOD-2 activity and increases oxidative stress in the kidney during ANG II-induced hypertension. Thus, there are site-specific effects of ANG II or salt loading on SOD isoform expression within the kidney and blood vessels.
Oxidative stress in the renal medulla has been assigned an important role in the development of hypertension (41). Increased medullary ROS induced by prolonged SOD inhibition are associated with reduced medullary blood flow (MBF), reduced Na+ excretion (UNaV), and hypertension (175, 176).
Collectively, these studies demonstrate that a defective renal SOD system contributes to increased renal oxidative stress and hypertension, but the patterns of altered SOD isoform expression vary with the hypertensive model.
Genetic deletion of SOD isoforms has provided excellent models to study the effects of a prolonged compartmental increase in O2 −• on BP and renal function. Mice with genetic deletion of SOD-1 or SOD-3 do not show obvious phenotype changes, but are more sensitive to tissue damage during oxidative stress despite some compensatory upregulation of other SOD isoforms (89, 304). In contrast, SOD-2 (−/−) mice have severe phenotypes, with embryonic or neonatal lethality. The absence of SOD-2 is not compensated by overexpression of SOD-1 (40). Thus, SOD-2 is essential for life (155, 163, 257). Viable SOD-2 (+/−) mice have ∼50% reduction in SOD-2 activity in the kidney (289) without compensatory upregulation of the other SOD isoforms (290).
Basal levels of BP are normal (28, 144) (Fig. 1A, panels a and b) or modestly reduced (53) (Fig. 1A, panels c and d) in SOD-1 (−/−) mice, but are not changed significantly in SOD-2 (+/−) mice (Fig. 1B, panels a and b) (35, 243). The BP is unchanged (49, 128) (Fig. 1C, panels a and b) or modestly elevated (Fig. 1C, panels c and d) (304) in SOD-3 (−/−) mice.
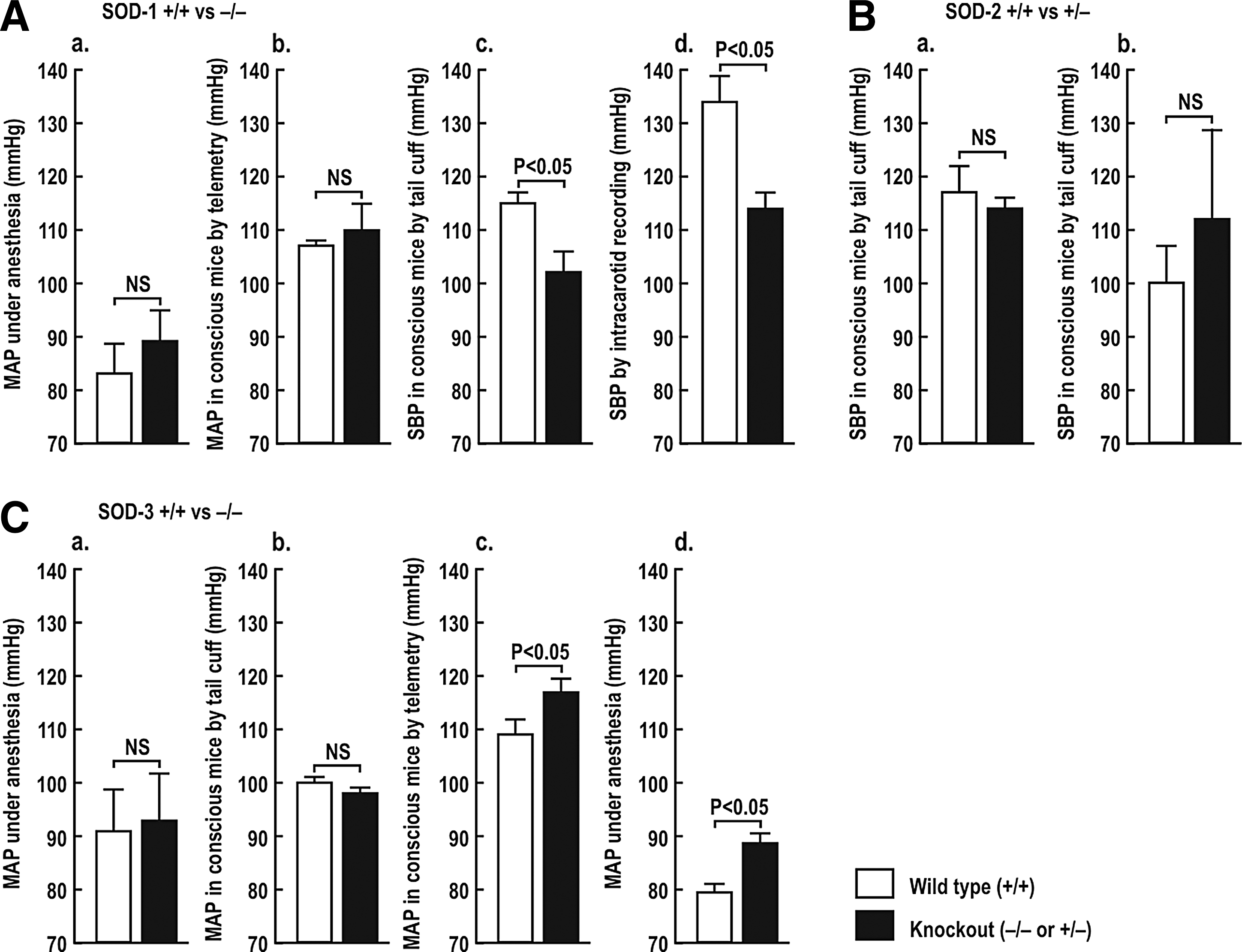
Several studies have reported the BP response to a prolonged infusion of ANG II in the SOD gene-deleted mice. Carlstrom et al. (28) reported an unchanged basal mean arterial pressure (MAP), but a much accelerated rise of MAP during a slow-pressor infusion of ANG II into SOD-1 (−/−) mice, although the final levels of BP after 2 weeks were not different from SOD-1 (+/+) mice (Fig. 2A). SOD-2 (+/−) mice had similar BP as wild-type mice after ANG II infusion (38, 243)(Fig. 2B). SOD-3 (−/−) mice had a higher basal level of MAP, but a similar response to ANG II in one study (304) (Fig. 2C), but an enhanced rise in BP to a rather high-rated ANG II infusion in another (89) (Fig. 2D). The RVR also was significantly higher in anesthetized SOD-3 (−/−) mice (304).
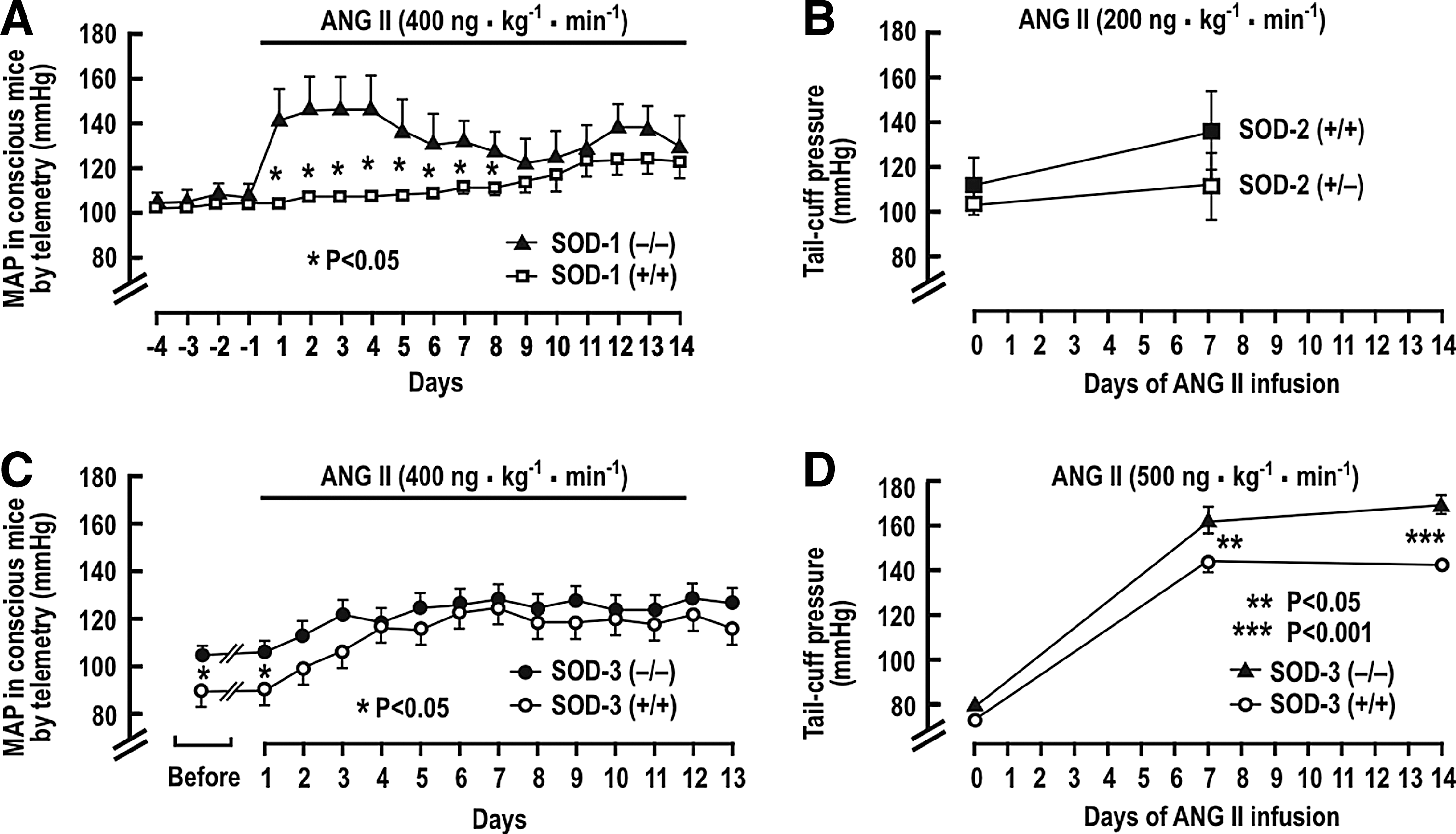
Jung et al. (128) studied the effect of SOD-3 gene deletion on the development of ANG II-induced and in 2K,1C hypertension in mice. After renal artery clipping, both SOD-3 (−/−) and wild-type mice had impaired endothelium-dependent vascular relaxation, reduced vascular NO, and elevated O2 −•. However, SOD-3 (−/−) mice had higher BP after 2 and 4 weeks of renal artery clipping, and after 1 week of ANG II infusion at a pressor rate. These effects were ameliorated by human recombinant SOD-3 and by the SOD mimetic tyron. This study demonstrated that the increased BP and limited NO bioavailability in the absence of SOD-3 were due to enhanced O2 −•. These data demonstrate that a primary increase in tissue O2 −• could enhance BP and RVR, and the rate of BP rises with ANG II, but the final degree of change is modest. Moreover, some studies did not detect a rise in basal BP in SOD-1-, SOD-2-, or SOD-3-gene deleted mice. Thus, these studies in aggregate demonstrate only modest and inconsistent effects of lifelong enhanced O2 −• produced by gene deletion of SOD isoforms in mice. They indicate that prolonged increases in O2 −• do not necessarily lead to hypertension, but may accelerate the development of hypertension, for example, with ANG II infusion.
