
Abstract
Introduction
I
Oxidative Stress and MPI
An imbalance between the generation of ROS and the detoxification of the reactive intermediates is defined as oxidative stress. Under aerobic conditions, ROS are generated as by-products of the oxygen metabolism during oxidative phosphorylation in the mitochondria (55, 74, 89). Apart from their deleterious effects, ROS functions as signaling regulators. ROS can modulate mitochondrial functions via regulating electron transfer chain enzymes and mitochondrial membrane potential (121, 196). ROS are crucial for various cellular processes, including cell growth (18), apoptosis (2, 82, 184), cell adhesion and immune responses (38). Some ROS also act as second messengers in redox signaling (52). When the normal redox state of a cell is impaired, highly reactive peroxides (hydrogen peroxide [H2O2]), and free radicals (O2 •−) with the ability to damage any component of the cell, including proteins, lipids, and DNA, accumulates (68). Disruption of the ROS production/detoxification cycle, contributes to the development of human pathologies, including age-related diseases, such as cancer (58, 68, 80, 178), cardiovascular (38, 139), neurodegenerative (178), and genetic disorders (33, 88). ROS can also be generated by exogenous sources like ionizing radiation (IR) (75, 110), through direct interactions with either cellular targets or with water, resulting in DNA damage and gene mutations (45, 69) or cell death (110). However, under oxidative stress, ROS induces the intrinsic antioxidant enzymes to protect cells from ROS-induced toxicity (64).
Adaptive response to oxidative stress
Adaptive response has been observed in both malignant (116) and nonmalignant (117, 118) cells that were exposed to thiol-containing drugs, oxidative stress, such as hyperoxia (80%–95% O2) or H2O2 (160) as well as reactive nitrogen species (RNS or RNOS), namely, nitric oxide (NO) (143). However, this review will focus on the adaptive response to IR; radioadaptive response. Chronic exposure of cells to IR induces an adaptive response that results in enhanced tolerance to the subsequent IR cytotoxicity (2, 47, 64). Exposure to low dose IR (LDIR) generates beneficial effects for mammalian cells with respect to the maintenance of genomic integrity and the ability to repair damaged DNA (49). Radioadaptive resistance is a feature of many other species, including E. coli, algae, higher plant cells, and insect cells (94). In mice, whole-body preirradiation with LDIR reduced the incidence of thymic lymphoma from 46% to 16% with a challenging high dose of radiation, indicating the radioadaptive response (13). Similarly, in rabbits, Liu et al. reported LDIR-induced radioprotection (98). Human lymphocytes exposed to LDIR showed fewer chromatid breaks when later exposed to larger doses (156). Pretreatment of normal (3, 47) or cancer cells (64) with LDIR (1-10cGy) induced the radioadaptive response by elevating the expression and activity of antioxidant enzymes, primarily MnSOD (116). The above data and many other reports suggest that radioadaptive response is conserved in all species (2); and is also a feature of tumor cells, in which therapeutic radiation induces adaptive response (64, 75, 79), contributing to the overall ineffectiveness of cancer radiotherapy. However, the exact mechanism of MnSOD induction governing the radioadaptive response of tumor cells is still largely unknown.
Mitochondria
Mitochondria generate cellular fuel for aerobic cells to maintain normal functions (150, 153). The mitochondrial bioenergetics and the number of mitochondria per cell are related to the overall energy demands of the cells. In mammalian cells, about 90% of the ATP is generated by the mitochondria via oxidative phosphorylation. As a result of their role in metabolism, mitochondria are the primary source of ROS in aerobic cells. Apart from this critical function in cell respiration, mitochondria are also involved in other fundamental cellular functions, such as apoptotic response (2, 58, 80), cardioprotection (178), and neurodegenerative disorders (38, 178). To perform and regulate such a wide range of cellular roles, mitochondria are engaged in an extensive communication with their cytoplasmic environment via a constant flux of inorganic ions, metabolites, and proteins that traffic between the cytosol and mitochondria.
Mitochondria contain their own genome, a single circular DNA, that encodes 2 ribosomal RNAs, 22 tRNAs and 13 polypeptides, which all integrate into the mitochondrial respiratory chain complexes (153). However, the majority of mitochondrial proteins are encoded by nuclear DNA, synthesized as precursors on cytosolic ribosomes, and then transported into the mitochondria. Such peptides are imported into the mitochondria by a complex multistep mechanism involving the recognition, translocation and membrane insertion of these precursors, which then become predominant regulators of mitochondrial functions (152). The number of polypeptides in a single mitochondrion had been estimated to be over 1000 (151) until recent proteomic analyses identified 500–800 proteins (153). Among these are the proteins involved in the biogenesis of mitochondria and in various other biochemical activities of mitochondria. It is important to note that the composition of the mitochondria and relative abundance of proteins in them is highly cell type/tissue/organism specific. The exact functions of many of these mitochondrial proteins are unknown.
Mitochondrial Protein Import/Influx
Mitochondria are the site of oxidative phosphorylation and carry the electron transport chain complexes in their inner membrane, but they also accommodate the Krebs cycle in the matrix, and are involved in fatty-acid metabolism, urea cycle, and programmed cell death. These diverse functions require the transport of metabolites and proteins to and from the cytosol. Mitochondrial influx of proteins is implicated in a wide spectrum of mitochondrial functions, including apoptosis and ATP production (78, 131); and the influxed proteins are targeted to all mitochondrial compartments (9, 72), including the cytoplasm-facing outer surface of the outer membrane (66). This review will focus on a specific cluster of MPI that translocate into the mitochondrial matrix under oxidative stress conditions to regulate MnSOD function.
Mitochondria are membrane-enclosed organelles with two membranes: an outer and an inner membrane. Other compartments include the intermembrane space, cristae space formed by the convolution and folding of the inner membrane, and the matrix (Fig. 1 insert). Proteins destined to be imported into mitochondria face the challenge of being routed to their correct sub-mitochondrial compartments. Moreover, the proteins targeted to the mitochondrial matrix, the innermost compartment, must be transported across two membranes. These challenges are overcome by two specific machineries on the outer and inner mitochondrial membranes. The transfer of mitochondrial proteins from their site of synthesis into the mitochondria occurs post-translationally through interactions with the mitochondrial import machinery located at the outer membrane (Fig. 1) (135). However, some precursor proteins enter the import channels cotranslationally (57). Mitochondria-targeted proteins can be divided into two main classes; proteins with presequences and proteins with internal targeting sequences (180). Presequences are N-terminal cleavable sequences, which form positively charged extensions that interact with the mitochondrial import receptors; whereas internal sequences are not cleavable, do not necessarily contain charged amino-acid residues, and are incorporated into the mature protein. Apart from positively charged amino-acids like arginine and lysine, presequences also contain hydrophobic residues and hydroxylated amino-acids, such as serine and threonine (146). The presequences are usually devoid of acidic residues (146); and they show a high tendency to form an amphipathic α-helix that presents one positively charged surface and one hydrophobic surface (145), both of which are recognized by distinct receptor subunits of the translocase of the outer membrane (TOM) complex. On the other hand, the internal sequences usually exist in clusters and can be charged or uncharged. The individual signals can function independently but the import is more efficient when they cooperate (135). Mitochondrial localization of proteins can be predicted based on the presence of N-terminal targeting sequences using bioinformatics approaches; however, the consensus sequence for N-terminal presequences is not completely satisfying and the internal sequences are yet to be characterized.
