
Abstract
Introduction
R
The strengths and weaknesses of tissue-specific knockouts are becoming more apparent, as are the options for creating a variety of transgenic animals. Thus, this review is aimed at examining the current state of MnSOD tissue-specific knockout animals, highlighting the methodologies and strategies considered most informative, and pointing out deficiencies in model animals that are inconclusive. Tissue-specific knockouts of MnSOD hold great potential for studying the role of oxidative stress and the contribution of MnSOD deficiencies in tissues, but only when the limitations of the system are carefully considered.
Mitochondrial oxidant production
Mitochondria utilize a series of redox processes to transfer pairs of electrons from O2 to H2O in an orchestrated, stepwise manner leading to the production of ATP. During the catalytic cycles of both the NADH-ubiquinone oxidoreductase of Complex I (31, 64, 106) and the ubiquinone:cytochrome c reductase of Complex III (6, 106) in the electron transport chain, single electrons leak to form O2 •−. The location of the electron transport proteins, embedded within the inner mitochondrial membrane, causes O2 •− to be released into the matrix and the intermembrane spaces. Production of ROS in close proximity to oxidatively sensitive proteins and abundant lipid targets in mitochondrial membranes creates a unique dependence on MnSOD within the mitochondrial matrix (Fig. 1).
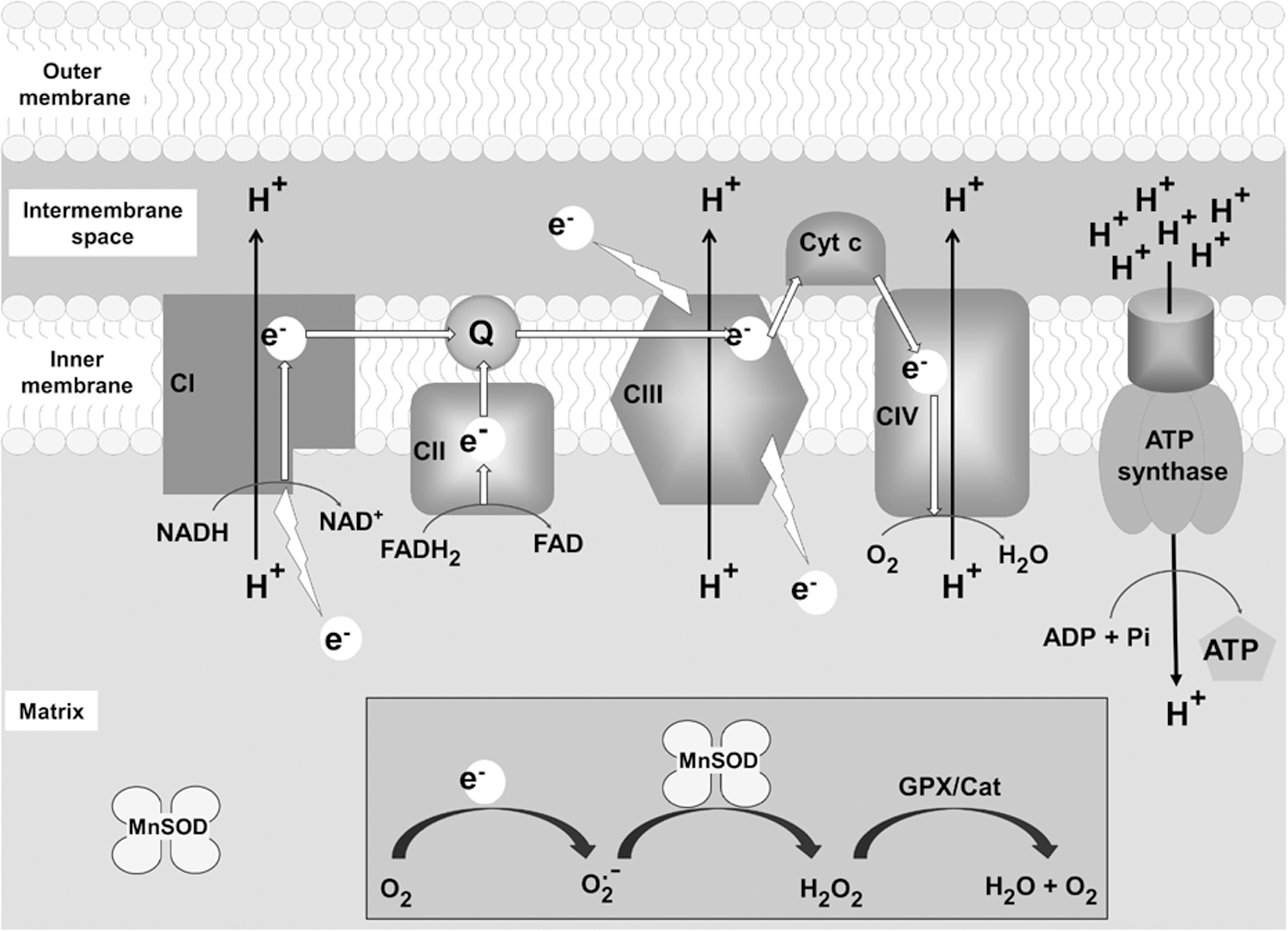
In addition, the Complex I-associated flavin containing α-ketoglutarate dehydrogenase and pyruvate dehydrogenase complexes are capable of producing O2 •− and hydrogen peroxide (H2O2) in the matrix, particularly in a low-energy state when the ratio of NAD+/NADH is low (99, 105). Additional sources of O2 •− originating in the mitochondria include redox active flavin-containing enzymes such as flavinprotein Q reductase (6, 98) and possibly acyl-CoA dehydrogenase (6), however, the contribution of these systems to the oxidant flux in resting or stressed mitochondria are not well known. In addition to participating in oxidant production, these flavinoproteins are particularly sensitive to inactivation by excess 4-hydroxynonenol derivatives (42, 43) as well as H2O2, peroxynitrite, and oxidants that lead to an elevated protein carbonyl content (84).
The exact cellular sources of nitric oxide (NO) are still unclear, although evidence supports the production of NO in the cytosol and possibly in the mitochondria (27, 49, 102, 110). Also, while NO is produced in puffs by some nitric oxide synthases (NOS) and continuously by others, a low steady-state level of NO exists more or less continuously. It readily diffuses across membranes to participate in reversible thiol group modification via S-nitrosylation (39), or reversibly bonding to iron containing proteins such as hemoglobin and many other heme-Fe proteins (15). Indeed, at nanomolar concentrations of NO, Brown and Cooper (7) demonstrated in isolated brain synaptosomes that NO competes with O2 for binding at the Fe center of the cytochrome oxidase to disrupt respiration. Other evidence suggests that mitochondrial respiration is dynamically regulated via reversible inhibition of cytochrome oxidase by NO (94). NO reacts with O2 •− at a diffusion-limited rate (81) to produce peroxynitrite (ONOO−), a highly reactive oxidant capable of damaging DNA, lipids, and proteins.
Of particular interest are the O2 •−- and ONOO−-sensitive iron–sulfur complexes in the matrix enzyme aconitase (12, 37, 103). The coordination of Fe+3 in these centers can be disrupted by O2 •− leading to the release of Fe+2 and the availability of free iron to participate in Fenton-type reactions leading to the production of the highly reactive hydroxyl radical, as well as the inactivation of the protein (17). This sensitivity to inactivation has been used as a sensitive biomarker for the production of mitochondrial O2 •− and ONOO− flux (26).