Overexpression of specific SOD isoforms mitigates oxidative stress and hypertension in some (28, 63, 160, 198), but not all, studies (5, 63). Adenoviral gene transfer of human SOD-3 (AdECSOD) into SHR increased SOD-3 expression in the blood vessels and in the kidney, reduced the vascular O2 −• (39), and improved the endothelial function and vascular reactivity. AdECSOD reduced the BP and Na+ balance, implying that both renal and vascular extracellular oxidative stress contributes to hypertension in the SHR. AdECSOD also improved endothelial function and increased vascular NO availability in the SHR stroke-prone (SHRSP) model (63). The injection into SHR of human recombinant SOD-1 containing a heparin-binding domain to mimic the vascular binding of SOD-3 decreased BP, whereas SOD-1 lacking this domain had a little effect (198). Overexpression of SOD-2 improved endothelial function in deoxycorticosterone acetate (DOCA) salt-hypertensive rats (160) and protected PT cells in vitro from mitochondrial injury and apoptosis induced by adenosine triphosphate (ATP) depletion (43). Perfused afferent arterioles isolated from transgenic mice overexpressing SOD-1 had a reduced sensitivity and contractile responsiveness to ANG II (28).
These studies demonstrate that maneuvers that increase SOD activity could prevent oxidative stress and mitigate the development of hypertension (319). The importance of each SOD isoform depends on its location, the origin of the oxidative insult, and the species studied, but most studies concur in identifying O2 −•as the villain. Therefore, SOD mimetic agents might represent a rational strategy to prevent renal oxidative stress, high BP, and its consequences (319).
Other antioxidant enzymes
Catalase, GPX, Trx, and Prxs metabolize H2O2 to water and O2. Their dysregulation leads to the accumulation of H2O2, which interacts with transition elements to form the highly reactive OH−• by Fenton reaction. H2O2 is a mediator of cardiovascular and renal dysfunction and hypertension (36, 267, 268) and produces vasodilation (80), vasoconstriction (81), or a biphasic effect depending on the vascular bed (35, 44, 80).
Catalase is a 240-kDa homotetrameric heme-containing protein located predominantly in the peroxisome and expressed abundantly in the liver, lungs, and kidneys. Catalase deficiency results in overexpression of mitochondrial ROS and functional mitochondrial impairment (118). Catalase overexpression protects against H2O2 toxicity, whereas catalase deficiency exacerbates oxidative injury (110, 118, 140, 331).
Basal levels of H2O2 in the rat renal medulla are twofold higher than in the renal cortex (36). Exaggerated levels of H2O2 are reported in the renal medulla in several models of hypertension. They are implicated in the rise of BP, since intramedullary microperfusion of catalase reduced the H2O2 concentrations and moderated the hypertension, whereas intramedullary microperfusions of tempol or PEG-SOD were not fully effective (36, 267, 268).
Reduction of cellular glutathione (GSH) concentrations in rats by administration of buthionine sulfoximine caused progressive hypertension (292, 321). GPX not only metabolizes H2O2 but also converts toxic lipid peroxidation products into lipid alcohols in the presence of GSH. The reaction of O2 −• with arachidonate (AA) forms lipid peroxides, termed isoprostanes (240). Isoprostanes, prostaglandin (PG) endoperoxides, cyclooxygenase (COX) metabolites of hydroxyeicosatetraenoic acid (HETE), the stable mimetic U-46,619, and perhaps even prostacyclin (61), in addition to thromboxane A2 (TxA2) all activate the TP. The TP is expressed in ECs and VSMCs and in the kidney (291). ANG II infusion in the mouse increases the generation of isoprostanes and TxA2. Remarkably, studies in TP (−/−) mice demonstrated that this receptor mediates not only the ANG II-induced increases in BP and RVR but also the oxidative stress (133). Increased production of renal eicosanoids activating TP receptors enhances Cl− reabsorption in the LH (311) and augments TGF responses (311). Thus, these multiple effects indicate that ligands for this receptor or oxidative stress increase TP activity (287) that may blunt the pressure natriuresis and contribute to salt sensitivity (321, 346).
The Trx system comprises Trx, thioredoxin reductase (TrxR), and Prx. These abundant proteins from different family members are widely distributed through the cytoplasm, mitochondria, and other cell compartments (59, 211). This system has been implicated in the regulation of the cellular redox state, DNA synthesis, cell proliferation, and apoptosis (74). Three Trxs have been identified in mammalian cells: Trx1, located in the cytoplasm and the nucleus; Trx2 in the mitochondria; and Trx3 in spermatozoa (211). The TrxR is a selenocysteine-containing flavoprotein with three isoforms that control the activity of Trxs and thereby the cellular redox state. TrxR1 is located in the nucleus and the cytoplasm, whereas TrxR2 and TrxR3 are located in the mitochondria (211). The proteins of the Trx system are expressed in both ECs and VSMCs (59). Trx reduces Prxs, which in turn reduce H2O2 to H2O (59). Prxs compose a large antioxidant gene family that uses conserved cysteine (Cys) residues at the sites of peroxidation. The Prx1 and Prx2 isoforms are expressed in the cytoplasm, Prx3 and Prx5 in the mitochondria, and Prx4 in the endoplasmic reticulum (281). Mitochondrial Trx systems (Trx2, TrxR2, and Prx3) protect cells from mitochondrion-dependent ROS and apoptosis (59, 74, 314). Trx proteins are abundant in the renal tubules (52), especially in PTs and distal tubules (DTs), while lower levels are reported in glomerular cells (54, 211).
There are only limited studies of Trx and TrxR in hypertension. The Trx system is upregulated (58) or downregulated (277) in hypertension, depending on the tissue, the stimulus, and the model. Decreased Trx expression was observed in the aorta, heart, and kidneys of SHR and SHRSP and was related to the hypertension (277). In contrast, Trx expression was increased in the hearts of mice infused with ANG II at a slow-pressor rate (58). Trx1 expression in human umbilical vein endothelial cells was enhanced by low concentrations (10–50 μM) of H2O2, but was degraded by higher concentrations (100–500 μM) (98, 99). Transgenic mice overexpressing EC-specific Trx2 (Trx2Tg) have reduced mitochondrial and total oxidative stress and increased NO activity and endothelium-dependent relaxations and lower basal BP. An infusion of N
ω-Nitro-
In summary, the Trx system has been a focus of study in vascular biology where it has been shown to preserve NO bioavailability and endothelial function and to improve BP in ANG II-induced hypertension (74, 314, 347). Studies in the kidney are more limited, but generally concur in concluding that an impairment of the renal Trx system contributes to the impairment of NO activity associated with hypertension (277).
ROS and Renal Hemodynamics
Action of renal ROS on hemodynamics
Renal blood flow (RBF) determines GFR and tubular Na+ delivery and thereby is an important component of body salt and fluid homeostasis. The regulation of RBF by ROS has been studied extensively. A reduced RBF, or an altered intrarenal blood flow distribution, has been implicated in the development and maintenance of hypertension (236, 266, 317). Increased RVR attributable to ROS has been reported in genetic hypertension in the SHR, renovascular hypertension in the 2K,1C Goldblatt model and in the reduced renal mass (RRM) model of CKD, and in the model of ANG II-infused rats and/or mice (134, 251, 303, 305, 317). Infusion of ANG II at a slow-pressor rate increases the RVR in rats and mice before hypertension (134, 317) and is accompanied by increased renal and vascular ROS, increased renal excretion of lipid peroxidation products, and increased renal tyrosine nitration, implying ONOO− generation (29, 103, 133, 134, 216, 294, 312, 317). The coadministration of tempol prevents those events, reinforcing the evidence for involvement of O2 −• in ANG II-dependent hypertension (134, 301 –303).
ANG II acting on AT1Rs enhances the generation of O2 −• by NADPH oxidase and the release of AA by phospholipase A2. AA is metabolized by COXs to PG endoperoxides (PGG2 and PGH2), which are further metabolized by enzymes, including thromboxane synthase to TxA2, or oxidized by O2 −• to 8-isoprostane F2-alpha (8-iso). PGH2, TxA2, and 8-iso all activate TP (191). Welch and colleagues (33) reported that hypertension was maintained in rats at the ANG II-dependent phase of 2K,1C Goldblatt hypertension by AT1R and by COX-1 products that activated the TP (311). The TP also is implicated in the ANG II slow-pressor response, since rats given TP antagonists or TP (−/−) mice do not develop oxidative stress, renal vasoconstriction, or hypertension (133, 308). These prohypertensive effects of TPs were hypothesized to include increased pre- and postglomerular resistance and enhanced TGF responses (133).
Therefore, renal hemodynamics and GFR depend on the interaction of several elements, including ANG II, PGs, and TP activation, all of which can interact with ROS and NO to regulate the tone of the afferent and efferent arterioles.