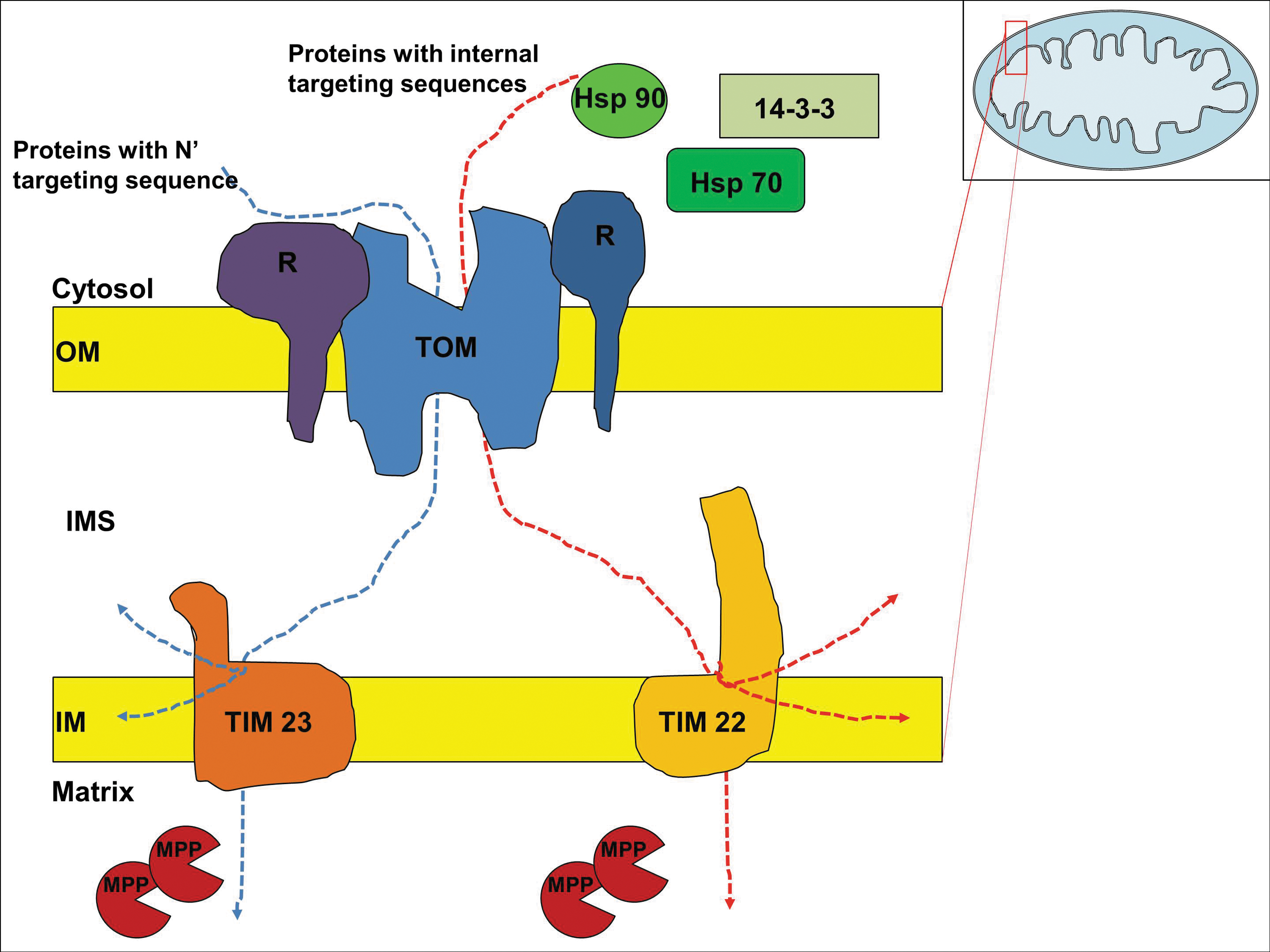
Nuclear-encoded proteins cross the outer membrane of the mitochondria at the TOM complex, from where they are directed to one of two functionally different translocase of the inner membrane (TIM) complexes to transport across the inner membrane. TIM 23 guides the import of proteins with N-terminal presequences and TIM 22 is involved in the import of proteins with internal targeting sequences (34). The TOM complex consists of seven subunits; three receptors Tom20, Tom22, Tom70; the channel forming protein Tom40; and three small proteins Tom5, Tom6, Tom7 (180). Proteins with presequences are recognized by the receptor proteins within the TOM complex, whereas the proteins with internal targeting sequences usually interact with cytosolic chaperons, Hsp70 and Hsp90, which dock onto the receptors of TOM complex (193). Three other cytosolic factors that act on mitochondrial preproteins have been described: mitochondrial fusion protein targeting factor from yeast (24), presequence binding factor from rabbit (114) and mitochondrial import stimulating factor from rat (67). Among these, mitochondrial import stimulating factor is a member of the 14-3-3 protein family, a ubiquitous eukaryotic protein family with a wide range of chaperone properties (5). 14-3-3 proteins may form complexes with Hsp70 to facilitate the mitochondrial localization of cytosolic proteins (109). The transfer of proteins across the inner membrane also requires the mitochondrial membrane potential (Δψ) (106). The mitochondrial processing peptidase (MPP) cleaves off the presequences from the proteins in the matrix before the protein folds into its active form with the help of chaperons (71). Once the proteins pass through TOM and TIM complexes and the presequences are cleaved off, they are sorted according to the proteins' sorting signals into inner membrane, intermembrane space, outer membrane, or the matrix.
Although the mechanism and the machinery responsible for mitochondrial protein import have been extensively studied, little is known about the factors that affect the influx of proteins under physiological or pathological conditions. It has long believed that MPI is an unregulated event, until recent evidence from plant studies showed that mitochondrial internalization of proteins is influenced by light-dark cycle and developmental cues (39). The next section will address the regulation of MPI in oxidative stress conditions.
Regulation of MPI under oxidative stress
Studies in mammalian systems have shown that oxidizing conditions decrease the mitochondrial protein import and cause the accumulation of precursor proteins outside mitochondria. The precursor proteins that have failed to be imported end up being degraded by proteasomes (187). Interestingly, Tom 20, a core component of the mitochondrial outer membrane protein import receptor, was also among the proteins that are degraded by proteasomes under oxidative stress (187). These results suggested that oxidizing conditions lead to a mitochondrial influx lesion resulting in the decreased number of mitochondrial proteins. This decrease may represent the mitochondrial response to the oxidative stress. A relatively recent study showed that IR affects the mitochondrial protein import based on the radiation dose. At lower doses (10cGy), IR enhanced the mitochondrial import; while at higher dose (4 Gy), mitochondrial import was decreased (134). These results suggested that altered mitochondrial protein import by LDIR may be linked to the adaptive response of the cells, where increased protein import is aimed to protect the cells. Translocation of proteins across the inner membrane requires potential difference (Δψ) across the membrane. IR may disrupt the membrane potential and affect the MPI. Though, changes in membrane potential alone were not enough to explain the mitochondrial import deficiencies observed in this recent study (134). Further studies showed that some components of the mitochondrial import machinery may be damaged by IR, resulting in deficient protein import (10). Defective protein import, in turn, is shown to amplify the effects of oxidative stress and cause several health defects, including degenerative diseases and metabolic disorders.