Mitochondrial antioxidant defenses
The primary enzymatic antioxidant system in the cell is the family of O2 •− scavenging metalloenzymes known as the superoxide dismutases (SODs). Three isozymes catalyze the dismutation of O2 •− to H2O2 and O2 at a near diffusion-limited rate (29). In a solution, the spontaneous dismutation of O2 •− is relatively fast (k=2×105 M −1 s−1), but because O2 •− is reacting with itself, the rate slows exponentially as the concentration of O2 •− decreases. The reaction of O2 •− with SOD is at least 10,000-fold faster (2×109 M −1 s−1) (28). The Cu- and Zn-containing SOD (Cu,ZnSOD or SOD1) mainly resides in the cytosol, although reports have a small fraction of Cu,ZnSOD being imported into the mitochondrial intermembrane space in a sequence-independent manner (53). The tetrameric manganese-containing SOD (MnSOD or SOD2) localizes to the mitochondrial matrix and is transcriptionally (20) and post-transcriptionally regulated (113) in response to cytokines and inflammatory stimuli (20). Recent reports indicate that a significant fraction of MnSOD localizes in the protein-mtDNA nucleoid complexes (55) not only to protect the mtDNA from O2 •−-mediated damage, but also to possibly prevent nitration-mediated inactivation of sensitive residues in the mitochondrial DNA polymerase γ (Polγ) (3). Our laboratory has previously shown that MnSOD is susceptible to tyrosine nitration and oxidation leading to irreversible inactivation, resulting in an increase in the flux, or steady-state level of mitochondrial O2 •−, due to decreased scavenging (67, 68). The extracellular SOD is a Cu- and Zn-containing enzyme that is exported from cells (25) and has been shown to play a role in protecting the extracellular milieu from oxidant stress (65, 86), and may be particularly important in the endothelium (65).
In the mitochondria, the primary antioxidant systems responsible for the removal of H2O2 are the glutathione peroxidase isozymes, which utilize glutathione (GSH) as a cofactor (22), the thioredoxin peroxidase, which uses thioredoxin (Trx) as a cofactor (16), and catalase [found primarily in the heart and liver mitochondria (87, 92)]. Importantly, both systems require electrons from cytosolic pools of NADPH to reduce and regenerate GSH and Trx. The concentration of GSH in the mitochondrial matrix is high, measured to be∼11 mM (30, 44, 107) making it a particularly abundant cofactor for a number of mitochondrial GSH-dependant enzymes, as well as a direct scavenger of several ROS (44). The GSH/oxidized glutathione redox potential is a useful biomarker of oxidative stress and ROS and NOS production in the mitochondria since it is particularly sensitive to a number of pathological and toxicological insults (36).
Genetic manipulations of MnSOD
Since pharmacological inhibitors specific to SOD isoforms have not been identified, the study of SOD activity in vivo has relied on genetic manipulations through overexpression via transgenic techniques, as well as gene ablation using knockout technology. Multiple mouse lines of MnSOD overexpression have demonstrated protection from oxidant damage in models previously suspected to involve the rapid increase in O2 •− flux during hyperoxic lung injury (112), adriamycin-induced cardiotoxicity (114), and ischemia-reperfusion damage (13). While the overexpression of MnSOD decreased biomarkers of aging and age-related oxidative stress, there was no statistical effect on the lifespan (46).
Characterization of the original MnSOD knockout model revealed that a 50% reduction of MnSOD (MnSOD+/− mice), had no overt phenotype (109) and a normal lifespan (46). However, these mice did show decreased GSH, accelerated accumulation of oxidized mtDNA markers, and increased susceptibility to developing cancers over their lifetime (108). When subjected to oxidative stress using the cerebral ischemia model (56), 50% MnSOD deficiency led to increased mitochondrial O2 •− and an exacerbation of the cerebral damage, suggesting that halving the amount of MnSOD activity resulted in a state of mitochondrial oxidative stress under pathophysiological conditions. Lowering the threshold for oxidative damage could turn a subclinical injury into a clinical one, or make a reversible injury irreversible.
When MnSOD was completely knocked out (MnSOD−/−), differing phenotypes were observed, depending on the background strains, although all mouse lines showed lethality within 18 days of birth (41, 59, 62, 70). The MnSOD−/− mice originally developed by Li et al. (62) showed severe cardiovascular pathologies, including a dilated left ventricular cavity, significant steatosis in the liver and muscles, yet no apparent neurological pathology. The MnSOD−/− mice on a mixed B6/129 background described by Lebovitz et al. (59) also developed cardiomyopathy, but also presented with histological evidence of neurodegeneration. When MnSOD was knocked out in mice from the C57Bl/6J background, Huang et al. (41) observed dramatic prenatal dilated cardiomyopathy with embryonic or perinatal lethality. However, animals on the DBA/2J or the backcrossed B6D2 background lived up to 2 weeks longer, but developed cardiac abnormalities and metabolic acidosis (41). Thus, depending on the background strain of the mice, differing spectrums of anatomical and metabolic abnormalities emerged. Because of the evidence of significant oxidant damage and the short lifespan, Melov et al. (70) treated the MnSOD−/− animals with the catalytic antioxidant MnTBAP, resulting in a significant protection from the cardiac deformities and an increase in lifespan, but a worsening in the neurological pathologies. Ultimately, all of these models supported the idea that some amount of MnSOD is essential for the preservation of mitochondrial viability and ATP production, particularly in tissues with high energy demand. The total ablation of MnSOD produces a severe and rapid phenotype, making it difficult to dissect the defects responsible for lethality. In the global MnSOD−/− mouse models, major organ systems, including the heart (41, 59, 62) and brain (59, 70) are severely impaired, either of which might ultimately prove lethal, but the short lifespan of these mice makes it difficult to characterize the contribution of different organs and tissues to the mechanisms involved with mitochondrial damage in the absence of MnSOD. This can only be done with organ- or tissue-specific MnSOD knockouts.