Tubuloglomerular feedback
TGF regulates the glomerular arteriole resistance. There is a vasoconstrictive response of the renal afferent arteriole during increased NaCl delivery to, and reabsorption by, the MD segment. The proximal tubular fluid flow that yields 50% activation of TGF (set point) is close to the ambient rate measured by micropuncture. Thus, a reduction in tubular fluid flow will reduce TGF and vasodilate the afferent arteriole. TGF is a unique renal mechanism whereby vasoconstriction is regulated by tubular NaCl transport, and thereby the renal hemodynamics is adjusted to maintain the body fluid volume and BP. The MD cells at the junction of the thick ascending limb (TAL) of the LH are activated by reabsorption via the luminal Na+/K+/2Cl− transporter type-2 (NKCC2). This leads to the elaboration of positive (notably adenosine, ATP and vasoconstrictor PGs) and negative (notably NO and vasodilator PGs) signaling molecules, which adjust the tone of the renal afferent arteriole. Ren et al. (238) reported an efferent arteriole vasodilation in response to reabsorption of fluid by the MD that was mediated by adenosine receptors type-2. Thus, activation of TGF may reduce single-nephron glomerular filtration rate (SNGFR) both by increasing afferent arteriole vasoconstriction and by decreasing efferent arteriole vasoconstriction.
The myogenic response is a stretch-activated contraction of VSMC in the interlobular and afferent, but not efferent, arterioles. Myogenic and TGF responses together coordinate renal autoregulation whereby the RBF and the GFR remain stable despite changes in the renal perfusion pressure within a physiological range. ROS interact with and/or mediate both the myogenic and the TGF components of renal autoregulation and RVR, hypertension, and associated renal damage.
TGF- and MD-mediated renin release both have been implicated in the short- and long-term regulation of renal hemodynamics and in salt and water balance. Activation of TGF increases the afferent arteriolar resistance and reduces the SNGFR. Resetting of TGF by neuronal NOS (nNOS) in the MD during changes in BP or salt intake likely contributes to efficient excretion of excessive NaCl excretion (17, 282).
NaCl reabsorption in the MD stimulates nNOS-derived NO production, which puts a brake on TGF responses (217, 288, 322). Microperfusion of the tubular lumen of the MD of Sprague-Dawley or Wistar Kyoto (WKY) rats with nonspecific NOS inhibitors (NG-methyl-
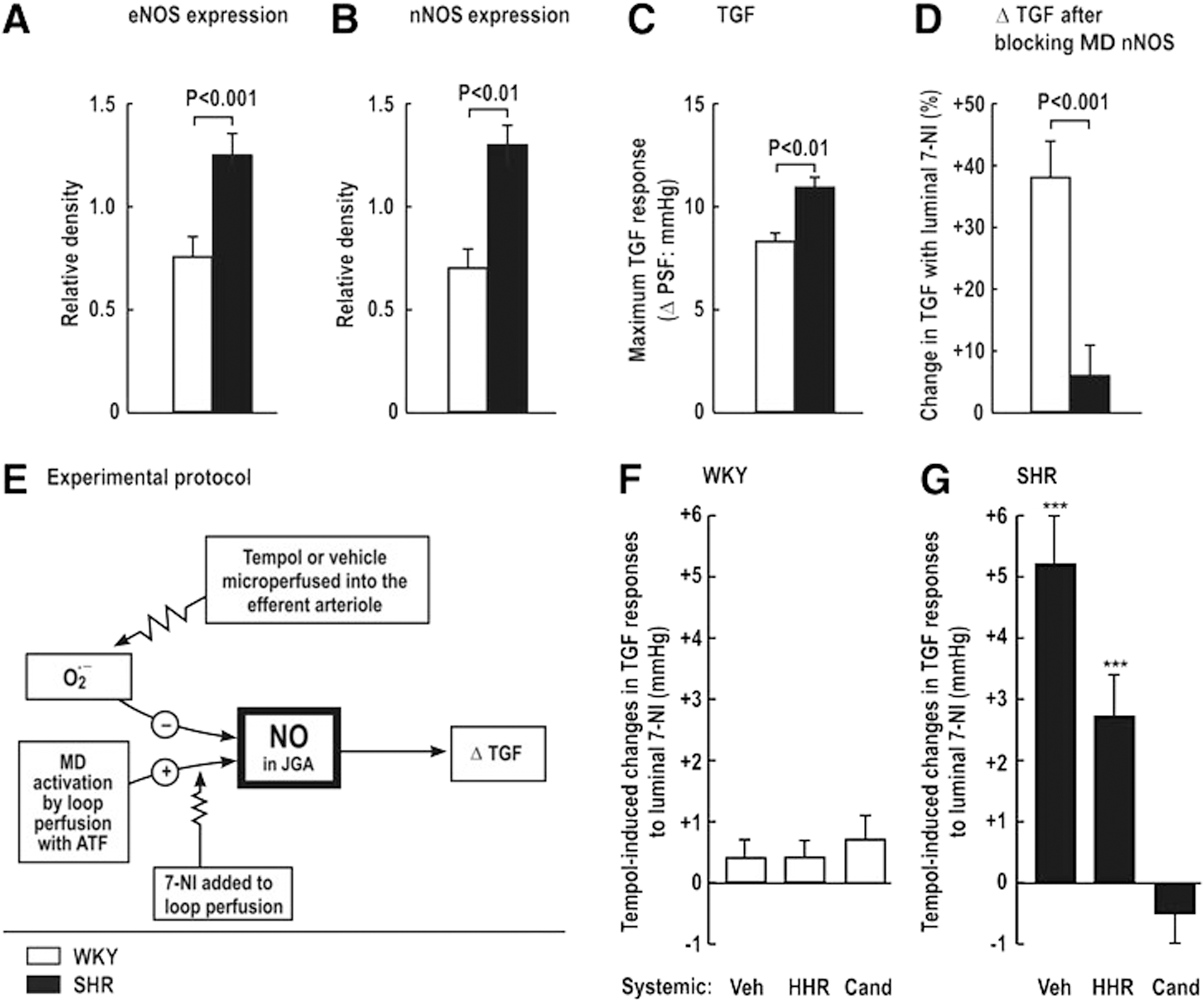
Increased MD levels of O2 −• that enhance TGF responses contribute to renal vasoconstriction, salt retention, and hypertension, for example, during ANG II infusion or in the SHR. Thus, SHR, compared with WKY, have increased renal expression of eNOS and nNOS (Fig. 3A, B), yet paradoxically enhanced TGF responses (Fig. 3C) and diminished blocking of TGF by juxtaglomerular apparatus (JGA)-derived NO, as indexed from the increase in TGF during blockade of nNOS by luminal 7-NI added to the artificial tubular fluid (Fig. 3D). Tempol or vehicle was perfused into the star vessel (efferent arteriole) to reach the peritubular capillaries surrounding the test nephron to correct oxidative stress (Fig. 3E). The effects of tempol on NO signaling in the JGA again were assessed from the changes in TGF during luminal 7-NI. There were no significant tempol-induced changes in the response to luminal 7-NI in normotensive WKY rats, implying no interference in NO signaling by O2 −• (Fig. 3F). In contrast, tempol microperfused into the efferent arteriole and peritubular capillaries of the SHR increased the TGF by circa 40%. This effect in SHR nephrons was prevented by 2 weeks of administration of the ANG II type-1 receptor blocker (ARB) candesartan, but not by equivalent lowering of BP with hydralazine, hydrochlorothiazide, and reserpine (Fig. 3G). Welch et al. (310, 311) conclude that AT1R-mediated increases in O2 −• in the JGA of the SHR enhance TGF by preventing its buffering by MD-derived NO from nNOS. This reflects NO bioinactivation by O2 −• in the JGA of the SHR. In other studies, systemic silencing of the p22phox gene during ANG II infusion reduced renal ROS, and prevented progressive hypertension (191) and moderated the TGF responses (207). Fu et al. (70) reported that activation of AT1R by ANG II mediated NOX2-dependent O2 −• production in the MD cells, whereas basal O2 −• generation was sustained by NOX4. Thus, increased NOX2-derived O2 −• may inactivate nNOS-derived NO in the MD and eNOS-derived NO in the afferent arteriole in hypertensive models. This could account for the enhanced TGF responses that contributed to renal vasoconstriction and renal Na+ retention (70, 312, 316, 320, 321).
Arteriolar constriction and dilation
ROS regulate renal hemodynamics both directly, through vasoconstriction, and indirectly, through reducing NO activity (3, 175, 250). ROS are generated in microvessels and in genetic models such as the SHR and in the kidney in response to both short-term and prolonged infusions of ANG II, or other vasoconstrictors such as ET-1 or TP agonists (29, 226, 294). Thus, vasoconstriction produced by the TP activator U-46,619 on rabbit isolated, perfused afferent arteriole is moderated by coincubation with tempol, but enhanced by L-NAME (251).
ROS have been implicated in the acetylcholine-stimulated release of COX-1 and COX-2-derived EDCFs in renal afferent arterioles of rats or rabbits with ANG II or 2K,1C renovascular hypertension (250, 293, 294, 305). The EDCFs activates the TP on afferent arteriolar VSMCs to cause vasoconstriction. Tempol, COX-2 inhibitors, or TP antagonists, all improved endothelium-dependent relaxations and eliminated endothelium-dependent contractions in these models. Prolonged administration of a COX-2 antagonist or tempol also reduced the BP, increased the RBF, and restored the endothelial function of renal arteries of the 2K,2C renovascular hypertensive rats (283). These studies implicate intrarenal and afferent arteriolar ROS in the generation of an EDCF that caused TP-dependent renal vasoconstriction and contributed to hypertension.