Superoxide Dismutases
Under normal circumstances, the deleterious effects of ROS are avoided by several antioxidant enzyme systems that detoxify ROS produced in the cells (92). Among the antioxidants, the SOD scavenger enzymes convert superoxide radicals into H2O2 and molecular oxygen (2O2 •−+2H+→H2O2+O2) (56). The resultant H2O2 is largely removed by peroxiredoxins (Prx) (125), glutathione peroxidases (104) and catalase (56, 69). There are three SOD enzymes expressed in mammalian cells. Copper/zinc-containing superoxide dismutase (Cu/ZnSOD, SOD1) is primarily localized to cytosol (159), although it has been found in the intermembrane space of mitochondria (128, 179). MnSOD (SOD2) is the mitochondrial antioxidant that exists in homotetramers (19, 140, 183) and localizes in the mitochondrial matrix (128, 179). The third SOD is the extracellular superoxide dismutase (ECSOD, SOD3) is localized to the extracellular space (51). The presence of SOD within the mitochondria suggests that superoxides generated in the mitochondria do not readily cross membranes; and due to their deleterious effects on biomolecules, they must be dismutated immediately by a SOD, preferably at the site of their production.
Manganese Superoxide Dismutase
Human MnSOD is a tetrameric enzyme with four identical subunits each harboring a Mn+3 atom (19, 183). Human MnSOD gene is located on the 6th chromosome, 6q25.3 region (174) encoding a ∼223 amino acid/26 kD precursor monomer containing a mitochondria targeting sequence of 26 amino acids (183) that is required for mitochondrial localization. Human MnSOD resides in the mitochondrial matrix (19, 183), where the mitochondrial oxidative phosphorylation complexes that generate superoxide are accessible. MnSOD is a highly conserved protein with over 40% sequence homology among human, yeast and E. coli (11). Each MnSOD monomer has two distinct domains: an N-terminal helical hairpin domain and a C-terminal a/b domain, containing five alpha helices and three-stranded antiparallel beta sheets (19). As MnSOD is taken up by the mitochondria, the mitochondria targeting sequence is clipped off, leaving a 22 kD monomer, which later incorporates an Mn+3 ion and assembles into an 88 kD homotetramer in the mitochondria (183). Residues D159, H163, H26, H74, and a water molecule from each subunit contribute to the manganese metal-binding site, namely, the active site (19).
Mitochondrial import/processing of MnSOD
MnSOD is encoded by nuclear genome, synthesized in the cytosol, imported into the mitochondrial matrix post-translationally via TOM and TIM23 complexes and assembled into active enzyme with the incorporation of a manganese ion in the mitochondrial matrix (183). Human MnSOD is translated as a precursor peptide with a leader sequence that is proteolytically removed by MPP in the mitochondrial matrix during the processing of the mature enzyme. Similar to other mitochondrial leading sequences, the presequence of MnSOD has been shown to form amphipathic α-helices with positive charged and hydrophobic residues on opposite faces of the helix (146, 183). This amphipathic helix mediates its interaction with the mitochondrial import machinery and facilitates the import process. Furthermore, the mitochondrial transport of MnSOD precursor has been long known to be energy dependent (183). Energy derived from ATP is required for uptake and processing of not only MnSOD, but also most precursor mitochondrial proteins (146). It is believed that mitochondrial proteins integrate cofactors as a terminal step of their assembly because cofactors recognize the tertiary structure of the proteins (144). MnSOD is no exception to that; it is imported into the mitochondria as an inactive apoprotein, which later incorporates its cofactor Mn2+ ion in the matrix.
A human genetic polymorphism encodes for either alanine (Ala) or valine (Val) in the mitochondrial targeting sequence of MnSOD protein and is shown to modulate MnSOD activity and its mitochondrial influx (165, 166). This polymorphism is at codon 16 of the human precursor protein, which is also located at position −9 of the mature protein; therefore, it is often referred to as either the Ala9Val or Ala16Val MnSOD dimorphism (166). Initial studies on the effect of this dimorphism in MnSOD mitochondrial influx were based on the computer models that can predict the configuration of polypeptides from their amino acid sequences. A partial α-helix configuration was predicted for Ala-MnSOD, while a single β-sheet structure was predicted for the Val-MnSOD. The expanded β-sheet structure was estimated to physically slow down the import of MnSOD through the TIM23 channel and cause partial stalling of the Val-MnSOD in the inner membrane (158). Further studies showed that mitochondrial import and activity of MnSOD is affected by the Ala9Val dimorphism in its MTS (107, 165, 166). The Ala-MnSOD variant with an α-helical MTS is easily imported into the matrix and has high mitochondrial activity, whereas the Val-MnSOD variant with a β-sheet structure in its MTS is arrested within the inner membrane import complex TIM23 and partially degraded in the proteasome. Therefore, the Val-MnSOD variant showed lower enzymatic activity (165, 166). However, conflicting data have been reported from Bastaki et al. and Martin et al., showing that Val-MnSOD variant has greater MnSOD activity than the Ala-MnSOD variant (107). The discrepancy in these reports may stem from different in vitro and in vivo models used in these studies, suggesting a cell type/content dependent effect for this dimorphism. Nevertheless, more studies are needed to elucidate the reasons for the differential results, and to further understand the effect of the dimorphism in the MnSOD activity. Most importantly, the clinical relevance of this dimorphism and whether it could modulate susceptibility to various human diseases requires further investigation.
Oxidative stress-mediated regulation of mitochondrial processing of MnSOD
Mitochondrial processing of human precursor MnSOD was shown to be compromised by superoxide generating compound paraquat (186), which is a redox-cycling agent that transfers electrons to molecular oxygen to form superoxide (54). It has been demonstrated that the mitochondrial processing of human MnSOD from precursor to the mature form was inhibited by paraquat treatment. This, in turn, resulted in lower MnSOD activity. The mechanism of paraquat-mediated inhibition is not known; however, it is suggested that paraquat induces a specific lesion in the MnSOD processing machinery (186). It is believed that paraquat treatment leads to oxidation of critical sulfhydryl groups (cysteine [Cys] residues) of the proteins of the mitochondrial import machinery (185). Hyperoxia is another well-known condition that increases superoxide generation in the mitochondria (53). Similarly, hyperoxic conditions were shown to inhibit MnSOD processing by interfering with the import machinery. These studies suggest the mitochondrial import machinery as a susceptible cellular target of superoxide radicals and support a role for ROS in decreasing mitochondrial import and activation of MnSOD enzyme under oxidizing conditions.
Regulation of MnSOD enzymatic activity within mitochondria under oxidative stress
MnSOD is essential and biologically significant to aerobic cells. Studies from the early 1970s using E. coli and yeast as model organisms provide the early evidence for the importance of MnSOD expression for surviving in aerobic environments (61). Later studies demonstrated clearly that total MnSOD gene knockout is lethal in mice (182) and fruit flies (40); and heterozygous mice with lower MnSOD activity are more susceptible to oxidative injury (169, 171). These, along with numerous other studies, indicated the crucial role of MnSOD in protecting against oxidative stress (85, 123, 195), including radiation insult (47, 64, 79, 130, 195). Although the exact mechanisms associated with the overexpression and/or activation of MnSOD are not fully understood, there are a great deal of reports exploring the role of post-translational as well as transcriptional and post-transcriptional regulation of MnSOD in governing its superoxide dismutase activity in the adaptive response against oxidative stress.