Gene knockout in specific tissues: the Cre/loxP system
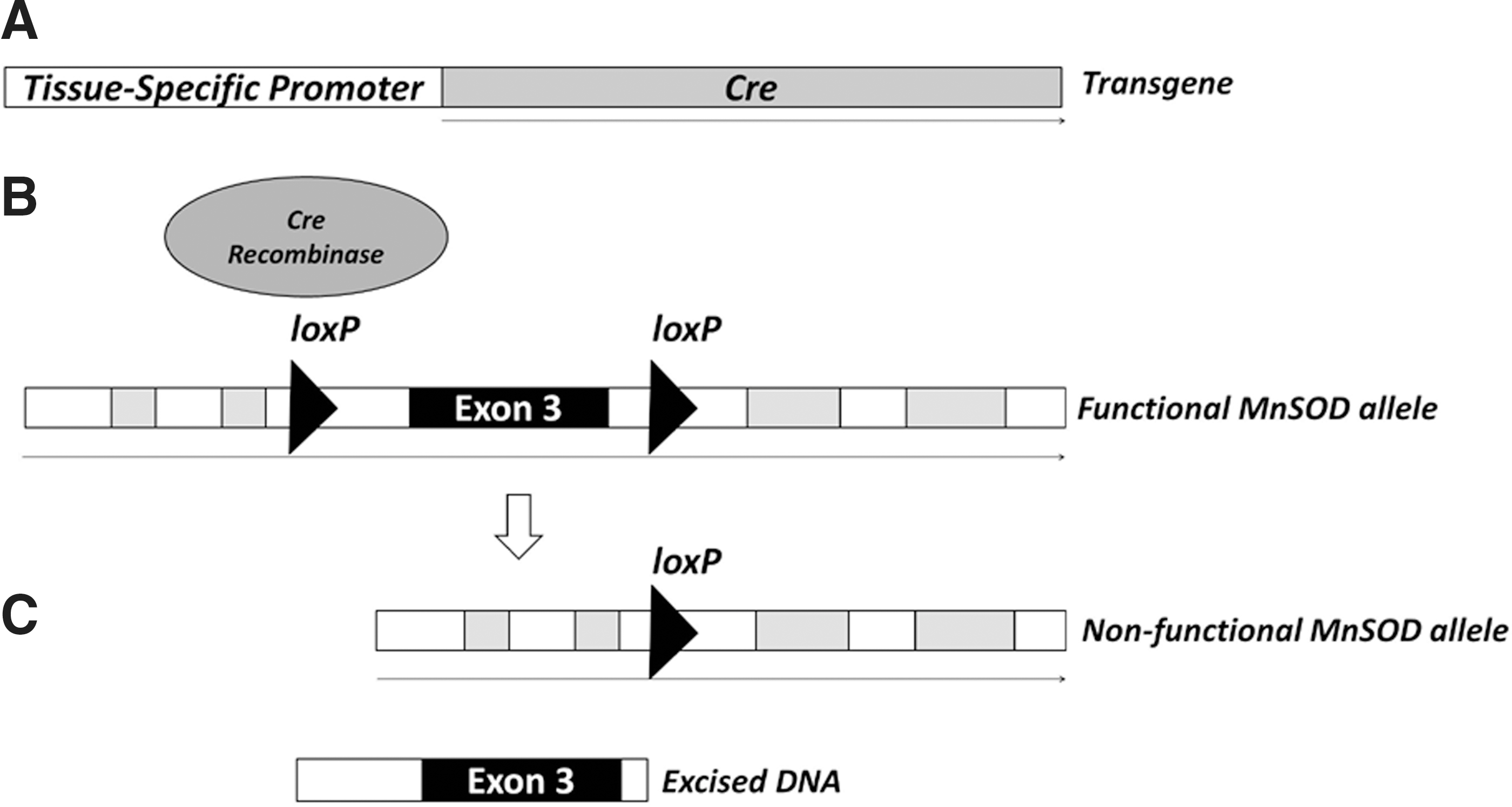
The Cre recombinase from the P1 bacteriophage directs DNA recombination between loxP sites (35), leading to a permanent rearrangement of genetic material. A better understanding of various tissue-specific promoter behavior and transgenic technology has led to a number of optimized Cre-expressing cell lines as well as transgenic mouse lines (5). In addition, the advancement of conventional gene targeting techniques facilitated the introduction of loxP sites within specific genes of interest to generate mice containing a functioning gene that is poised for rearrangement in the presence of Cre. By crossing Cre-expressing mice with animals having the introduced loxP loci, the resulting progeny have the gene, or a portion of the gene of interest, knocked out in only those tissues that recognize the specific promoter used to express Cre (Fig. 2). Thus, by compartmentalizing the necessary factors into separate mouse constructs, investigators have better control of the timing and location of the specific gene deficiencies imparted to the whole animal. For instance, to generate multiple models of MnSOD deficiencies in different tissues, careful breeding of a single strain of mice containing loxP sites flanking a critical exon in the MnSOD gene with different mice having Cre expression controlled by different tissue-specific promoters led to the loss of MnSOD in targeted tissues and organ systems (see Table 1.).
Jackson Laboratory mouse line.
Unexpected observation.
AFP, alpha-fetoprotein; ATP4b, noncatalytic β-subunit of H+, K+-ATPase; Col1α2, α2 chain of collagen type I; HSA, human skeletal actin; IF, immunoflorescence; IHC, immunohistochemistry; Ksp-cadherin, cadherin 16 promoter; Lck, proximal lymphocyte-specific kinase; MCK, muscle creatine kinase; MMTV, mouse mammary tumor virus; TnIFast, inhibitory subunit of troponin; VAChT, vesicular acetylcholine transporter; WB, Western blot; ?, unknown/not measured.
The requirements of the loxP sites for Cre-mediated recombination are a 34 bp sequence containing an 8 bp core site flanked by two 13 bp palindromic sequences (35). The power of the system is demonstrated by the fact that recombination does not depend on the orientation of the loxP loci; placing the sites in the same orientation along the same DNA strand leads to excision, while the opposite orientation leads to an inversion. Nagy (76) pointed out that the expectation of a random appearance of the loxP site would require a genome of 1018 bp, while the size of the mammalian genome is orders of magnitude smaller at∼3×109 bp, suggesting that the potential for the random appearance of the loxP site in the genome is considerably unlikely. While two loxP sites are necessary for Cre-mediated recombination, the introduction of too many sites leads to chromosomal instabilities and chromosomal loss (61, 76). Although the loxP recognition sequences are small, preliminary in vitro genetic experiments using the loxP-flanked (floxed) region of the gene must be performed to ensure fully functioning alleles, and that upon introduction of Cre, the resulting gene rearrangements result in a null gene product without adversely affecting other off-target genes. Nevertheless, given the relative ease of targeted mutagenesis followed by the introduction of exogenous DNA into discrete regions of the genome, careful strategies and selective breeding could lead to increasingly complicated scenarios in which, multiple elements are added and removed within the same animal or tissue.
The desired tissue location for expression of the Cre recombinase, however, has significantly more caveats and requires more awareness of the inherent limitations of the promoter systems used to develop a sufficient model system, as well as the ability to appropriately interpret the information derived from the tissue knockout animals. While the Cre system requires no cofactors for activity (76), and known tissue selectivity of well-characterized promoter systems driving the Cre expression is a great strength, the potential for off-target Cre expression and toxicity (47, 77) is a confounding factor to include in the interpretation of the results from tissue-specific knockout experiments. If the promoter has some leaky expression in the germ lines, recombination of the floxed allele may occur before fertilization (5) resulting in a global knockout of the respective allele. Such complications may be avoided by carrying the Cre transgene exclusively in the male or female line. Ectopic expression of Cre before organogenesis will lead to similar results and necessitate the use of an alternate Cre transgenic mouse line.
While our understanding of the activity of promoters has greatly accelerated the development of tissue-specific Cre expressing lines, we still must recognize that our breath of knowledge is incomplete. The selection of a promoter is dependent on the application desired, and the specific question being studied. Often, the use of the Cre/loxP system has been described as an organ-specific model of gene knockout, however, Cre expression from a particular promoter, and hence recombination, may not extend to all cell types in an organ or organ system. For instance, Shao et al. (96) reported a kidney-specific Cre mouse line under the control of the cadherin 16 promoter (Ksp-cadherin) with expression in the distal tubules (DTs), collecting ducts (CDs), and the ascending limbs of the Henle's loop, but not in the glomeruli, blood vessels, or the proximal tubules. Additional Cre expression was detected in the developing genitourinary tract, including the developing ureter and mesonephrotic duct (96). However, the utility of the Ksp-cadherin Cre animal in developing a model of renal disease was demonstrated by Lin et al. (63), who used these mice to knockout the KIF3a subunit of kinesin II leading to polycystic kidney disease. While application of this Cre line for the tissue-specific knockout of a gene of interest in the study of kidney physiology is possible, the limitation of Cre expression within specific renal cells, and the additional expression (and hence, MnSOD knockout) in the genitourinary tract must be taken into consideration when interpreting results.