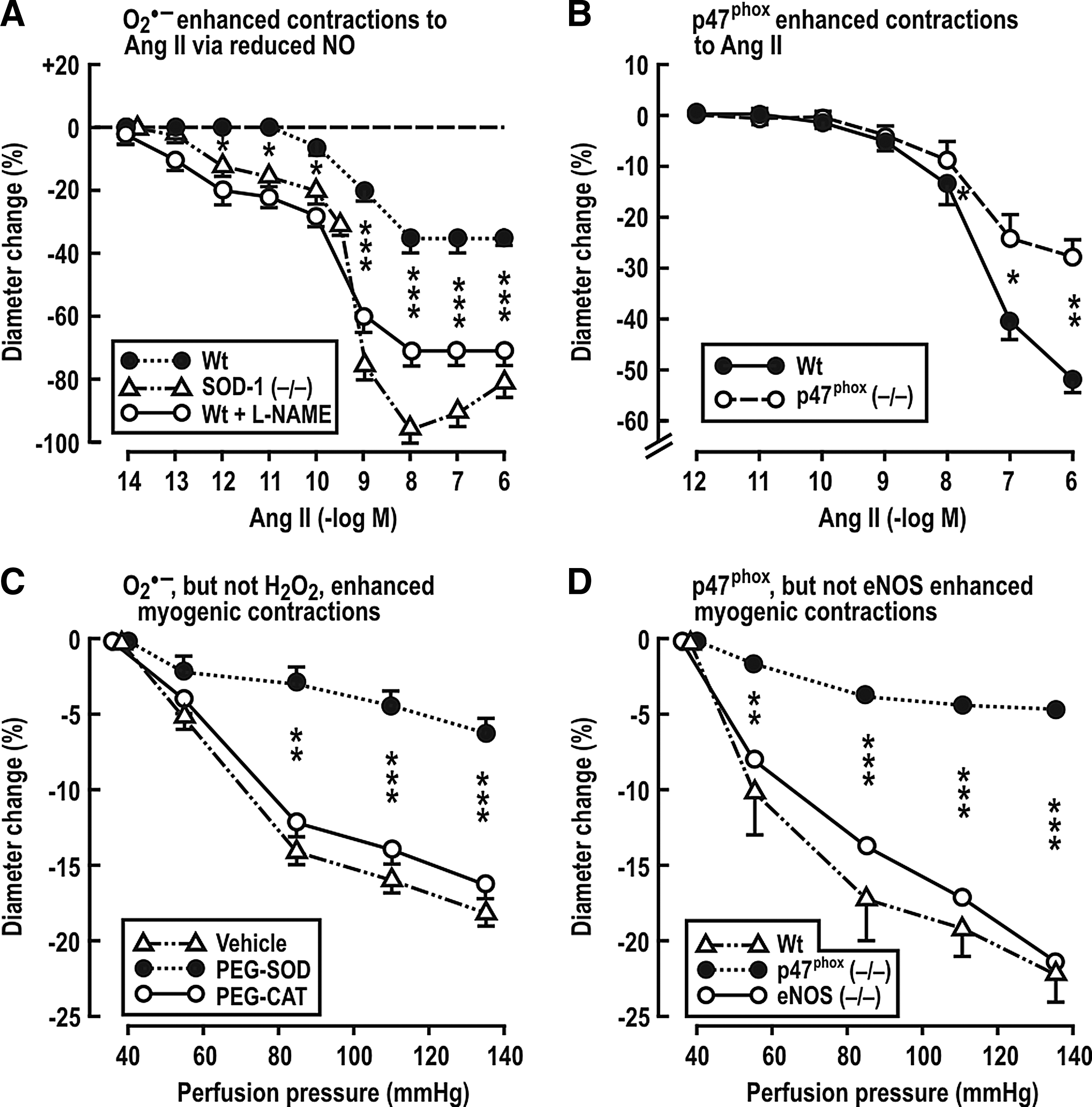
Adenosine receptors type-1 (A1AR) on afferent arterioles is required for TGF responses (21). Adenosine enhances the long-term constrictor effects of ANG II on preglomerular vessels (27, 69, 156). Lee et al. (156) reported that the infusion of ANG II at a slow-pressor rate in mice leads to hypertension and increases Na+ reabsorption that depends in part on A1ARs. A1AR (−/−) mice have blunted arteriolar and BP responses to L-NAME and ANG II (79). Gao et al. (79) propose that NO decreases, and ROS increases, adenosine production, and that adenosine mediates some of the renal effects of O2 −• vasoconstriction. The p47phox component of NADPH oxidase is required for a full hypertensive response to ANG II (93). Likewise, Lai et al. demonstrated that p47phox also is required for increased BP and RVR in mice infused with ANG II (153). Studies by Carlstrom et al. (28) and Lai et al. (153) have investigated the roles of O2 −•, H2O2, and NO on mouse isolated afferent arterioles. Both ANG II and increased perfusion pressure augmented afferent arteriolar ROS generation (153). Genetic deletion of SOD-1 increased O2 −• and enhanced the response to ANG II by 89% and the sensitivity by >1000-fold (Fig. 4A). This was attributed to bioinactivation of NO, since, in the presence of L-NAME, ANG II contractions became similar in SOD-1 (+/+) and in (−/−) arterioles (28). The O2 −• generated by ANG II requires the p47phox component of NADPH oxidase, since arterioles from p47phox (−/−) mice had diminished response to ANG II (Fig. 4B) and diminished ROS generation (153). Similarly, myogenic contractions of mouse afferent arterioles to increased perfusion pressure were diminished by PEG-SOD, but not by PEG-catalase (CAT) (Fig. 4C), and by maneuvers that blocked NADPH oxidase (154). However, unlike the response to ANG II, neither blockade of NO with L-NAME nor genetic deletion of eNOS affected the myogenic contractions (Fig. 4D) (153). These studies indicate that eNOS-derived endothelial NO potently inhibits the response to ANG II, but that this blunting is potently inhibited by O2 −•. In contrast, myogenic responses do not depend on the endothelium and therefore are modulated by O2 −• independent of NO (28). Carlstrom et al. (27) reported that NOX2 is required for contractile responses of the afferent arteriole to ANG II and/or adenosine and is implicated in prolonged ANG II-induced hypertension in mice. Ren et al. (236) reported that NOX2-derived O2 −• contributes to the enhanced myogenic response, and hence to the high preglomerular resistance, of SHR, but these authors did not detect an effect of O2 −• on myogenic responses of afferent arterioles from WKY rats.
In summary, inactivation of NO by increased renal O2 −• production enhances afferent arteriole tone, Na+ reabsorption, and TGF responses, all of which contribute to hypertension (178 –180, 310). ROS antagonizes the effects of eNOS-derived NO to increase renal plasma flow by dilating the glomerular arterioles. MD O2 −• prevents the effects of NO formed via nNOS in the MD to blunt TGF and reduces afferent arteriole vasoconstriction. Therefore, an increase in the intrarenal ROS can have importance on renal hemodynamics that could lead to hypertension (284, 350).
Afferent arterioles from a rat model of type-1 diabetes mellitus (T1DM) are dilated (286). Pharmacological studies demonstrated that this could be ascribed to activation of ATP-sensitive K+ (KATP) channels and Kir1.1/Kir3.x inward-rectifier K+ channels in VSMCs. Activation of membrane K+ channels should hyperpolarize the VSMCs, inactivate the voltage-gated Ca2+ channels, and thereby lead to vasodilation. The activation of the K+ channels in T1DM rat arterioles is attributed to oxidative stress, which is prevented in rats given tempol (286). This is an important result, since it provides a mechanism for diabetes hyperfiltration. It is unusual in demonstrating that prolonged ROS can cause afferent arteriole vasodilation, and provides a novel mechanism to explain it.
Renal autoregulation
RBF and GFR are efficiently buffered against changes in the renal perfusion pressure by a coordinated response of the renal afferent arteriole and the interlobular artery. At least three mechanisms participate in autoregulation of RBF: a fast myogenic response, a slower TGF response, and a third, very slow component of uncertain causality (131, 256). Just and Arendhorst (130) demonstrated that the myogenic response in the mouse kidney contributed 60%, TGF 40%, and the third mechanism 5% to RBF autoregulation. Apocynin attenuated the myogenic responses, which implicates O2 −•, and this was independent of NO. Similar conclusions were drawn by Lai et al. (154) (Fig. 4C, D).
Renal autoregulation is impaired in the RRM, DOCA/salt, and DS rat models of hypertension and in salt-supplemented SHRSP (109, 169, 194, 230, 247, 252). The glomerular capillary hydraulic pressure (PGC) and the blood flow in cortical glomeruli of SHR are maintained during elevation of the renal perfusion pressure by highly effective autoregulation (121, 233, 317). This suggests that SHR have increased preglomerular vascular resistance, which is consistent with the studies showing narrowed afferent arterioles (206, 317), with enhanced myogenic vasoconstriction (120) and augmented TGF responses (206, 310, 317). The connecting tubule tubuloglomerular feedback (CTGF) might modulate RBF autoregulation. The CTGF is activated by Na+ reabsorption in the connecting tubule (CNT) and leads to vasodilation the afferent arteriole (297). Thus, the CTGF counteracts the vasoconstrictive effects of macula densa TGF by decreasing the afferent arteriole resistance in responses to increased NaCl delivery (296). NaCl reabsorption and consequently activation of the CTGF are mediated by the thiazide-sensitive Na+/Cl− cotransporter and by the amiloride/benzamil-sensitive epithelial Na+ channel (ENaC) in the CNT (297). The role of the CTGF has not been investigated in hypertensive animals. However, Ren et al. reported that the intratubular perfusion of ANG II enhanced the CTGF through activation of AT1R, increased NaCl reabsorption mediated by the ENaC (235), which led to dilation of afferent arterioles mediated by protein kinase C (PKC)-dependent NOX2-derived O2 −• production. Coperfusion of ANG II with tempol, apocynin, or a NOX2 inhibitor blocked the effect of intratubular ANG II (237). Thus, activation of the CTGF by NOX2-dependent generation of tubular O2 −• could limit ANG II-induced vasoconstriction of afferent arterioles and preserve the GFR by activation of NADPH oxidase. This would constitute an unusual effect of renal ROS to limit renal vasoconstriction, autoregulation, and the development of hypertension.