MnSOD activation via MPI-mediated phosphorylation
Post-translational modifications of proteins control many biological processes through a variety of mechanisms that include the changes in protein activity, interactions and subcellular localizations. Post-translational modifications of mitochondrial proteins by reversible phosphorylation/dephosphorylation events play essential roles in a number of cell signaling pathways regulating the mitochondrial respiration as well as the apoptosis (77, 132). The significance of reversible phosphorylation in cellular function can be estimated by the amplitude of the genome dedicated to kinases and phosphatases (6). The involvement of mitochondrial proteins in phosphorylation events has been studied for years and more than 60 mitochondrial proteins have been identified as phosphoproteins (9, 72). The recent advances of mass spectrometry along with the development of new reagents, including phospho-specific antibodies and dyes enabled the characterization of the post-translational modifications of MnSOD and many other mitochondrial phosphoproteins (77).
The first evidence suggesting MnSOD is phosphorylated came from the studies with bacteria (7, 173). Mass spectrometric analysis of phosphoproteins in Campylobacter jejuni identified SOD as one of the cytoplasmic phosphoproteins (173). Archambaud et al. reported that MnSOD from the cytoplasmic Listeria monocytogenes was phosphorylated on serine and threonine residues and the phosphorylated MnSOD was less active when the bacteria were in stationary phase of growth (7). This study was not only the pioneering work showing that MnSOD can be phosphorylated, but also the first to indicate that phosphorylation of MnSOD controls its activity, although the relevant biological effects of this activity remained to be investigated. Likewise, later studies predicted mitochondrial MnSOD as a target of phosphorylation. Phosphorylation of MnSOD was first reported in potato mitochondria but no activity measurements were conducted (21). Hopper et al. observed phosphorylated MnSOD in porcine heart mitochondria using phosphoproteome technique (77). Rat MnSOD is also subjected to phosphorylation shown by in vitro kinase assays using mitochondrial extracts as the kinase source (25). Taken together, these initial studies suggested that MnSOD is regulated by phosphorylation in a large scale of species, from bacteria to mammals.
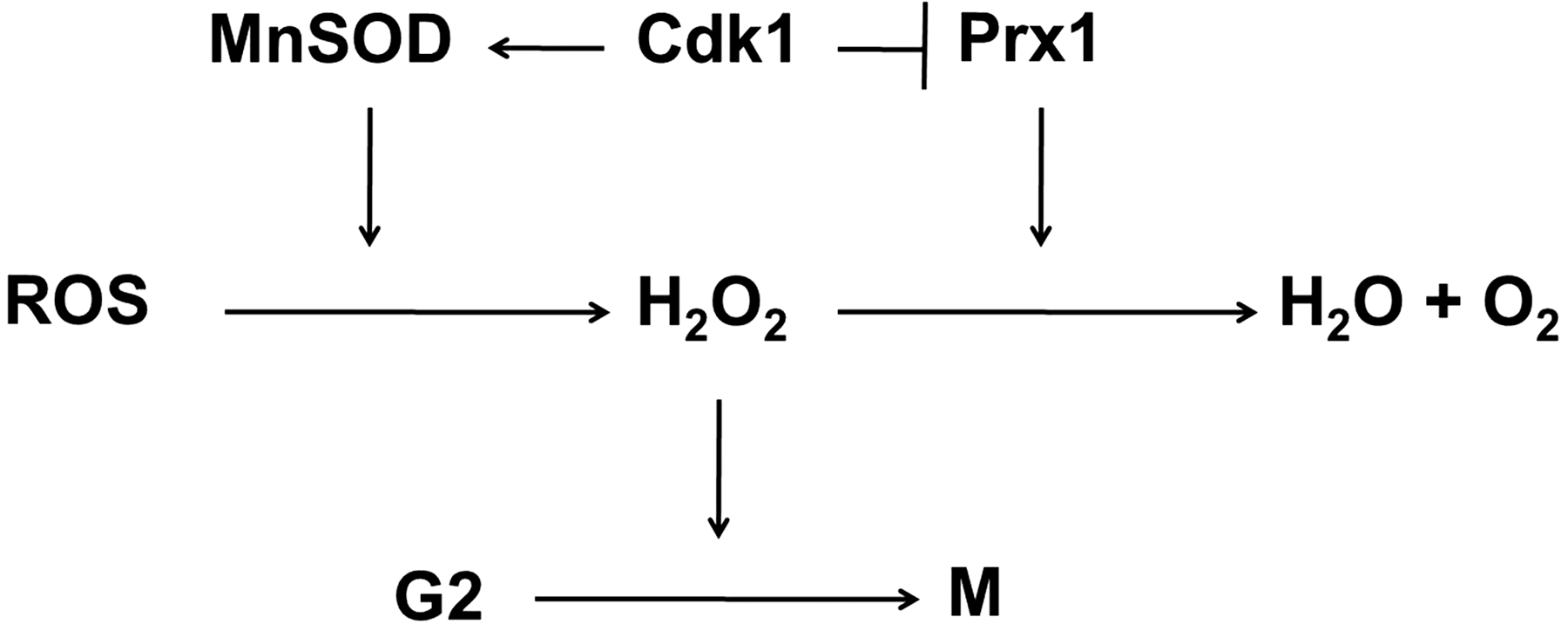
Parallel studies demonstrate that the phosphorylation of antioxidant proteins is linked with the growth in mammalian cells (27). Chang et al. showed that phosphorylation of the human antioxidant peroxiredoxin I (Prx 1) by cyclin-dependent kinase 1 (Cdk1) resulted in the inactivation of this antioxidant and affected cell cycle progression (27). Prx are the enzymes involved in the elimination of H2O2 and are present in organisms from all kingdoms (142). At least six Prx enzymes are present in mammals (142, 155) and these are distributed differentially within cells: Prx I and II are localized to the cytosol (125), where Prx III and V to the mitochondria (155). Prx I contains a consensus site for Cdk1 phosphorylation and the phosphorylation of Prx I at Thr90 reduces its peroxidase activity (27). In addition to Cdk1; Cdk2, Cdk4, and Cdk6 can also phosphorylate Prx I in vitro. The regulation of Prx I with Cdk-mediated phosphorylation may shed light onto the regulation of MnSOD antioxidant enzyme through similar mechanisms.
Human MnSOD contains a minimal Cdk1 phosphorylation consensus sequence (Serine/Threonine-proline [Ser/Thr-Pro]) (170) at Ser106. The fact that Ser106 is very accessible in the final homotetrameric conformation of MnSOD encouraged the further analysis of the possibility of phosphorylation at this residue (Fig. 2). We recently found that substantial amounts of CyclinB1/Cdk1 (22, 120) localized into the mitochondria following LDIR. The presence of any canonical MTS within these proteins was explored using the web-based software MitoProt II (

Additional results suggest that cells may utilize Cdk1-mediated phosphorylation of MnSOD in normal progression of cell cycle: the transient inhibition of Prx 1 activity via Cdk1-dependent phosphorylation is thought to cause H2O2 accumulation, which then stimulate the transition from G2 to M phase (27). Thus, it is logical to assume that the enhanced activity of Cdk1-phosphorylated MnSOD will further contribute to the accumulation of H2O2, assisting the progression through G2 phase (Fig. 3). Nevertheless, further studies are needed to fully understand the role of Cdk1-mediated phosphorylation of MnSOD in cell cycle progression and other critical cellular outcomes.