Particularly important in the development or selection of a Cre-expressing animal is a thorough cataloging of Cre tissue expression, and the elimination of off-target recombinase expression. Several mouse lines have become available from The Jackson Laboratory (
Although not discussed in this review, multiple variations and control systems for the Cre expression are continuously being developed. The well-characterized tetracycline (tet)-inducible system has been employed in the Cre/loxP system (4) as well as the tamoxifen-inducible system (23) in which, a chimeric protein containing the Cre protein and the ligand-binding domain of the estrogen receptor are expressed. The fusion protein remains in the cytoplasm until it binds with exogenously added tamoxifen leading to nuclear translocation and Cre access to the loxP sites. These systems have their own unique sets of advantages and limitations, and more discussions of these systems are available elsewhere (5, 76, 88).
Knockout of MnSOD in various tissues
As described above, the Cre/loxP system is the ideal technology for studying the focal elimination of MnSOD during development, normal growth, and when exposed to insults producing oxidative stress. To date, we have identified 14 studies utilizing the Cre/loxP system to knockout MnSOD from tissues with varying success (Table 1), and given the power of the system, additional mouse models are certain to follow. In this review, we wish to examine these studies and highlight models that provide useful data, while also pointing out their caveats and limitations. We suggest some guidelines to compare results with different models, and hope this discussion will be useful to readers who might wish to generate a tissue-specific MnSOD knockout.
Myocardium
In MnSOD−/− mice and in studies of heart failure (80), increased levels of ROS have been suspected to contribute to the development of cardiac pathology, particularly dilated cardiomyopathy (80). Since the total knockout mice did not survive to maturity, mechanisms involved in the progressive damage to heart tissue and specific aging processes that may also play a role in oxidant-mediated damage were difficult to dissect from the contribution of significant damage in other tissues.
During the development of a model of congestive heart failure and age-related cardiac dysfunction, Nojiri et al. (78) used a Cre/loxP system controlled by the muscle creatine kinase (MCK) promoter to develop a knockout of MnSOD in myocytes and the striated muscles. The animals survived significantly longer than the MnSOD−/− mice (up to 22 weeks) and developed significant morphological and biochemical markers of oxidant stress in the mitochondria in addition to a progressive loss in cardiac performance. The authors utilized a combination of Western blots and immunohistochemistry (IHC) to confirm the loss of MnSOD in specific tissues, steps that are critical in pinpointing the affected tissues. In addition, the authors observed a partial, but significant improvement in the cardiac function and physical exercise capacity in 12-week-old animals treated from the age of 8 weeks with daily injections of MnTBAP, suggesting that the amelioration of oxidant stress may partially rescue impaired muscle contractility in the absence of MnSOD.
The authors' selection of Cre-expressing MCK-Cre mouse highlights one of the important caveats inherent in the system. Since Brunning et al. (8) showed extensive Cre activity in the skeletal muscles as well as the myocardium when the MCK-Cre mouse was first developed for a study of insulin-dependent signaling in skeletal muscles, off-target Cre expression and MnSOD deficiency in skeletal muscle would be expected. While the authors recognized that the MnSOD deficiency was not limited to a single tissue, they go on to suggest a strong correlation between the loss of MnSOD in the myocardium and the histological and functional evidence of dilated cardiomyopathy and progressive heart failure.
Strassburger et al. (100), however, reported oxidant stress in the myocardium through an inadvertent result from the Cre/loxP system. In pursuit of an aging model of MnSOD deficiency in the epidermis, the authors crossed an in-house generated floxed MnSOD (100) mouse strain with a Cre mouse under control of the human keratin 14 promoter resulting in all of the progeny being heterozygous for MnSOD in all tissues as a result of the promoter being active in the oocyte (100). Therefore, the animals were genetically similar to previously reported MnSOD+/− mice (109). Curiously, when the Cre transgene was carried in the females, the expected epidermal expression pattern was noted, although not reported. The complete inactivation of the paternal MnSOD allele would be expected since these mice previously showed distinct expression as an oocyte protein (32). This selection of the Cre-mouse highlights another common pitfall with the Cre/loxP system: the unanticipated activity of promoter elements driving Cre expression, and the appearance of phenotypes in off-target tissues. Nevertheless, the authors report significant oxidant stress manifested in the myocardium in the form of increased nitrotyrosine and lipid peroxidation products as well as structural abnormalities of the mitochondria in the heart.
Skeletal muscle
The further refinement of the skeletal muscle MnSOD knockout mouse was accomplished by Kuwahara et al. (58), who used a mouse strain containing a modified human α-skeletal actin promoter construct to direct Cre expression to the cells in the striated muscle lineage without ostensibly affecting MnSOD expression in cardiac tissue (71). The loss of MnSOD was determined via Western blotting, although the authors did not report any IHC data for either MnSOD or Cre expression to ensure both on- and potential off-target recombinational effects. Using the chemiluminescent probe 2-methyl-6-p-methoxyphenylethylnyl imaidazopyazionone, the authors demonstrated that muscle tissues had decreased the electron transport activity and increased oxidant generation in isolated mitochondria. In addition, consistent with decreased mitochondrial function and reduced ATP generation, these mice showed severe exercise intolerance and a more rapid progression to fatigue, although without severe disturbances to the total muscle mass. Interestingly, a single administration of the putative antioxidant organomanganic compound EUK-8 provided a sustained recovery of muscle functions and exercise capacity in these mice. As in reports of myocardial/myocyte MnSOD deficiency (78, 100), the authors did not detect any appreciable increase in apoptosis in the muscle tissues, and speculate that damaged muscle cells die through necrosis rather than apoptosis.
Lustgarten et al. (66) reported a refinement of skeletal muscle MnSOD knockout with the use of a Cre expression system developed in-house using a well-described quail-derived promoter from the inhibitory troponin (TnIFast), a strategy that reportedly provides greater expression in type IIB than in type I, IIA, and IIX skeletal muscle fibers (33, 34). While MnSOD knockout in multiple tissues was confirmed using SOD native gel electrophoresis and double IHC that colocalized the loss of MnSOD with the expression of the myosin heavy chain family of proteins, the authors did not offer any data involving the specificity of the Cre-expression in the tissues of the animal. Consistent with other muscle tissue knockouts (58) and the MnSOD+/− animals, these muscle cell mitochondria had significantly increased oxidant production and lipid peroxidation, and mitochondrial oxidant stress primarily in glycolytic muscle fibers appeared to adversely affect muscle function and the exercise capacity. Importantly, however, the reported variations in the efficiency of knockdown as demonstrated by the residual MnSOD activity in both the Type IIB glycolytic fibers, as well as the Type I+IIA muscle fibers, suggest an incomplete knockout of MnSOD, or populations of cells within the tissues that may have failed to express the Cre recombinase.