Overall, these data imply that increased renal ROS in hypertensive animals enhance myogenic and macula densa TGF responses and the contraction of afferent arterioles to ANG II, ET-1, and TP activation. Oxidative stress leads to the elaboration of an EDCF that activates TP on VSMC whose activity also is enhanced by ROS. This is accompanied by blunted EDRF/NO (28, 79, 151, 293) and augmented TGF responses (207, 310, 312), all of which contribute to a renal vascular prohypertensive pathway activated during oxidative stress.
Intrarenal distribution of blood flow
Renal medullary ROS generation contributes to the development of hypertension in several models by direct effects of H2O2 and by limiting NO bioavailability. Medullary ROS restrict the medullary perfusion and Na+ excretion, and increase BP (3, 36, 52, 175, 176, 197, 209, 217, 222, 267, 284). Vascular H2O2 enhances eNOS expression and enhances the activity of NOS and NO generation by cultured ECs (23, 56). NO generation was markedly increased in the medulla of rats infused with ANG II at a subpressor rate (195, 351). However, this response was blunted in the medulla of DS rats, which have an impaired NOS expression and activity (275). The unchanged levels of O2 −• found in the renal medulla of rats during ANG II infusion were ascribed to the reaction of O2 −• with the high level of NO to form ONOO− (52, 222, 348). Hypertension in DS rats is accompanied by a reduced medullary expression of SOD-1 and SOD-2 that could contribute to enhance O2 −• (187). ANG II infusion increases O2 −•generation in the mTAL, which interacts with NO in the vasa recta to mediate a tubulovascular crosstalk that culminates in a reduced MBF and reduced UNaV (52).
Further studies implicate medullary H2O2 in salt-sensitive hypertension. Thus, the intramedullary infusion of SOD inhibitors, such as diethyl thiocarbamate, reduces the MBF and raises the BP (175), yet the intramedullary infusion of tempol did not reduce the BP unless coinfused with catalase to reduce H2O2. (36, 268). Although catalase expression is increased in the kidneys of SHR (265, 346), its activity is decreased (346). Catalase overexpression reduced the pressor response to ANG II in mice (331), and intramedullary catalase infusion prevented the hypertension accompanying an intramedullary infusion of H2O2 (175).
Sousa et al. (267) reported a selective increase in AT1R and NOX4 expression and in medullary H2O2 in rats infused with ANG II at slow-pressure rates. Intraperitoneal administration of PEG-CAT caused only a transient fall in BP that was attributed to a normalization of renal angiotensinogen (AGT) levels, which are rate limiting for ANG II generation in the kidney (141). Apparently, elevated levels of H2O2 generated during prolonged ANG II stimulate the intrarenal RAAS, but inhibit the systemic RAAS (175, 268, 331). Mice overexpressing AGT in the PT are hypertensive independent of the plasma renin concentration, reinforcing the concept that a selective activation of the renal RAAS causes hypertension probably by enhancing ROS, renal vasoconstriction, and Na+ retention (65).
An elevation of renal perfusion pressure during hypertension may directly increase ROS production in the renal medulla, MBF and NaCl delivery to, and reabsorption by, the TAL (125). Jin et al. (125) reported that a short-term increase of renal perfusion pressure enhanced the production of both NO and H2O2 in the outer medulla of rats. The increased NO was attributed to the pressure-induced increase in MBF that enhanced vascular shear stress and to the increased H2O2 with higher flow rates through the mTAL. These studies suggest that increased renal perfusion pressure creates oxidative stress within the outer medulla of the kidney that leads to generation of the medullary H2O2 that contributes to salt-sensitive hypertension, while increased levels of medullary NO counteract these effects and restrain the salt sensitivity and hypertension (125). Thus, H2O2 in the renal medulla rather than O2 −• has been identified as the major ROS mediating hypertension.
ROS in the Glomerulus
The direct effects of ANG II on kidney cells have been studied extensively (16, 168, 329). Both podocytes and mesangial cells (MC) generate ANG II in response to overproduction of ROS, whereas ROS are produced by ANG II. Indeed, ANG II is implicated in glomerular hypertension, disruption of the glomerular filtration barrier (GFB), and proteinuria in several hypertensive models (73, 239, 246, 274, 330). This provides a potential for an ANG II-initiated, feed-forward production of glomerular ROS (249, 329). On the other hand, podocytes express angiotensin-converting enzyme 2 (ACE-2), which can metabolize ANG II to angiotensin-(1 –7) (102). Thus, ACE-2 may provide a brake on this feed-forward process.
Podocyte injury or dysfunction has been implicated as an initial event in glomerulosclerosis and proteinuria (313). Although ANG II-induced oxidative stress contributes to podocyte injury, the mechanisms have not been elucidated completely. ANG II enhances ROS generation in podocytes, promotes podocyte autophagy through mitochondrial generation of ROS, decreases nephrin expression, and induces apoptosis (55, 124, 239). Prolonged infusion of ANG II into rats at a slow-pressor rate induced A1AR-dependent hypertension, proteinuria, and podocyte injury and decreased nephrin expression and caveolin-1 phosphorylation (239). Thus, ANG II-induced ROS generation could interrupt the crosstalk between caveolin-1 and nephrin that is required to maintain the integrity of podocytes and the GFB.
Aldosterone also has been implicated in ROS-dependent podocyte injury and MC proliferation (20) and also may be involved in a feed-forward signaling between ROS and the RAAS (20, 139, 202, 212). Tempol preserved podocyte function and attenuated glomerulosclerosis in rats with aldosterone plus salt-induced hypertension (202). Mice lacking the guanylyl cyclase-A (GC-A) receptor for natriuretic peptides and given aldosterone and a high-salt diet increased oxidative stress, podocyte injury, and albuminuria, which were ameliorated by tempol or an ARB (212). The authors attributed the renoprotective properties of the GC-A system to a local inhibition of the RAAS and oxidative stress in podocytes, independent of BP.
Although the major site of action of aldosterone is the CD, it also activates mineralocorticoid receptors (MR) on podocytes. Shibata et al. (259) reported proteinuria and decreased nephrin and podocin expression in uniphrectomized rats infused with aldosterone and fed a high-salt diet. The BP, renal ROS production, NOX2, p47phox, p67phox, Rac-1 and Sgk1 (an effector kinase of MR) all were elevated by aldosterone. This was related to ROS, since all these effects were normalized by tempol but not by nonspecific correction of hypertension.
ROS mediate the MC proliferation, migration and production of extracellular matrix (ECM) induced by ANG II or aldosterone (73, 90, 168, 189). ANG II increases NADPH oxidase-dependent O2 −• generation in MC and increases production of ECM proteins such as fibronectin, which are implicated in the development of glomerulosclerosis (115). MR activation by aldosterone induces NADPH-dependent ROS generation in MC that leads to translocation of p47phox and p67phox. MR antagonists ameliorate glomerular injury and decrease proteinuria in several animal models of hypertension, independent of changes in BP (189, 202, 241). Thus, intrarenal ANG II and aldosterone signaling via the MR both elicit renal oxidative stress that is widely implicated in the pathogenesis of hypertension and associated renal damage in animal models.
ROS and Renal Tubular Transport
Proximal tubule
NADPH oxidase-derived ROS can reduce renal tubular Na+ transport in the PT either directly or by reducing NO activity. Although there is enhanced Na+ and fluid uptake in PT of young SHR (4, 280), Panico et al. (224) reported reduced reabsorption in the PT of adult SHR and relate this to increased intratubular NADPH oxidase-derived O2 −•. An intratubular perfusion of tempol, apocynin or systemic silencing of p22phox with a siRNA restored PT reabsorption in adult SHR nephrons, thereby implicating NOX-derived O2 −• production in the reduced PT fluid reabsorption (224). This was ascribed to increased expression of the Na+/H+ exchanger regulatory factor (NHERF2), which inhibited the Na+/H+ exchanger (NHE3) activity (Fig. 5). The authors suggest that downstream sites of Na+ reabsorption, increased by O2 −•, would compensate for the dysfunction in the PT and thereby account for the similar Na+ and water excretion in both WKY and SHR. While NOX4 is the primary source of O2 −• in the PT (87, 88), the increase in PT reabsorption in the SHR by luminal apocynin implicates other NOX isoforms that required p47phox, such as NOX1 or NOX2 (224). Inhibition of PT fluid reabsorption by O2 −• may be secondary to limitation of NO, since fluid reabsorption is enhanced by NO and reduced by the intratubular perfusion of a nonspecific or by an nNOS- or an iNOS-specific inhibitor (10, 199). Moreover, iNOS (−/−) and nNOS (−/−) mice but not eNOS (−/−) mice had reduced PT reabsorption (298, 299).