Recent studies indicate that several cytosolic kinases translocate into the mitochondria (76, 78). A phosphorylation site identification program, NetPhosK, (
Adapted from (131).
IMS, intermembrane space; MnSOD, manganese superoxide dismutase.
MnSOD regulation via other post-translational modifications in mitochondria
As recent developments in mass spectrometry enabled detailed structural analysis of covalent modifications of proteins, the identification of post-translational modifications are largely facilitated as well. MnSOD is one of the many proteins whose covalent modifications are widely studied, especially due to its vital role as the mitochondrial antioxidant and the urgent need to elucidate the regulatory mechanisms of its dismutase activity. Besides phosphorylation, the post-translational modifications of MnSOD discovered so far include nitration, acetylation, glutathionylation, methylation and metal incorporation. The amino acid residues of MnSOD that are involved in post-translational modifications, along with the executing molecules, are summarized in Table 2.
Nitration
The nitrogen monoxide molecule (•NO) is a ubiquitous cellular messenger, whose overproduction is associated with several diseases. The pathological effects of NO are linked to the generation of RNS, such as nitrogen oxides (NOx) and peroxynitrite (ONOO−), as a result of its reaction with oxygen and superoxide, respectively (41). Of these, peroxynitrite stands out as the biologically relevant nitrating agent (62). The reaction of proteins with nitrating agents results in the covalent modification of their tyrosine (Tyr) residues (41, 62). Tyr nitration has been shown to severely diminish the function of proteins when the nitrated Tyr residues are in the active site of the target peptide (41, 62). However, protein nitration is a highly selective process since a relatively limited number of proteins are preferential targets of nitration and within these proteins; only one or a few specific Tyr residues have been identified to be nitrated in vivo (8).
In situations of increased oxidative stress, NO is found to react with O2 •− and forms the highly reactive peroxynitrite (12). Peroxynitrite is well-established to inactivate MnSOD by nitrating a critical Tyr residue (Tyr 34) at the enzyme active site both in vitro and in vivo (102, 137, 141, 163). A recent study also reported Tyr nitration of MnSOD and resultant inactivation of the enzyme upon exposure to NO (162). MnSOD protects cells from deleterious effects associated with the overproduction of NO and peroxynitrite in two ways: (i) by reacting with RNS itself; (ii) via its active role in the detoxification of ROS, so it prevents the reactions of ROS with NO to form peroxynitrite (163). However, it must be re-emphasized that the reactions of MnSOD with RNS results in impaired enzymatic activity meaning that the nitration/inactivation of MnSOD operates as a positive feedback loop for enhanced mitochondrial peroxynitrite formation, which will eventually lead to the amplification of oxidative stress by allowing the accumulation of O2 •− and trigger apoptosis (138). Besides, peroxynitrite also causes lipid peroxidation, DNA damage and nitration of proteins other than MnSOD. The importance of MnSOD in preventing the nitration of proteins can be better comprehended by looking at the pathologies associated with oxidative stress and overproduction of NO (65, 189). In these pathologic conditions, MnSOD itself is Tyr nitrated and inactivated (103).
Acetylation
The discovery of the mitochondrial localization of at least three sirtuin family of NAD+-dependent protein deacetylase enzymes (SIRT3, SIRT4 and SIRT5) suggested the existence of sirtuin substrates in the mitochondria (157). With the addition of several lines of evidence linking SIRT3 to metabolism (157), lysine acetylation has recently emerged as an important post-translational modification to regulate mitochondrial proteins (99). When studies with Sirt3 knockout cells showed increased mitochondrial superoxide levels upon exposure to IR (4), the researchers suspected altered regulations of MnSOD in Sirt3-lacking cells. Additional support came from the studies observing significantly increased MnSOD acetylation and corresponding decrease in the MnSOD superoxide dismutase activity in Sirt3 knockout cells (168). Tao et al. further identified Lys 122 of MnSOD as a reversibly acetylated residue (168). These pioneering investigations on the regulation of MnSOD activity by changes in specific lysine acetylation are followed by the reports on SIRT3-mediated regulation of MnSOD activity. Qiu et al. found that calorie restriction induces SIRT3-mediated MnSOD activity resulting in reduced oxidative stress (136). In the same study, Lys 53 and Lys 89 were identified as acetylated residues that control enzymatic function. Moreover, Ozden et al. demonstrated that SIRT3 is activated by oxidative stress induced by IR as well as by cellular nutrient status to deacetylase Lys 122 of MnSOD and regulate MnSOD enzymatic activity (129). Taken together, MnSOD acetylation represents an alternative mechanism of post-translational regulation of MnSOD, which may contribute to the overall MnSOD activity in stress response.
Glutathionylation
Glutathionylation is the post-translational modification of protein Cys residues by the addition of glutathione. It is promoted by oxidative and nitrosative stress, although, some also occur in unstressed cells (31). Human Cu/ZnSOD can be glutathionylated at Cys 111, resulting in a reduction in its activity and promoting its aggregation (181). Iron superoxide dismutase (FeSOD) from a eubacterium species was also shown to be gluthathionylated at Cys 57, which did not result in a significant change in its enzymatic activity; however, this modification protected the enzyme from Tyr nitration via peroxynitrite and was enhanced upon cell exposure to oxidative stress (26). In mammalian cells, protein glutathionylation is involved in redox signaling and the defense against oxidative stress. In mitochondria, the high concentration of glutathione has been related to the presence of several glutathionylated proteins (83). The known sensitivity of human mitochondrial MnSOD to reducing agents (108) has been associated with a highly reactive cysteine residue (Cys 196) exposed on the surface of the tetrameric enzyme. So far, glutathionylation of MnSOD was only found for rat recombinant MnSOD grown in E. coli using mass spectrometry analysis (25). On the basis of high similarity between rat and human MnSOD, the candidate cysteine residue, Cys 196, was identified as the target of glutathionylation (25). Accumulating evidence suggests glutathionylation as an important mechanism of cellular response to oxidative damage and redox signaling (133). Future research is needed to elucidate the biological function of glutathionylation of MnSOD as well as other mitochondrial targets in vivo and the possible role of such post-translational modification in enzyme activity and cellular outcomes.
Methylation
A recent study with mass spectrometry revealed a complex pattern of lysine and arginine methylation of MnSOD in both quiescent and proliferating mouse embryonic fibroblast (MEF) cells (150). Methylation of MnSOD was detected at lysines 68, 89, 122, and 202; and arginines 197 and 216. Lysine 68 was shown to be dimethylated during quiescence and monomethylated during proliferation states of fibroblasts. Lysines 89 and lysine 202, on the other hand, were monomethylated during quiescence and unmethylated in proliferating cells. Arginine 197 was shown to be dimethylated, whereas arginine 216 was monomethylated in quiescent cells; and the arginine 197 dimethylation did not change between quiescent and proliferative growth states. These results indicated a dynamic regulation of MnSOD via methylation during cell cycle. In the same study, mutagenesis analysis determined that lysine 89 methylation is important for MnSOD activity, while lysine 202 methylation is not. Further computational modeling simulations based on the mass spectrometry data demonstrated that lysine and arginine methylation of MnSOD may increase the accessibility of the active site to superoxide during quiescence in MEF cells.