Kidney
The post-translational inactivation of MnSOD in models of renal disorders as reported by our laboratory (18, 67, 69) suggests that an increase in mitochondrial-derived ROS plays a critical role in oxidant-mediated tissue damage. Our approach was to utilize the Cre/loxP system to develop a kidney-specific knockout animal that would closely mimic the dramatic MnSOD inactivation we observe in renal tissues following transplantation (67, 69) and ischemia/reperfusion (18, 89). We chose the Cre mouse under the control of the Ksp-cadherin since Shao et al. (96) reported the Cre expression pattern to be exclusively limited to the kidney, particularly in the CDs and ascending limbs of the loops of Henle, as well as the developing genitourinary tracts. As depicted in Figure 3, Ksp-cadherin-Cre expression in renal tissues led to MnSOD deficiency in specific tissues, while preserving mitochondria oxidant protection in others. At the time, we needed to develop this mouse model (per NIH grant reviewer request), and the only Cre mouse available with expression in the kidney was the Ksp-cadherin. If money and time were unlimited, we would have chosen a mouse with Cre expression in proximal tubules only, or in multiple cell types within the kidney to attempt to develop a renal failure model.
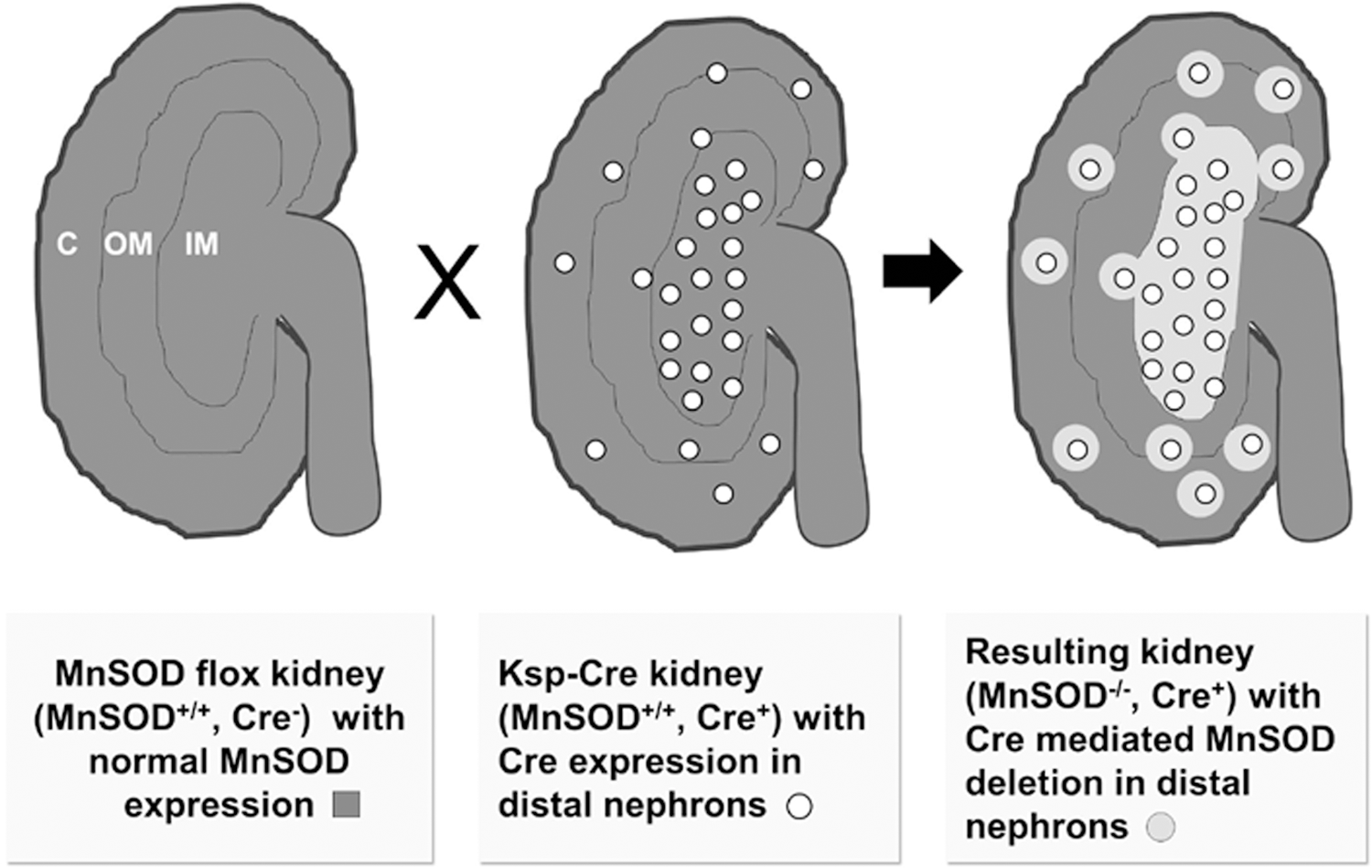
In generating the renal tissue-specific homozygous knockout, we devised a breeding scheme to generate Cre-expressing control mice as well as heterozygous and homozygous kidney-specific MnSOD knockout mice (83). To ensure that the Cre-expression in our mice reproduced the pattern previously reported (96), and that the floxed MnSOD fragment was efficiently excised in the target tissues, we localized both Cre and MnSOD expression in the kidney using IHC and found good agreement with the expected loss of MnSOD protein in tissues expressing Cre. While Cre was previously reported to be ectopically expressed in the developing genitourinary tract, our Ksp-Cre and Cre-MnSOD-/-mice had no overt urinary tract defects and were fertile with no observable effect on the litter size (NP and LAMC unpublished data).
Unlike the MnSOD−/− mice and the myocardium-specific MnSOD knockouts, we observed no effect on survivability (at least to 22 months of age) in the MnSOD-deficient mice. Body weights of the knockout animals were significantly decreased, although curiously, a smaller body size did not translate to decreased vital organ weights suggesting a defect elsewhere, possibly in the musculoskeletal system or bone mineral deposition. In the absence of MnSOD, oxidant stress as measured by the localization of nitrotyrosine formation was increased in a gene dose-dependent manner (83). As suggested by the pattern of expression from the Ksp-cadherin mice, the MnSOD protein present in the proximal tubules and glomeruli of the cortical area was not altered, and most likely accounted for the residual MnSOD activity in the kidney homogenates. These results highlight the incredible interpretive value of confirming the loss of MnSOD with IHC rather than the sole reliance on measuring homogenate SOD activity, especially when the Cre-expression pattern is not uniform throughout the organ.
Of particular interest are the observations that MnSOD deficiency significantly altered the structure of the kidney, including dilated DTs with significant swelling without any significant effects on serum creatinine, blood glucose, or systolic blood pressure (83). While these results demonstrate that the loss of MnSOD in a particular tissue system is not always detrimental under normal physiological conditions, the model remains a potentially useful tool for the study of the downstream effects of MnSOD deficiency in specific cell types of the kidney. It is entirely possible that small, even subclinical, insults are exaggerated or become irreversible in the absence of MnSOD. Studies examining the extent of damage and/or renal dysfunction following ischemia are ongoing in our laboratory.