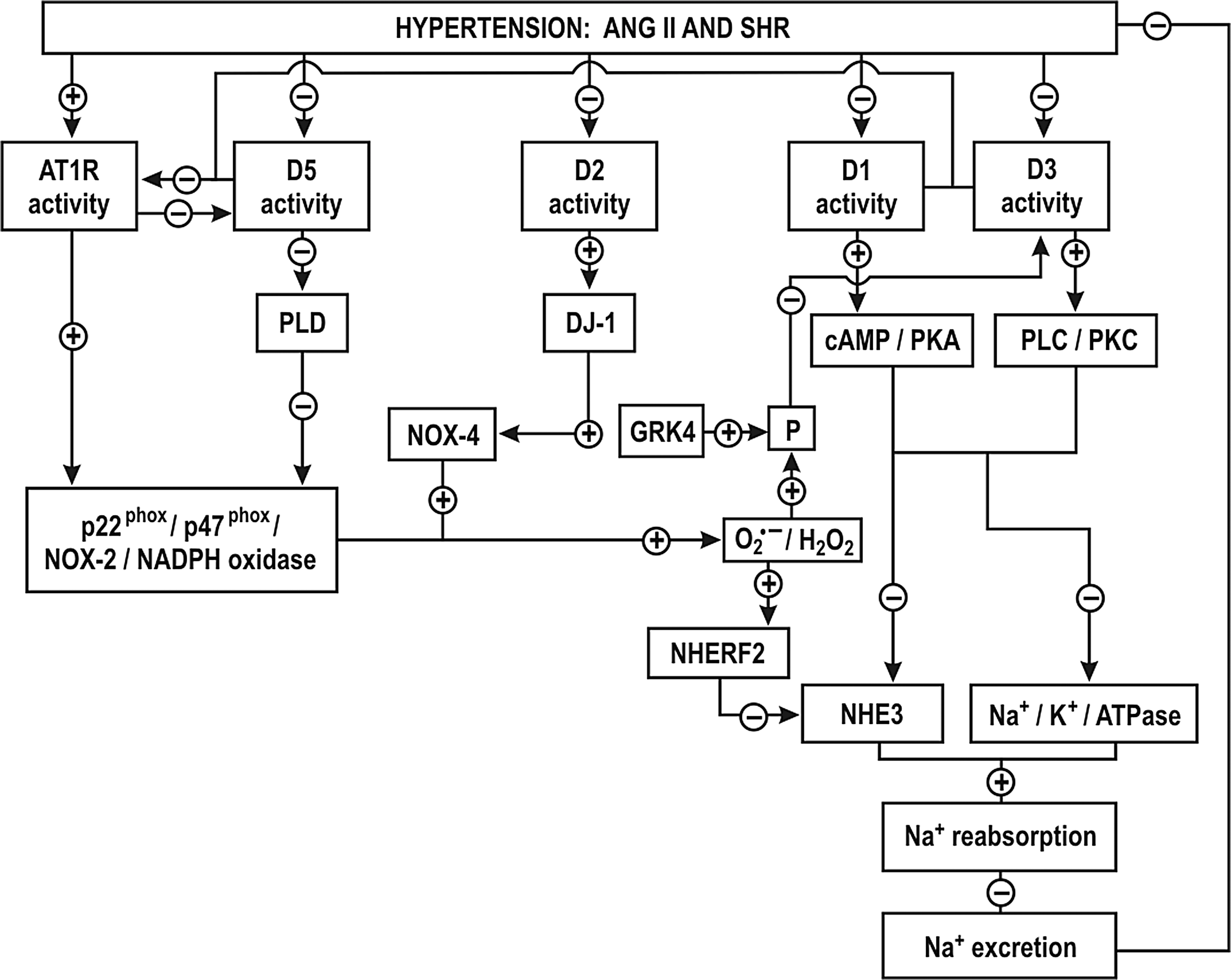
There are normal distributions of NHE3 and sodium-phosphate-cotransporter (NaPi2) proteins in the PT of young SHR, while in adult SHR, and in other some hypertensive rat models, the transporters are relocated from the luminal brush-border into the cytoplasm. This may contribute to decreased PT Na+ and fluid reabsorption (174, 185, 334, 338).
It is not yet clear whether O2 −• and/or NO regulate luminal expression of sodium transporters in the PT. While hypertension causes a pressure-natriuresis, the infusion of ANG II causes an anti-natriuresis (157, 158, 185, 349). The infusion of a nonpressor concentration of ANG II enhanced renal ROS production (29) and translocated NHE3 and NaPi2 to the apical microvilli in the PT, which should increase Na+ and water reabsorption (157, 158). On the other hand, the infusion of pressor concentrations of ANG II had the opposite effect (185). A nonpressor infusion of ANG II was confirmed to simulate NHE3 activity in the PT brush border and in TAL cells (149, 328).
Blockade of A1ARs reduces Na+ transport in the PT and leads to natriuresis. Thus, the increased expression and function of A1ARs in the PT of rats during salt restriction may contribute to enhance PT reabsorption. (146, 323). Activation of A1AR augmented Na+ transport in human immortalized PT cells (HK-2) (276). H2O2 increased A1AR expression in smooth muscle cells of hamsters by activating NFκ-B (200). It will be important to determine the precise role of ROS and other related systems such as adenosine on tubular Na+ reabsorption.
A defect in the dopaminergic system of the PT has been implicated in the development of hypertension. Dopamine acting on D1-like and D2-like receptors decreases Na+ transport in volume-expanded states. Nevertheless, D2-like receptor activation can increase Na+ transport in volume-depleted states. The exact mechanisms whereby D2-like receptors regulate ion transport have not been established. There is reduced renal expression and/or impairment of dopamine receptors in several models of hypertension, such as the DS rats and the SHR (12, 270, 339, 343). D-1-like receptor function was impaired in the PT of volume expanded SHR and was implicated in a decreased natriuresis and the development of salt-sensitive hypertension (270, 343). This impairment was attributed to oxidative stress since it was corrected by tempol (12, 127). Indeed, D1, D3 and D5 were all impaired in the PT of SHR (Fig. 5) (127, 343 –345). Increased renal O2 −• and G protein-coupled receptor kinase type 4 (GRK4) in SHR induced the phosphorylation of D1 and D3 receptors in the PT, uncoupling them from their G protein effector protein and thereby preventing their activation (Fig. 5) (343). The activation of AT1R by ANG II impaired the D5 receptor (345). ANG II via AT1R stimulated the NADPH oxidase-dependent O2 −• production in the PT of SHR and increased the expression of the NHE3 inhibitor factor (NHERF2), which decreased Na+ reabsorption in the PTs of SHR by inhibition of NHE3 (Fig. 5) (224). Since the D1, D3 and D5 receptors negatively regulate AT1R and oxidative stress (Fig. 5), their impairment in the in the PT of SHR would result in enhanced effects of ANG II that would account for the decreased Na+ reabsorption.
The interaction of D1-like with D2-like receptors regulates Na+ reabsorption in the PT and Na+ excretion (60, 335). The impairment of D1 receptor function in the PT and in the TAL of SHR (117, 201, 343) leads to increased luminal NHE3 and NaPi2, and basolateral Na/HCO3 − co-transporters and Na+-K+-ATPase that together account for enhanced PT reabsorption (117, 127, 201, 343).
D2 (−/−) mice have hypertension that is in part dependent on oxidative stress. There is increased renal expression of NOX1, NOX2 and NOX4 and NADPH oxidase activity in D2 (−/−) mice (8). DJ-1, a multifunctional protein with antioxidant properties is required for the D2 receptor signaling (Fig. 5). Renal silencing of DJ-1 gene in mice increases NOX4 expression and NADPH oxidase activity and increases the BP (45). D5 (−/−) mice have hypertension that is dependent on oxidative stress (335). There is increased renal expression of NOX2, NOX4, p47phox and enhanced NADPH oxidase activity (335). D5 (−/−) mice also are salt sensitive and have higher expression of AT1R in the kidney (345). D3(−/−) mice also have increased renal AT1R expression but only a mild hypertension (344). This is attributed to a renal upregulation of D5 receptors, which prevents oxidative stress in D3 (−/−) mice (300). Thus, stimulation of D1-like and D2-like receptors activates antioxidant pathways while their impairment during hypertension exacerbates the oxidative stress induced by ANG II and its effects on Na+ reabsorption.
Loop of Henle
The apical Na+/K+/2Cl− cotransporter type-2 (NKCC2) in the TAL accounts for 70–80% of transcellular Na+ reabsorption whereas the NHE3 accounts for the remainder. The energy is provided by the basolateral Na+-K+-ATPase that maintains a low intracellular Na+ concentration. O2 −• activates a signaling pathway in the mTAL that includes PKC-alpha and NKCC2 to enhance Na+ transport (217, 264). Although NADPH oxidase is the main source of O2 −• in TAL, xanthine oxidase (XO), COX and CYP 450 enzymes also are expressed in this segment and might contribute to O2 −• production (86). NOX2 and p47phox were identified as the main sources of O2 −• generation in the TAL in response to ANG II or increased luminal flow (114, 254, 264). Feng et al. (62) reported that disruption of the gene for p67phox decreased medullary NADPH oxidase-dependent O2 −• production and attenuated salt-sensitive hypertension in DS rats. However, Hong and Garvin (113) reported that flow-induced production of O2 −• in the TAL was dependent on NOX4 but not on NOX2.
NO is a major regulator of NaCl transport in the TAL. Studies in isolated, perfused TAL demonstrated that NOS-3 derived NO inhibits NKCC2 and NHE3 at this site (85, 218, 219). The NKCC2 inhibition was mediated by a phophodiesterase-mediated degradation of 3′-5′-cyclic adenosine monophosphate (cAMP) whereas the inhibition of NHE3 was attributed to direct effect of NO (85). A high salt diet decreases NaCl reabsorption in the TAL. This is attributed both to an increased medullary osmolality and to release of ET-1, which through ETB receptors increased NOS-3 expression and NO-dependent inhibition of NKCC2 (108). Impairment of NO has been shown to increase NaCl reabsorption at this nephron segment in models of salt sensitive hypertension, which may thereby contribute to salt-dependent increases in BP (82).
T1DM is often associated with hypertension, salt sensitivity and oxidative stress (45, 171, 271). Studies report increased Na+ transport and oxygen consumption in diabetic rats (11, 231). Yang et al. (333) reported increased Na+ transport and O2 consumption in the mTAL of diabetic rats. This was ascribed to a NADPH oxidase- and PKC-dependent increase in O2 −•, which entails NKCC2 and Na+ -K+-ATPase.
Renal production of 20-HETE is elevated in SHR, DOCA-salt or ANG II-infused rats and contributes to hypertension (111, 123, 196, 324). Roman and colleagues (196) reported that reduced 20-HETE production in the TAL of DS rats contributed to elevated Na+ reabsorption. The reduction of 20-HETE is dependent on ROS (111). Induction of 20-HETE synthesis improved pressure-natriuresis and attenuated the development of hypertension in DS rats (324). Thus, ROS generally enhance Na+ reabsorption in the TAL, in part via reduction in 20-HETE production and contributes to hypertension.