Metal incorporation
Insertion of the catalytic metals into the SOD is necessary for full activation of the enzymes and occurs post-translationally. After the precursor polypeptide MnSOD enters the mitochondria matrix, the mitochondria targeting sequence is cleaved off and manganese is subsequently inserted into the MnSOD. Two membrane transporters have been studied as facilitators of manganese insertion to MnSOD in yeast: (i) Smf2p, a manganese transporter critical for cell surface uptake of manganese (101); (ii) Mtm1p, a member of the mitochondrial carrier family of transporters residing in the mitochondrial inner membrane and important for the exchange of solutes between mitochondria and cytosol (100). Studies with Smf2 mutants showed accumulation of an iron-substituted form of MnSOD in the mitochondria (192). Monomethylamine permease (Mtmp1) mutants exhibited lost MnSOD activity, albeit accumulation of high levels of mitochondrial manganese occurred (100). Iron incorporated MnSOD was also detected in Mtmp1 mutants (100). Biological significance of iron misincorporated MnSOD is not fully understood, but several indications have emerged from in vitro metal substitution studies (191). Iron-substituted MnSOD is inactive but showed more thermal stability than the native MnSOD and a H2O2-mediated •OH (hydroxyl free radical) generating activity (191), pointing to a multitude of disadvantages for cells: a loss of enzymatic activity, a gain of radical generating ability and increased stability of iron-misoncorporated MnSOD. Considering iron excess in the mitochondria has been linked to various diseases (105), it would not be surprising to see the presence of iron-misincorporated MnSOD in these diseases.
The regulation of MnSOD via mitochondria-localized p53
p53 is a homotetrameric transcription factor with well-established roles in tumor suppression via induction of apoptosis or cell cycle arrest by post-translational and transcriptional mechanisms (97, 194). Diverse biological actions of p53 in regulating cellular functions is cell type- and stimulus-dependent, complicated, and beyond the scope of this review. However, this review will address the evidence linking mitochondrial p53 to oxidative stress and MnSOD.
Mitochondrial translocation of p53 induces apoptosis by promoting changes in mitochondrial membrane potential, cytochrome c release and caspase activation (111, 126). p53 exerts its apoptosis-inducing role by binding to multiple targets on the outer membrane of the mitochondria, such as B-cell lymphoma (Bcl)-2/Bcl-XL (111), p53-regulated apoptosis inducing protein 1 (p53AIP1) (126) and Bcl-2–associated X protein (Bax) (28). Studies by Zhao et al. showed that p53 also localizes to the matrix of the mitochondria (195). In this same study, p53 was found to physically interact with MnSOD and inhibit its superoxide scavenging activity. This was the first study to show a direct role for mitochondrial p53 in the regulation of MnSOD activity and oxidative stress. Importantly, the mitochondrial translocation of p53 can be enhanced by radiation insult in wild type p53-bearing tumor cells (120, 175). Analysis of downstream effects and possible modulators of p53-MnSOD interaction will largely improve our knowledge of the molecular mechanisms of cancer progression and may provide novel therapeutic targets for cancer treatment. Among the upstream effectors of p53 is CyclinB1/Cdk1 complex, which can phosphorylate p53 at Ser315 (16). Establishment of mitochondrial localization of both p53 and CyclinB1/Cdk1 complex (120) suggested that this phosphorylation may also occur in the mitochondria. Nantajit et al. showed that mitochondrial Cdk1 interacted with and phosphorylated mitochondrial p53 at Ser315 in colon cancer cells upon exposure to IR (120). Interestingly, the phosphorylation of p53 at Ser315 by mitochondrial Cdk1 resulted in increased survival of the cells, suggesting an antiapoptotic role for mitochondrial phospho-p53. Further elucidation of the role of mitochondrial matrix p53 and the effect of Ser315 phosphorylation on proapoptotic functions of p53 is needed. However, we propose that the enzymatic regulation of MnSOD via p53 and Cdk1 induces two competing mechanisms and results in two opposite outcomes potentially depending on the severity of the oxidative stress and the resultant cellular damage: (i) proapoptotic response in case of severe injury via p53/MnSOD interaction and (ii) antiapoptotic response in case of minor fixable injury via Cdk1-p53-MnSOD pathway. Nevertheless, our current knowledge presents that the interaction of p53 with MnSOD in the mitochondria inhibits MnSOD scavenging activity and induces apoptosis, while the Cdk1-mediated phosphorylation of p53 in the mitochondria inhibits apoptosis, possibly by interfering with p53-MnSOD interaction. The studies demonstrating mitochondrial localization of p53 and CyclinB1/Cdk1 in oxidative stress conditions, together with our recently published findings that Cdk1 can directly phosphorylate and enhance MnSOD activity (22), may point to a Cdk1-p53-MnSOD regulatory mechanism in oxidative stress response (Fig. 4).
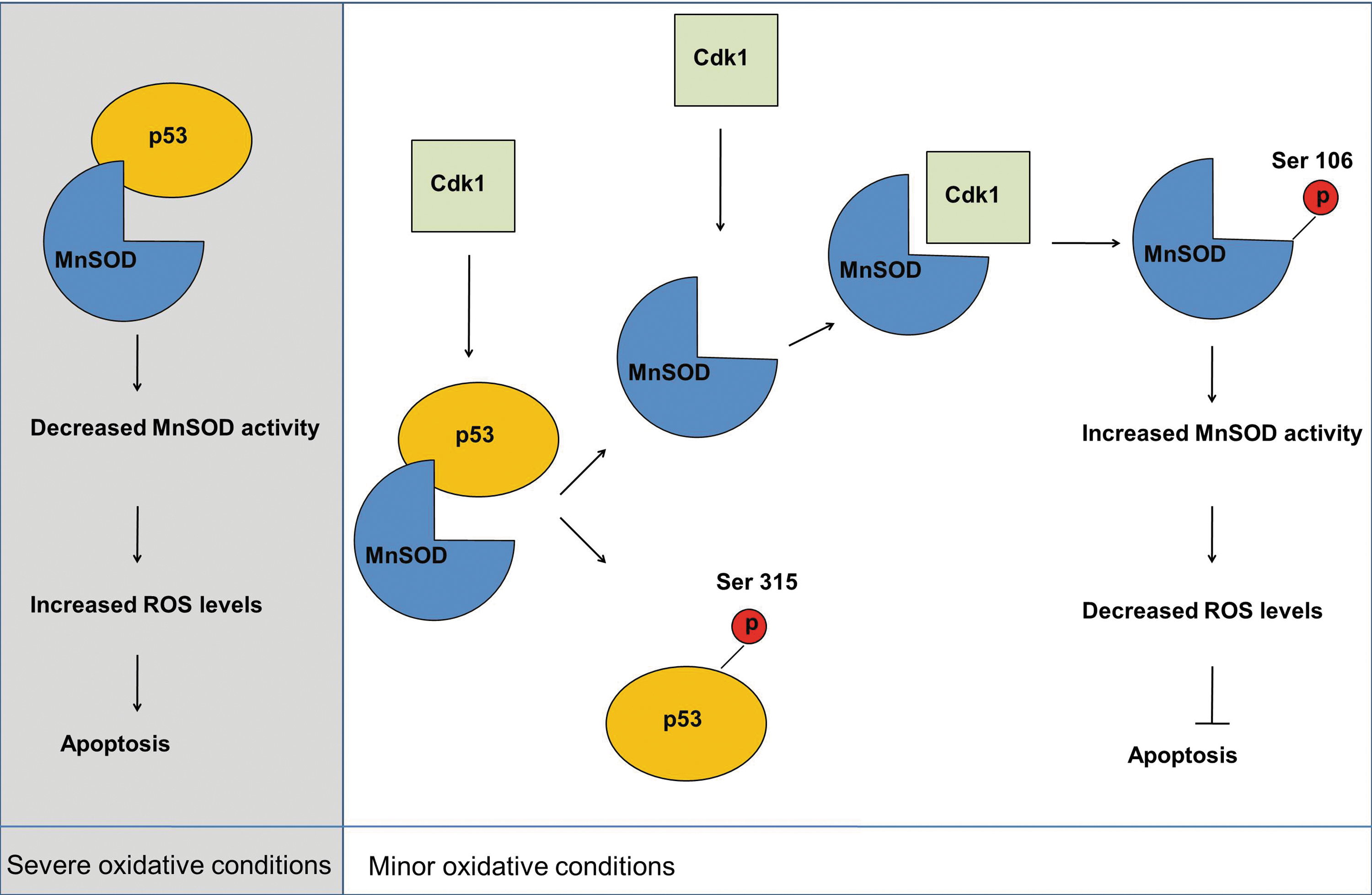
All in all, identification of MnSOD as a direct target of p53 is of great importance for tumor biology and holds enormous potential for clinical applications. p53-mediated regulation of MnSOD at the transcriptional level has also been implicated and will be addressed in the next section of this review.