T-cells
The involvement of ROS and RNS in the immune system has been well studied, especially with the activation of the oxidative burst in leukocytes in response to pathogens. Case et al. (11) pointed out that the role of oxidants and MnSOD activity in the development of the adaptive immune system and T-cells was wholly unexplored. The authors employed the readily available floxed MnSOD mouse (45) in the Cre/loxP system with mice expressing Cre under the control of the proximal lymphocyte-specific kinase promoter, which is active specifically during the CD4−/CD8− to CD4+/CD8+ transitional development stage of αβ-T cells. Loss of MnSOD was confirmed by reverse transcriptase (RT)-polymerase chain reaction (RT-PCR), Western blot, and SOD activity assays, and dihydroethidium (DHE) oxidation as an indicator of ROS production was increased in isolated T-cells. The developmental defect in T-cells from MnSOD deficiency severely exacerbated the susceptibility of mice to immunological challenge from influenza. However, the pretreatment of MnSOD−/− mice with the oxidant scavengers TEMPOL or mito-CPO from the time of weaning significantly delayed morbidity or rescued the animals from the lethal effects of the viral challenge. Changes in the structural integrity of T-cell mitochondria suggested that the organelles were severely damaged, and concomitant dysfunction was confirmed by a loss in the aconitase and succinate dehydrogenase activity. Interestingly, electron microscopy and Western blot data suggested an upregulation of mitochondrial autophagy (mitophagy) in the face of decreased MnSOD and/or mitochondrial dysfunction. These data agree with our submitted manuscript characterizing the kidney-specific MnSOD knockout showing increased autophagy and expanded mitochondrial biogenesis, at least under normal physiological conditions.
Brain
ROS and RNS have been implicated in a number of neurodegenerative diseases, including Parkinson's disease, Alzheimer's disease, and amyotrophic lateral sclerosis (2, 74, 75, 93), although the specific role of mitochondria-generated oxidants in the central nervous system has not been well studied. The MnSOD−/− mice described by Melov et al. (70) developed a severe neurological phenotype with the prolongation of lifespan by the oxidant scavenger MnTBAP, although once again, the global nature of the MnSOD knockout confounded the interpretation of the results. To date, three Cre/loxP animals have been reported with partial or complete knockout of MnSOD in brain or motor neurons. The model reported by Misawa et al. (72) expressed Cre recombinase in postnatal motor neurons with subsequent loss of MnSOD in a subset of somatomotor neurons. Extensive IHC confirmed the loss of MnSOD in the expected tissues; however, no activity data were reported. Enhanced levels of mitochondrial oxidants in the knockout tissues were demonstrated using in situ DHE oxidation measurements. The animals had little accumulated oxidative damage at 1 year of age, although distal nerve axons were susceptible to disorganization following injury (72). Overall, the knockout of MnSOD in the motor neurons produced a mild, if not barely detectable phenotype. While it is tempting to preclude MnSOD as a player in the protection from oxidant stress, questions remain. The use of IHC as the sole method for determining MnSOD knockout from cells is somewhat problematic since residual or incomplete knockout may preserve sufficient MnSOD activity for a minimal amount of protection. Nevertheless, the lack of phenotype does suggest that the motor neurons may have a particular resistance to mitochondrially derived oxidants.
Two groups studied the tissue-specific loss of MnSOD in the brain using mice expressing Cre from the nestin (nes) promoter, however, the background of the animals were very different. The nes-Cre mice used by Sasaki et al. (95) were on the C57Bl/6J background, whereas Oh et al. (79) used nes-Cre mice on the BKS.db+ strain, an established model of type II diabetes. Both animals developed severe neurological abnormalities manifested as an early abnormal gait and a progressive difficulty in righting themselves, and survived to approximately 4 weeks of age. Sasaki et al. (95) subsequently examined the site of O2 •− generation in brain slices during simulated hypoxia and reoxygenation in vitro, demonstrating a significant increase in oxidant production in the MnSOD-deficient mice. Unfortunately, no additional characterization of the brain histology was reported.
Although the stated purpose of the study by the Oh's group was to examine the MnSOD-dependent development of diabetic neuropathy (79), the animals failed to survive to the age of diabetes onset, therefore, the authors performed multiple histological analyses to determine the pathological basis for the rapid neurodegeneration. Interestingly, the authors describe a variable amount of MnSOD knockdown rather than a complete knockout in the cervical and lumbar spinal cords, as well as in the dorsal root ganglia. Pronounced vacuolization in the form of spongiform degeneration, as well as mitochondrial structural damage was evident in the cortical sections of the MnSOD-deficient mice, but not in the heterozygous or wild-type animals. There was no histological evidence of damage to the surrounding neuropil, microglia, or astrocytes, however, examination of the sciatic and sural peripheral nerves revealed prominent myelin delamination and disrupted mitochondria in myelinating Schwann cells. From the data presented in their manuscript, however, the determination of the specificity of the nes-Cre-mediated MnSOD knockout was difficult to assess. The variability of MnSOD knockout in the brain tissue suggests that nes-Cre animal may be insufficiently characterized to provide useful insight into the role of MnSOD in the development of neurodegenerative diseases.
Liver
Because the global MnSOD−/− reported by Li et al. (62) reportedly had significant lipid droplet formation in the liver and skeletal muscle, the liver was an early target for tissue-specific MnSOD knockout using the Cre/loxP system. Indeed, the first reported crossing of the floxed MnSOD mouse was with an albumin-Cre-expressing mouse to generate a liver-specific MnSOD knockout mouse (85). However, their study, as well as one reported by Lenart et al. (60) using an α-fetoprotein-Cre mouse, showed no sign of steatosis or liver damage. Both studies demonstrated hepatocyte MnSOD knockout using Western blot analysis of tissue homogenates, but not IHC localization. Lenart et al. noted that liver weights were significantly reduced (∼50%), although the level ROS or lipid peroxidation was variable or modestly elevated (60). Also, they observed a loss in the periportal pattern of zonation and deficiencies in gluconeogenic enzymes (60), although this defect was not directly correlated with the loss of MnSOD, nor was it demonstrated to lead to any additional structural or functional changes in the liver. The range of results from no detected phenotype [Ikegami et al. (45)] to some phenotype [Lenart et al. (60)] highlights the difficulty of assessing the role of MnSOD deficiency in the same tissue system without a clear indication of the specific localization of Cre expression and MnSOD knockout. Although the two Cre-expressing promoters are active in the liver, the reports fail to demonstrate their specificity, thus confounding the comparison of similar liver MnSOD knockout models.
Connective tissue
In the pursuit for a model of accelerated aging, Treiber et al. (104) crossed the floxed MnSOD mice (100) with the Cre mouse controlled by the promoter from the collagen type (I)-α-2 gene (24), leading to expression in fibroblasts and cells of the mesenchymal lineage. The loss of MnSOD in the connective tissue throughout every organ led to a complex phenotype, including skin atrophy, osteoporosis, and muscle degeneration. Using skin fibroblasts, the measure of oxidant production with MitoSOX showed significantly increased fluorescence. In addition, the accumulation of carbonyl proteins was also increased. Electron microscopy of the fibroblasts showed severely disrupted mitochondria with the loss of intramitochondrial structures.
The extensive IHC characterization of the specificity of Cre expression and the loss of MnSOD in representative tissues coupled with the complex phenotype, including decreased survival, although with a biphasic pattern of survivability, makes this an interesting model for the study of mechanisms of connective tissue-related accelerated aging resulting from the loss of MnSOD. The main challenge in interpreting mitochondrial dysfunction in this model, however, is the ubiquitous nature of the MnSOD knockout in the connective tissues of every organ system.