DT and CD
The ENaC in the distal nephron and cortical collecting duct (CCD) is responsible for much of the Na+ reabsorption in this segment. It provides the final renal tubular adjustments of Na+ reabsorption that are important for Na+ balance and thereby play a critical role in the regulation of BP (273). ANG II activation of AT1Rs in the CCD of rats increased ENaC activity by NOX-dependent ROS generation (272). Aldosterone increased ENaC activity in A6 distal nephron cells by O2 −• generation, which reduced the inhibitory effects of NO on the ENaC (340). H2O2 generated in the CCD during a high NaCl intake stimulated the ENaC by diminishing its inhibition by arachidonic acid (272). These limited data imply that ROS-induced stimulation of the ENaC might be the result of diminishing the inhibitory factors acting on this Na+ channel. ET-1 inhibits Na+ transport in the CD via activation of ETB receptors and reduces ENaC expression and activity and Na+-K+-ATPase and thereby Na+ reabsorption (84). This entails the generation of NOS-1 dependent NO and accumulation of prostaglandin E2 (22, 143). ET-1 production in the CD was increased by elevated Na+ intake and associated with natriuresis (173). Paradoxically, medullary levels of ET-1 and ETB receptor expression were lower in DS rats than in DR rats on a high salt diet (269). However, renal expression of the ENaC was increased in DS rats (7) and this could represent a mechanism for the development of salt-sensitive hypertension.
Pressure-natriuresis
The kidney maintains the extracellular volume and BP by modulation of the pressure-natriuresis mechanism. An increase in pressure-natriuresis is accomplished by a vascular component, which has been related to an increased MBF leading to a decreased medullary solute gradient and an increased renal interstitial hydrostatic pressure both of which inhibit Na+ reabsorption and a primary tubular component, which has been with associated decreased Na+ reabsorption in the PT and TAL (83, 91, 244)
A shift of the pressure-natriuresis relationship to higher pressures occurs in many models of hypertension. The pressure-natriuresis was the outcome of opposing effects of ROS and NO, particularly in the renal medulla (41, 209). Inhibition of oxidative stress with tempol increases pressure-natriuresis and diminishes salt-sensitivity (94, 186, 312), whereas prolonged inhibition of NOS with L-NAME inhibits pressure-natriuresis and causes salt-sensitive hypertension (341). Thus, the balance between O2 −• and NO determines the effectiveness of pressure-natriuresis and thereby the set point of BP.
ROS, tubular transport, and hypertension
Collectively, the studies linking ROS to tubular transport and hypertension suggest that O2 −• decreases Na+ and fluid reabsorption in the PT largely by decreasing NO availability, while O2 −• increases Na+ reabsorption in TAL and distal segments. NADPH oxidase was identified as the main source of O2 −• in the tubules. However, increased Na+ delivery or ANG II both enhanced H2O2 production in medullary segments (213) and increased Na+ reabsorption in the mTAL (213) and CDs (272). NOX2 and NOX4 generate O2 −• in the kidney, but their specific roles and sites of action are not yet well defined.
ROS, Renal Oxygenation, and Hypertension
PO2 and TNa/QO2 in the renal cortex and medulla
A reduction in renal tissue PO2 in the models of hypertension has been attributed to increase O2 usage for Na+ transport (TNa/QO2) (150, 303, 305, 312, 318). Welch et al. (301) reported a lower PO2 in the kidney cortex and the outer medulla of SHR that was dependent on renal AT1R and O2 −•, since it was normalized by an ARB or tempol, whereas nonspecific correction of hypertension was less effective (57, 301, 302). The PO2 also was reduced in rats infused with ANG II at a slow-pressor rate (303) and in the clipped kidney of the 2K,1C rat (305). Tempol restored the PO2 levels and TNa/QO2 in both these models, whereas an ARB or an angiotensin-converting enzyme inhibitor (ACEi) had no effects in the clipped kidney of 2K,1C rats (223). Interestingly, baseline cortical PO2 was reduced in the clipped kidney in the early phase of 2K,1C, but it was restored in the chronic model, perhaps because a high BP and lack of autoregulation maintained the blood supply to the clipped kidney and the fall in the GFR limited Na+ filtration and reabsorption, thereby reducing renal work and O2 demands (223). Mice with 5/6 nephrectomy and a prolonged RRM also had reduced PO2 levels and TNa/QO2, especially in the renal medulla, which were corrected by tempol (150). The authors propose that tempol enhanced NO bioavailability in the renal medulla and thereby improved the mitochondrial efficiency and MBF (150). Prolonged inhibition of ANG II by an ACEi or an ARB in rats reduced age-related renal mitochondrial O2 −• production and improved O2 usage (47). These studies suggest that an enhanced mitochondrial O2 −• in several models of hypertension may have reduced NO sufficiently to disinhibit and uncouple mitochondrial respiration, increase QO2, but limit ATP production, and consequently to decrease TNa/QO2. Indeed, inactivation of NO signaling by O2 −• is reported in the kidney of the SHR (310) in the clipped kidney of the 2K,1C rat (9, 19, 262, 263, 317) and the kidney of the ANG II-infused rat (9, 19, 50, 262, 263, 317). Since physiological levels of PO2 limit NADPH oxidase-dependent O2 −• generation in the renal medulla (34, 301), the relative hypoxia may put a brake on severe medullary oxidative stress. Renal hypoxia also may contribute to the development of hypertension (184). Renal oxidative stress associated with upregulated Poldip2 and SOD accounted for the renal medullary hypoxia in a mouse model of RRM (150) and has been postulated to contribute to hypertension and progressive loss of function in patients with CKD (64).
Renal Nerves
Efferent and central effects
Activation of the sympathetic nervous system (SNS) raises the BP, enhances renal Na+ reabsorption, reduces RBF, and increases renin secretion (92). ROS increase SNS activity by decreasing NO bioavailability within the central nervous system (26, 31).
The efferent sympathetic renal nerves are distributed throughout the renal vasculature and tubular segments in the cortex and outer medulla, with high innervation along the afferent and efferent arterioles (145). β1-adrenergic receptors (β1AR) predominate on VSMCs of renal microvessels and MD epithelium, where they inhibit ROS generation and lead to vasodilation (18). Activation of β1AR by norepinephrine (NE) protected renal afferent arterioles of ANG II-infused rabbits from the enhanced contraction induced by increased ROS generation that was mediated by α-1 adrenergic receptors (295). Thus, α- and β-adrenergic receptor signaling can have opposite effects on renal microvascular ROS.
Afferent and renorenal reflex
The renal afferent nerves are mainly located in the renal pelvis, where they are activated by chemical stimuli, for example, high NaCl concentrations and physical stimuli, for example, increased renal interstitial pressure (145). The signals from the renal afferent nerves converge to brain centers involved in the control of arterial pressure (145). Unilateral renal denervation leads to ipsilateral natriuresis and contralateral increased renal sympathetic nerve activity (RSNA) and antinatriuresis. This has been termed the renorenal reflex.
Hypertension and renal nerves
Renal denervation prevents or attenuates hypertension in many models, including SHR, SHRSP, ANG II-induced, DOCA–salt, and 2K,1C hypertension (51). Renal denervation of rats early after renal injury prevented the enhanced NE secretion from the posterior hypothalamic nuclei (PH), the increased RSNA, and hypertension, thereby implicating the renal afferent nerves in centrally mediated neurogenic hypertension (31, 336). The central sympathetic activation after renal afferent nerve activation is dependent on ROS, since it is prevented by intracerebroventricular (ICV) infusion of tempol (31). A convenient model is produced by an intrarenal injection of phenol in rats. This enhances NE secretion from the PH and RSNA, and leads to hypertension that is prevented by denervation of the kidneys or by ICV injection of tempol or PEG-SOD (337). Several brain nuclei in this model have increased expression of NOX2, NOX4, p22phox, and p47phox, whereas interleukin-1β and nNOS are decreased (31, 42, 104, 225, 232, 337). A vitamin E-enriched diet attenuated activation of the SNS and hypertension initiated by intrarenal injection of phenol (25).
O2
−• is elevated in the sympathetic ganglia of DOCA–salt-hypertensive rats, and this is attributed to ET-1 stimulation of the ETB receptors (46). The authors propose that O2
−• could inactivate NO in the SNS and thereby increase the activity of sympathetic neurons and the release of vasoactive substances, resulting in vasoconstriction and hypertension. In other studies, an acute intravenous infusion of tempol caused a transient decrease in the BP, heart rate (HR), and RSNA in both DOCA–salt-hypertensive rats and SHR (26, 325
–327). In DOCA–salt rats, the effect of tempol on BP, but not on HR and RSNA, was attenuated after NOS inhibition with NG-nitro-
In contrast, ICV infusions of tempol do not decrease BP in WKY rats or SHR (261). These studies suggest that the ICV action of tempol on the SNS could involve NO-dependent and NO-independent pathways, while the systemic effects of tempol involved peripheral vasodilation and reflex activation of the SNS. Thus, ROS can activate the SNS both by increasing the central sympathetic drive and by facilitating peripheral SNS signaling. Microinjection of an AT1R antagonist into the rostral ventrolateral medulla decreased BP and peripheral sympathetic nerve activity in SHR, but not in WKY rats (6). These studies support the hypothesis that ROS may activate the SNS at the levels of renal afferents, central nuclei, and peripheral sympathetic nerves and thereby contribute to hypertension.
The Renin-Angiotensin-Aldosterone System
Renin and aldosterone release
Renin release is regulated by systemic and intrarenal signals. There is growing evidence that oxidative stress regulates renin expression and release (48, 77, 78, 119, 306). ANG II stimulates the production of cytokines, which are strong inhibitors of renin release (112). TNF-α inhibits renin expression and release through oxidative stress (48, 119).
The juxtaglomerular (JG) cells of the renal afferent arteriole are the primary site of storage for prorenin, activation of renin, and renin release. Renin release is stimulated by sympathetic activation of β1-adrenergic receptors on JG cells, decreased renal afferent arteriole stretch, and decreased NaCl delivery to the MD or by specific hormones and peptides (148, 229). The short-loop feedback inhibition of renin secretion by ANG II involves oxidative stress. Galle et al. (77) reported that JG cells isolated from mice had enhanced renin release when the tissue O2 −• was increased. (77, 78, 119). Itani et al. (119) demonstrated that renin-expressing JG cells exposed to TNF-α and H2O2 had a time- and dose-dependent reduction in renin mRNA and renin promoter activity that was corrected by an antioxidant N-acetylcysteine (NAC). Interestingly, an effective antihypertensive dose of tempol increased the plasma renin activity in SHR, implying that inhibition of renin release did not contribute to the fall in MAP. Overall, these studies suggest that O2 −• enhances, whereas H2O2 inhibits, renin expression and release, but further studies are needed due to the complexity of the factors involved in the JG cell function.