Transcriptional/post-transcriptional regulation of MnSOD
MnSOD regulation is complex and occurs at both pre- and post-translational levels. Here we will briefly discuss some transcriptional/post-transcriptional mechanisms of MnSOD regulation (Fig. 5). The examples include changes in the rate of MnSOD protein synthesis (70), mRNA stability (29), and promoter activity (127) as well as regulation of chromatin accessibility via the methylation of CpG islands upstream of the Sod2 gene (84) and histone modifications (73). Of the factors regulating MnSOD expression, we will address nuclear factor-kappaB (NF-κB), p53, mTOR and mitogen-activated protein kinases (MAPKs) (ERK and p38).
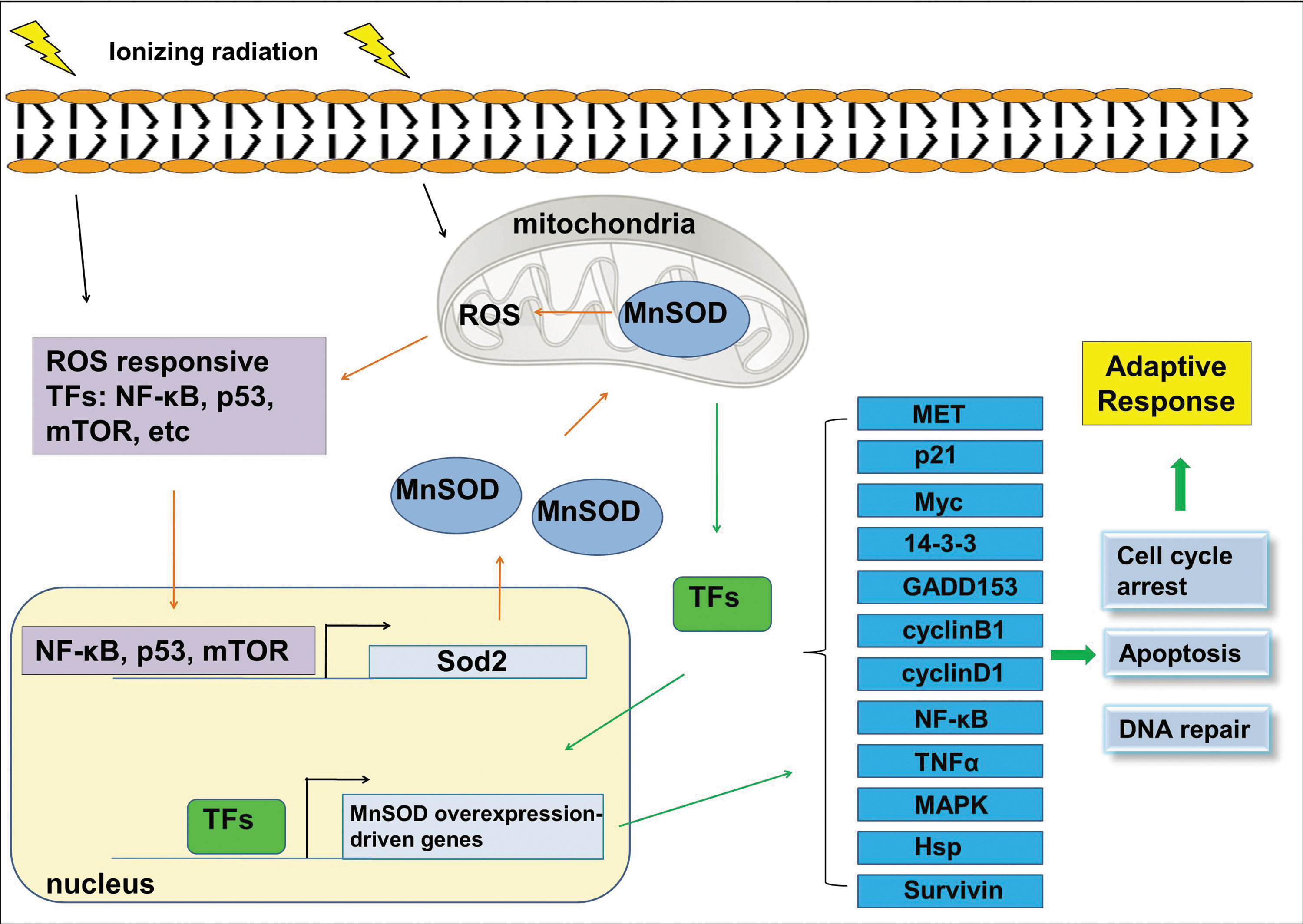
The connection between NF-κB and MnSOD has been observed in several studies on radioprotection compounds, where radioprotective thiols are shown to control MnSOD gene expression through NF-κB activation (32, 60, 117). In mammals, NF-κB family of transcription factors consists of five members: RelA (p65), RelB, c-Rel, p50/p105 (NF-κB1) and p52/p100 (NF-κB2). Although different combinations of homo- and heterodimers can be formed (172), the heterodimer of p50 and p65 is the most abundant form of NF-κB (119). NF-κB binding sites were found to be located in the promoter region (174, 190) as well as within the intronic enhancer element of the Sod2 gene (91). NF-κB is actively involved in the regulation of MnSOD expression in cells induced with various stimuli, including radiation (47, 64), thiol-containing drugs (117) and tumor necrosis factor, tumor necrosis factor alpha (TNFα) (32, 60). Moreover, NF-κB-induced MnSOD has been related to radioresistant phenotype of normal and cancer cells (64, 117, 124), pointing to MnSOD as a key NF-κB effector gene in radioadaptive resistance.