Mammary gland
Investigators from the same group that generated the T-cell-specific knockout (11) also produced a mouse model of mammary gland MnSOD deficiency (10) using a Cre expression cassette containing the mouse mammary tumor virus promoter (111). However, no discernible changes were detected in the development of the mammary gland, nor were any functional alterations observed with respect to the ability of the tissue to support adequate growth of normal-sized litters over multiple rounds of breeding. The evaluation of MnSOD knockout and oxidant stress was not as thorough as previous reports making the authors' interpretation that the mammary gland is more tolerant to oxidative stress than many other tissues somewhat weaker. While MnSOD knockout was evaluated by both RT-PCR and Western blot analysis, significant MnSOD mRNA and protein remained in the tissue. Unfortunately, the representative IHC images from lactating mammary glands were inconclusive. Thus, it is unclear if the residual MnSOD is a result of incomplete recombinase activity in the tissue, or a result of resident stem cell contamination in the samples, as suggested by the authors. In addition, no measures of mitochondrial function were made, and the only measure of oxidant stress was the qualitative IHC-based measurement of the oxidatively modified DNA metabolite 8-oxo-2′deoxyguanosine. While this model has multiple caveats, the lack of a distinct phenotype or structural modifications suggests that the loss of MnSOD has a lesser impact on the mammary gland, although more investigation is needed.
Gastric parietal cells
The parietal cells of the gastric mucosa are primarily responsible for the secretion of acid to aid in digestion, and are dependent on ATP generated by the mitochondria for proper function (14, 38). Parietal cell loss and increased oxidant damage has been shown to play a role in gastric diseases such as gastric hypertensive gastropathy (1, 52). Therefore, Jones et al. (48) developed a MnSOD knockout model using the Cre/loxP system under the control of elements of the noncatalytic β-subunit of the H+,K+-ATPase promoter shown to direct expression to gastric parietal cells (101). Although the mouse-to-mouse expression of the Cre-system was somewhat variable as evidenced by the presence of MnSOD in a lane in the Western blot and residual SOD activity in homogenates, the authors report a significant increase in O2 •− in isolated tissue slices, as well as increased nitrotyrosine and lipid peroxidation metabolites. Mitochondrial aconitase and ATP synthase activities were also decreased in the MnSOD-deficient tissues, while a small, but significant, increase in apoptotic cells was also detected. Importantly, gastric cells with decreased MnSOD have reduced basal and stimulated acid release, suggesting a significant functional deficit in the gastric mucosa, thus predisposing the tissue for oxidant-mediated injury.
Factors to consider when studying MnSOD tissue-specific knockouts
The use of the Cre/loxP system to generate alterations to specific regions of genetic material in vivo makes it a powerful tool for the creation of models of diseases that were once impossible. Two groups have reported loxP-containing MnSOD mice that have been utilized in the generation of the tissue-specific MnSOD knockout mice described herein (45, 100). Both are generated on the C57Bl/6 background, and use similar approaches to target and excise the critically important exon 3 of the mouse MnSOD gene (45, 100). Even though crossbreeding to a ubiquitously expressing Cre mouse strain confirmed the accessibility of the loxP sites to the recombinase (45), it is still possible that the promoter for the Cre mouse of choice may not drive expression of sufficient amounts of Cre protein in the targeted tissues leaving unmodified floxed allele(s), and a mosaic pattern of MnSOD expression within the tissue.
The great strength, as well as the great limitation, of the Cre/loxP system is the use of tissue-specific promoter elements to direct Cre recombinase expression in the desired location, and, with enough knowledge of the promoter activity, at the desired window of embryonic development. As with any transgenic animal, Cre mice are susceptible to potential problems with the integration sites, copy number, and stability of the transgene cassette in the genome [for reviews, see refs. (76, 88)]. The selection of the most appropriate Cre-expressing animal is critical for the successful development of a tissue-specific knockout animal. Fortunately, the CREATE Consortium (
The inducibility of the MnSOD gene expression and the modulation of MnSOD activity by oxidative and nonoxidative post-translation modifications have been extensively studied (20, 113) and reveal a complex regulation consisting of mRNA, protein, and enzyme activity, which are not necessarily correlated under normal or pathological conditions. In our kidney knockout model, for instance, we measured a significant amount of residual activity in the tissue homogenates. Careful IHC analyses of MnSOD expression in kidney slices revealed MnSOD expression in the glomeruli and proximal tubules, which is consistent with the lack of Cre expression in those structures. Without the activity and IHC data, our understanding of the model would have been incomplete. Because the MnSOD activity is efficiently and irreversibly inhibited by tyrosine nitration (67, 68), the lack of activity in a SOD homogenate assay does not always correlate with protein amounts in a tissue, and therefore cannot be easily interpreted as such, especially in models of increased mitochondrial oxidative stress. Indeed, nitrated MnSOD is a good oxidative marker in and of itself. Additionally, Western blot analyses measure the total protein content of the tissue, not the localization or activity. Thus, it is our view that multiple complimentary techniques to measure the MnSOD activity and protein localization are critical for complete characterization and interpretation of the tissue-specific knockout model.
The role of MnSOD is to protect the mitochondria from O2 •− produced from the electron transport chain, whether during the course of oxidative phosphorylation or when an exogenous insult short-circuits the process leading to an increase in the flux. In the last 10–15 years, great effort has been invested in demonstrating that MnSOD and other mitochondrial proteins, as well as mtDNA and repair processes, are susceptible to attack from ROS and RNS. In the absence of MnSOD, as is the case in the tissue knockout model, the affected mitochondria lack such protection and the direct determination of the steady-state levels of oxidants, as well as the assessment of multiple signatures of oxidative stress (carbonylation, nitration, lipid peroxidation, etc.) provide valuable insights into the mechanisms at play. Much debate and several reviews have been written describing the use and misuse of assays for the detection and quantification of ROS and RNS (9, 21, 50, 51, 97), therefore, care must be taken to select those that are most applicable and meaningful in the tissue of interest. Indeed, the same oxidative measure/marker must be used if direct comparisons between studies are to be meaningful.
The spectrum of phenotypes observed in tissue systems without MnSOD range from minimal or nonobservable, as seen in the liver and mammary gland, to severe perinatal lethality, as seen in the heart and brain (Table 1; Fig. 4). While it is tempting to dismiss the importance of MnSOD activity in models that, at best, have a minor phenotype, the conclusions cannot be supported without complete characterization of the model, including the extent of knockout of the protein, any residual MnSOD activity in the tissue or organ, and the consideration of compensatory mechanisms such as the mitochondrial repair pathway. It is not clear what threshold of O2 •− flux, or the minimal amount of MnSOD activity that is necessary to transition a tissue from a minimal to a mild phenotype or severely affected phenotype in the tissues (Fig. 5). An emerging area of mitochondrial research is the study of mitophagy, the cellular stress response to rapidly remove damaged mitochondria without the sacrifice of the entire cell by apoptosis (115). Conversely, stressed cells may have the cellular machinery and biogenic program primed for continuous replacement of mitochondria, at least until oxidative insults or demand for energy overwhelm the process. In our kidney model, we have observed that markers of autophagy are increased in the MnSOD knockout tissues (82). While this hypothesis requires additional experimentation to fully reveal the underlying mitochondrial responses to stress, the balance between mitophagy and biogenesis in response to damage is an exciting and promising avenue of study.