Aldosterone participates in the control of BP by central actions that lead to activation of the SNS, vascular actions that led to vasoconstriction, and renal actions that lead to enhanced Na+ reabsorption. Excessive aldosterone synthesis or release increases Na+ reabsorption by renal DTs and raises the BP (285). Aldosterone promoted renal and cardiovascular diseases associated with hypertension (20, 285). Aldosterone release from the zona glomerulosa of the adrenal cortex is driven by ANG II and [K+ ]. The pressor responses induced by prolonged aldosterone infusion and salt supplementation in rats were mediated by central AT1R and ROS, since they were inhibited by ICV administration of an ARB, apocynin, or tempol (285). Moreover, the pressor effects of a systemic infusion of low dose of ANG II also were dependent on central MR, since they were abolished by the ICV infusion of an MR blocker or an aldosterone synthase inhibitor (116). These studies disclose a crosstalk between ANG II and aldosterone in the brain to sustain hypertension that involved a central production of ROS. Fujita and Nagase (258) demonstrated that salt-dependent augmentation of ANG II effects in the kidney depended on renal MR/Rac1 activation. They proposed that a crosstalk between ANG II and aldosterone contributes to hypertensive kidney injury (135, 260). ANG II induced the synthesis and release of aldosterone in the adrenal cortex cells by activation of AT1Rs and increased CYP11B2 expression and activity (13, 205, 234) that were mediated by H2O2 derived from NOXs and mitochondria (234). Pretreatment with NAC, PEG-CAT, a NOX inhibitor, or an ARB prevented the ANG II stimulation of aldosterone release. These studies demonstrate that oxidative stress is an important regulator of the RAAS in the kidney, brain, and adrenal glomerulosa cells.
MD versus afferent arteriole mechanisms
The afferent arteriole is the effector site of TGF and renin secretion. ANG II enhances the afferent arteriolar constriction with TGF activation (188) and enhances the activity of the luminal NKCC2 in the MD (147), which generates the signal for TGF. Adenosine activity on AT1Rs enhances the afferent arteriolar response to ANG II (152). A1AR inhibition reverses the enhanced renal vasoconstriction in ANG II-infused rats (68). AT1AR (−/−) mice have an absent TGF response and attenuated effects of ANG II in reducing renal function and increasing RVR (79, 100, 253). Gao et al. (79) demonstrated that A1ARs increased afferent arteriolar contractility by increasing ROS generation. Therefore, an interaction between adenosine acting on A1AR and ANG II acting on AT1R modifies the degree of vasoconstriction induced by these agonists through increased arteriolar ROS. This would contribute to the development of hypertension by sensitizing TGF and the reactivity of preglomerular vessels.
Angiotensin type 1 and 2 receptor expression
Angiotensin II type 1 and 2 receptors (AT1Rs and AT2Rs) are demonstrated throughout the rat kidney (190, 221). AT1R protein is expressed on renal blood vessels, glomeruli, PTs, and DTs, especially in the outer medulla (106, 183). The highest expression of AT1R is found in the renal cortical vasculature and in the S3 segment of PT (190). Two AT1R subtypes are identified in rodents (122, 190, 248), AT1AR and AT1BR, but due to the high homology, their precise function in the kidney has not yet been fully defined. The mRNA for AT1AR and AT1BR is demonstrated in the glomeruli, tubules, and vessels from the renal medulla of rats (190). The AT1AR receptor has the highest homology with the human AT1R (42).
AT1AR (−/−) mice have a similar basal afferent arteriolar diameter to wild-type mice, but reduced constrictive responses to ANG II, whereas the diameter of efferent arterioles is higher in the AT1AR (−/−) mice, and they do not respond to Ang II (105). Kidney-specific AT1AR (−/−) mice infused with ANG II at a slow-pressor rate had no increases in BP. However, transplantation of a wild-type mouse kidney into systemic AT1AR (−/−) mice restored the responses to ANG II (42). AT1AR/AT1BR (−/−) mice have renal microvascular dysfunction, tubular injury, and interstitial inflammation despite a lower BP (220). These mice have enhanced renal expression of renin, AGT, NOX2, p40phox, p67phox, and XO, but decreased expression of NOX4. The authors attributed the increased NADPH oxidase components in the kidney to the infiltration of inflammatory cells. The cause for the paradoxical renal inflammation and dysfunction in mice lacking AT1Rs has not been determined.
AT2Rs are expressed on the renal vasculature, PTs, DTs, and CDs of rats, whereas glomeruli and mTAL are negative (190). AT2R regulates renal AT1R expression and function via the NO/cyclic guanine monophosphate (cGMP) pathway (332). Stimulation of AT2R reduced inflammation and increased renal production of NO and cGMP in the clipped kidney of 2K,1C hypertensive rats (182). Overexpression of AT2R in VSMC (126) or stimulation of AT2R in PT cells from rats (332) decreased AT1R expression. On the other hand, PT cells from AT2R (−/−) mice had increased AT1R expression. Inhibition of AT2R enhanced the upregulation of renal NADPH oxidase subunits by ANG II (29). Thus, there is extensive reciprocal regulation of AT1R and AT2R in the kidney that determines the expression of the NADPH oxidase isoforms.
AT2R (−/−) mice have high BP, decreased renal function, impaired water and Na+ handling, augmented vasopressor response to ANG II, and increased expression of AT1R in the kidney. AT2R activation in the TAL decreased Na+ reabsorption by an NO-mediated reduction of the NKCC2 cotransporter activity (107). NKCC2-dependent transport is impaired in the TAL of DS rats with oxidative stress, which may contribute to Na+ retention and salt-sensitive hypertension (113).
Recent studies have emphasized the importance of ANG II receptors in the nuclei and mitochondria of renal cells in modulating intracellular oxidative stress (1, 96, 162, 164, 227). Activation of AT1R in the isolated nuclei from the rat renal cortex was linked to increased ROS generation (228), whereas the activation of the AT2R and ANG II (1 –7) receptors increased NO generation (97). Studies employing immunoelectron microscopy demonstrated AT2R bound to ANG II in the mitochondria of mouse kidney cells (1). An AT2R agonist stimulated the production of NO in the renal mitochondria and inhibited respiration in the heart mitochondria (1). These findings reinforce the role of AT2R in buffering the intracellular actions of AT1R on ROS production and extended this to the mitochondria and nuclei. These recent studies identify an intracellular RAAS system in the kidney that modulates nuclear and mitochondrial oxidative stress in hypertension.
ROS, hypertension, and the RAAS
Activation of the RAAS causes a widespread increase in ROS in the kidney via NADPH oxidase, mitochondrial dysfunction, decreased NO availability, and decreased antioxidant enzymes. This is implicated in Na+ retention, vasoconstriction, and hypertension. Moreover, ACE inhibitors, AT1R blockers, and MR antagonists decreased oxidative stress and ameliorated hypertension and protected the kidney in several hypertensive models (14, 15, 29, 57, 73, 157, 223, 302, 305, 308, 312, 334).
Increased ANG II generation of ROS is implicated in the development of hypertension in several mouse or rat models, including ANG II infusion at a slow-pressor rate (134, 216), prolonged NOS inhibition (214), salt sensitivity (101, 187), SHR (301, 306), 2K,1C rats (305), and salt-dependent hypertension in DOCA-salt rats (328).
The renal AT1R is a key element for the development of hypertension. (177, 224, 294, 305). Activation of this receptor engages renal afferent and efferent arteriolar vasoconstriction and decreased relaxation, limits the GFR and RBF, increases Na+ and fluid reabsorption, decreases pressure natriuresis, and augments TGF (177, 224, 294, 305). Many of the AT1R signaling pathways are antagonized by AT2R. AT1R activation in the kidney enhances the expression and stimulated the assembly of NADPH oxidase subunits and decreases antioxidant enzymes, resulting in increased ROS (29, 191, 304), whereas AT2R activation stimulates NO production and counteracts the effect of AT1R activation (29, 274). Aldosterone causes glomerulosclerosis and proteinuria by activation of the MR linked to ROS. Salt intake potentiates the deleterious effects of ANG II and MR activation via increasing ROS in the kidney, even with a suppressed systemic RAAS (135). Thus, a crosstalk between ANG II and MR in the kidney that both engage ROS could contribute to the renal hemodynamic alterations and injury involved in hypertension.
Innovation
This Forum reviews the most recent publications in basic research focusing on the contribution of renal oxidative stress to hypertension. Studies have shown that, on one hand, NADPH oxidase/O2 −• is an essential requirement for hypertension during slow-pressor infusion of ANG II. On the other hand, prolonged O2 −• tissue excess from knockout of SOD-1 or SOD-3 either does not change BP or increase it modestly, but does enhance BP with ANG II infusion. These and other studies suggest a critical modulating, but not necessarily mediating, role for ROS in hypertension development.
Conclusions
The kidney plays a crucial role in the development of hypertension. Current treatments utilizing ACE inhibitors, ARBs, and renin inhibitors have been effective in reducing BP, but do not fully prevent the progressive loss of kidney function in CKD. The therapeutic role of effective antioxidant strategies in human hypertension and CKD remains to be explored.
Studies with isolated preglomerular microvessels or renal cells have distinguished between the direct vascular actions of ANG II and those exerted by the high BP. Studies of microperfusion of vascular and tubular segments in vitro or in vivo have defined a clear role for ROS in mediating many of the effects of the renal RAAS on renal Na+ transport, TGF, RBF, and GFR. The development of knockout mice with global or site-specific deletion of a target gene has demonstrated that a primary increase in ROS, independent of ANG II, can be a provocative stimulus to increase BP. Finally, studies of the renal signaling pathways and transcription factors mediated by ROS have expanded the knowledge on the role of oxidative stress in the kidney and hypertension.
Footnotes
Acknowledgments
This was supported by National Institutes of Health grants from the NIDDK (DK-049870 and DK-036079) and by the NHLBI (PO1 HL068686) and by funds from the George E. Schreiner Chair of Nephrology.