Likewise, there are p53 binding regions at 328 and 2032 bp upstream of the transcriptional start site of the Sod2 gene, mutations of which were shown to result in altered MnSOD expression (36, 37, 85). Apart from direct binding to the gene, p53 has been suggested to repress Sod2 gene expression by a number of mechanisms, including interfering with transcription initiation (85), blocking the access of gene activators at the promoter region by forming an inhibitory complex with them (48) and protein-protein interactions (96). In contrast, p53 can induce Sod2 gene expression (36, 96). The opposite effects of p53 on Sod2 expression is proposed to be cell type-, species- and concentration-dependent such that low concentrations of p53 increases MnSOD expression jointly with other transcription factors, such as NF-κB; and high concentrations of p53 suppresses MnSOD expression (37, 91). Interestingly, mutations of the NF-κB binding sites within the intronic enhancer element of the MnSOD gene inhibited p53-induced MnSOD gene transcription; and knocking down p65 via siRNA similarly resulted in reduced MnSOD gene transcription through p53 (91).
MnSOD upregulation through ROS-driven ERK-dependent activation of p53 has also been indicated (96). According to this study, ROS induced the translocation of ERK into the nucleus, where it interacted with and phosphorylated p53 at Ser15, resulting in the activation of p53 and subsequent increase in MnSOD expression. Involvement of another MAPK, p38, was also implicated in regulation of MnSOD expression (167, 184). A recent study with epithelial stem cells proposed that inhibition of mammalian target of rapamycin (mTOR) mediated the increased expression of MnSOD, resulting in the suppression of oxidative stress caused by the accumulation of ROS after radiation in normal human oral keratinocytes but not in head and neck squamous cell carcinoma (HNSCC) cells (86). Interestingly, the inhibition of mTOR did not increase the MnSOD mRNA levels, indicating that translational (such as increased rate of translation) or post-translational (such as enhanced protein stability due to protein modifications) mechanisms, rather than transcriptional events, may account for the increased MnSOD expression. Nonetheless, these results showed that mTOR inhibitors may reduce oxidative stress by inducing MnSOD overexpression and provided strong clinical implications for anticancer treatment.
Finally, the concept of MnSOD overexpression raises the questions regarding the levels of H2O2 that will accumulate as a result of such overexpression. However, mitochondria seem to co-regulate MnSOD expression with mitochondrial H2O2-removing capacity to prevent extreme fluctuations in redox status (90). The essence of maintaining optimal MnSOD protein levels is also supported by the findings that even though MnSOD mRNA levels were greatly elevated following the induction of gene expression, the resultant MnSOD protein levels were not as high (90). These observations implied a tight control over the expression of MnSOD protein despite the availability of mRNA for translation.
MnSOD-regulated gene expression
MnSOD has been shown to increase the expression of a number of genes participating in diverse functions related to radiation-induced adaptive responses (64, 118). These genes include MET, α-catenin, p21, v-myc myelocytomatosis viral oncogene homolog (Myc), 14-3-3 zeta, CyclinA, and CyclinB1. A more recent study with MnSOD-overexpressing HeLa cells identified MnSOD overexpression-driven expression of several other transcripts (79). This new set of genes includes GADD45, RAS homolog, tumor necrosis factor receptor, NF-κB, glucocorticoid receptor, heat shock proteins, cyclinD1, survivin and ataxin1. A similar study in MnSOD-overexpressing adult Drosophila revealed numerous cellular signaling pathways induced by MnSOD overexpression (30). Among these are both pro- and antiapoptotic genes as well as members of NF-κB, Jun N-terminal kinase (JNK), ERK, Janus kinase (JAK)/signal transducer and activator of transcription (STAT), cell cycle, and ubiquitin-mediated degradation pathways. These studies significantly contributed to the understanding of the downstream signaling molecules involved in the MnSOD-induced cell radiation-resistance phenotype. Regulation of a variety of cellular functions by MnSOD may encourage studies to determine specific targets to enhance radiation tolerance in normal cells (Fig. 5). Likewise, further studies to fully elucidate the mechanisms of MnSOD-mediated radioprotection may provide novel therapeutic targets to modulate tumor radiosensitivity during anticancer therapy as antioxidants, such as MnSOD are currently investigated for cancer treatment.
MnSOD interactome
Many hemostatic functions of cells, including the maintenance of the mitochondrial membrane (1, 2), activation of TNF signaling (115) and inhibition of aggressive phenotype of cancer cells (81, 94), can be induced by MnSOD under varied oxidative conditions (43, 46, 122). However, the exact MnSOD-mediated signaling network regulating the oxidative stress-induced adaptive response remains elusive. A recent study carried out in human skin keratinocytes (HK18) aimed to determine the signaling network associated with the MnSOD-induced radiation protection (42). In this study, a MnSOD-interacting protein profile was established in LDIR-treated HK18 cells. Analysis of the profiles of MnSOD-interacting partners before and after LDIR identified different patterns of MnSOD protein-protein interactions, suggesting a shift in the MnSOD-interacting partners following radiation exposure. Interestingly, many of the MnSOD-interacting proteins detected in this study are known to have functions related to mitochondrial regulation of cell metabolism, apoptosis and DNA repair (Fig. 6). These results led to the assumption that the role of MnSOD in the adaptive response is not only due to its dismutase activity, but also relates to its interactions/communications with many cellular/mitochondrial proteins, an interesting topic worthy of further investigation.

Innovation
Extensive communications between the mitochondria and cytoplasm regulate the activity of mitochondrial antioxidant MnSOD under oxidative stress. Proteins, such as cyclins, Cdks and p53 are imported into the mitochondria to alter MnSOD activity via post-translational modifications. Recent findings suggest phosphorylation of human MnSOD in mitochondria as a novel post-translational modification that enhances its superoxide scavenging activity in oxidative stress conditions. The regulation of mitochondrial import of MnSOD itself as well as of the proteins that modulate MnSOD activity was broadly discussed in this Forum review.
Conclusion
The mitochondrion imports and processes the vast majority of its proteins, including its structural elements and metabolic pathways from cytosol. This suggests that the regulation of MPI is key to the proper functioning of the mitochondria and of the cells. Here we discussed the specifics of mitochondrial protein import and its regulation, especially under oxidizing conditions. The mitochondrial superoxide dismutase MnSOD, the primary defense agent of mitochondria against oxidants, is of particular interest since its dysfunction is indicated in many pathological conditions. The mitochondrial import and processing of MnSOD itself as well as the mitochondrial influx of several proteins that regulate MnSOD enzymatic activity under oxidative insult reveals the importance of mitochondria, specifically its protein import apparatus, in cellular response to genotoxic environment. Along with the post-translational regulation of MnSOD activity, the transcriptional regulation of Sod2 gene and MnSOD-interactome profile in oxidative stress present a unique coordinated network that governs the adaptive response. Further studies will unfold additional regulatory mechanisms of this pivotal antioxidant enzyme and will instigate novel strategies to fight against diseases associated with defective MnSOD activity.
Footnotes
Acknowledgments
The authors would like to thank Dominik J. Green for his help in presentation of MnSOD protein structure and discussion on specific residues of MnSOD. The authors acknowledge Gayle Woloschak, David Grdina, Douglas Spitz, and Daret St. Clair for their communication and suggestions to the research in the author's lab. We would like to acknowledge the support of National Institutes of Health Grants RO1 CA133402 and RO1 CA152313, and the Department of Energy Office of Science Grant DE-SC0001271.