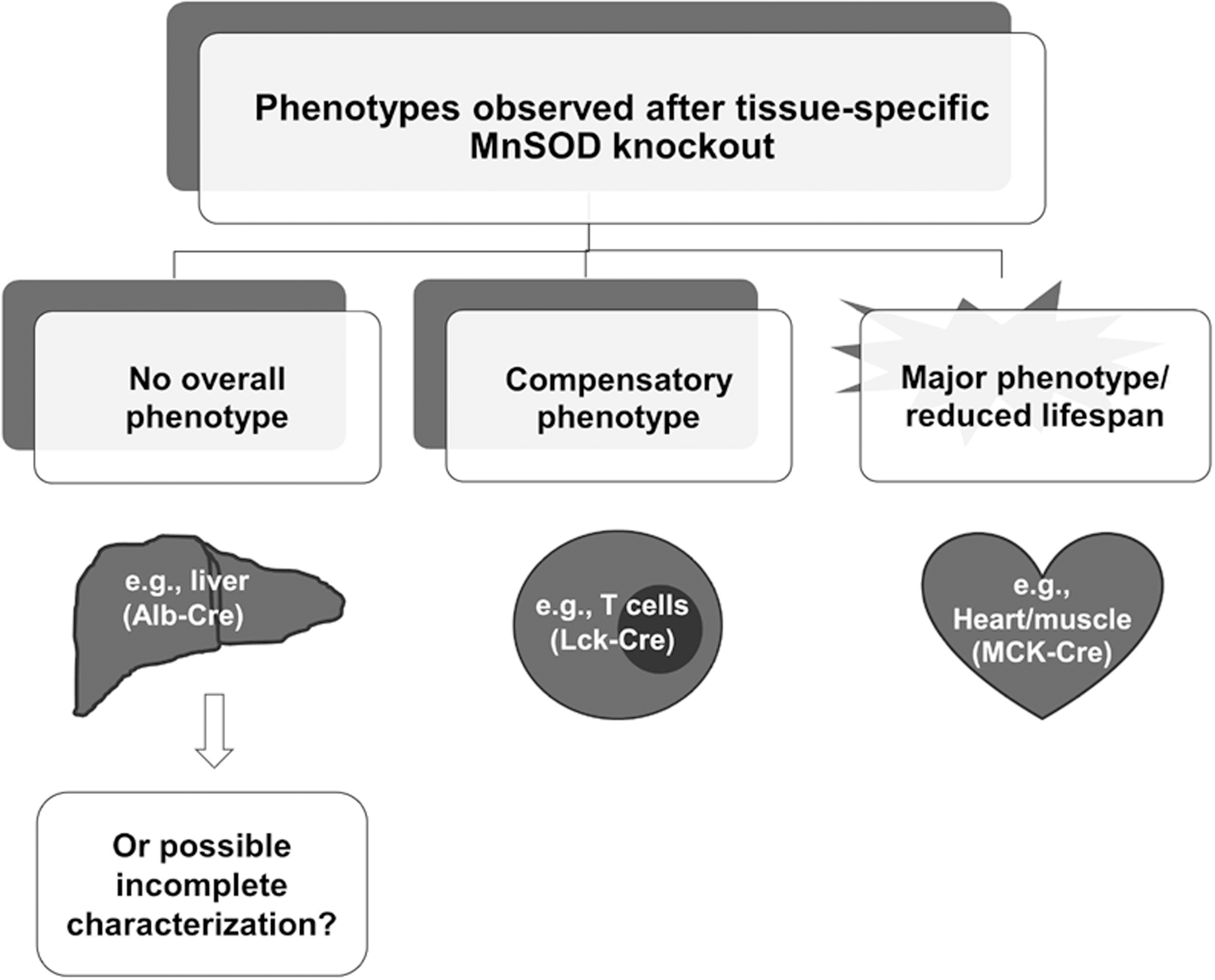

The hypothesis that a critical threshold of active MnSOD is essential for fully functional mitochondria is supported by a whole host of in vitro and in vivo data, and is the underlying reason for the development of disease models based on tissue-specific MnSOD deficiency. What is less clear, and the focus of much research, is the extent to which, endogenous compensatory systems interface with mechanisms that protect energy production in the face of acute or chronic stress. Much of the data from the MnSOD tissue knockout studies assessing mitochondrial function and the integrity of efficient ATP production involve indirect measures of the activity of proteins within the electron transport complexes. These measurements are often difficult to perform in situ, although the introduction of the Seahorse and Oxygraph instruments and newly developed protocols allow investigators to assess the functional integrity of affected mitochondria, as well as to dissect the contribution of individual complexes to oxidant-induced mitochondria dysfunction. Thus, we recommend that investigators with the access to such instrumentation take advantage of the power of these techniques so that we all may better understand the interplay between MnSOD and mitochondrial function in specific tissues under normal and stressed conditions.
Additional systems for site-specific and tissue-specific gene manipulations are in development, although not as widely adopted as the Cre/loxP system. The flippase (Flp/FRT) is the eukaryotic site-directed recombinase homologue to the Cre/loxP system derived from Saccharomyceses cerevisiae (57, 88, 90) and has been used with some success, although hampered by the decrease thermostability of Flp and lower recombinational efficiency in mammalian cells (57). The Streptomyces lividans bacteriophage ΦC31 utilizes directional site-specific integration with the recognition of the attB and attP sites, although it suffers from a low recombinational efficiency, as well as the tendency to recognize naturally occurring pseudo-attP sites resulting in unintended integrations. These emerging technologies show promise, however, have not been utilized for tissue-specific MnSOD knockout models.
Another area of active research and much debate is the usefulness of supplemental endogenous administration of small molecule SOD mimetics and catalytic antioxidants in models of disease and states of increased oxidant production as is seen in tissue-specific MnSOD knockouts. In the MnSOD−/− knockout mice (70), the putative SOD mimetic MnTBAP increased the lifespan of the animals only to unmask an underlying neurological deficit. Similarly, the small molecule antioxidants used by Case et al. (11) provided partial protection in T-cells from the susceptibility to infection, while Kuwahara et al. (58) and Nojiri et al. (78) restored exercise capacity in damaged skeletal muscles. Antioxidant supplementation in models of MnSOD deficiency is complex and fraught with unanswered questions involving the selection of an antioxidant(s), the specificity of antioxidants with ROS and RNS, proper dosing regimens, and the bioavailability in the mitochondrial compartment, to name a few. While supplementation data must be interpreted with caution, we feel that wider use and study of small molecule antioxidants in models of tissue-specific MnSOD knockouts will enhance our understanding of the role of MnSOD in diseases.
Summary
Tissue- and organ-specific knockout of MnSOD provides support for a role of functioning antioxidant systems for the protection from oxidant stress, especially within mitochondrial compartments. Because the complete knockout of the MnSOD gene is lethal in mice precluding the study of protective mechanisms within localized tissues most often stressed or damaged during the course of disease development, the recombinase-mediated Cre/loxP system addresses some of these issues and allows tighter control of MnSOD knockout in specific cell types and tissues. Even so, proper interpretation requires that both Cre and MnSOD be visualized by IHC in all tissues and cell types, given the potential for variable Cre expression. MnSOD Western blots can only provide information regarding total tissue expression of the protein, and provide no information regarding activity. The use of additional tools employing genetic manipulations to probe the involvement of the antioxidant genes and proteins at the level of tissue development and within cellular processes during homeostasis and the development of diseases is rapidly expanding with even more precise techniques. The Cre/loxP system and its reliance on multiple factors, including a clear understanding of the behavior of promoters, however, comes with many caveats and limitations that are crucial to address in the interpretation of data arising from its use, as we have attempted to highlight in this review. It is expected that the number of reported tissue-specific MnSOD knockout mouse models will continue to grow so as to expand our understanding of oxidant- and antioxidant-mediated processes in human physiology and disease.
Footnotes
Acknowledgment
This work was supported by National Institutes of Health grant 1RO1DK0789361.