
Abstract
Introduction
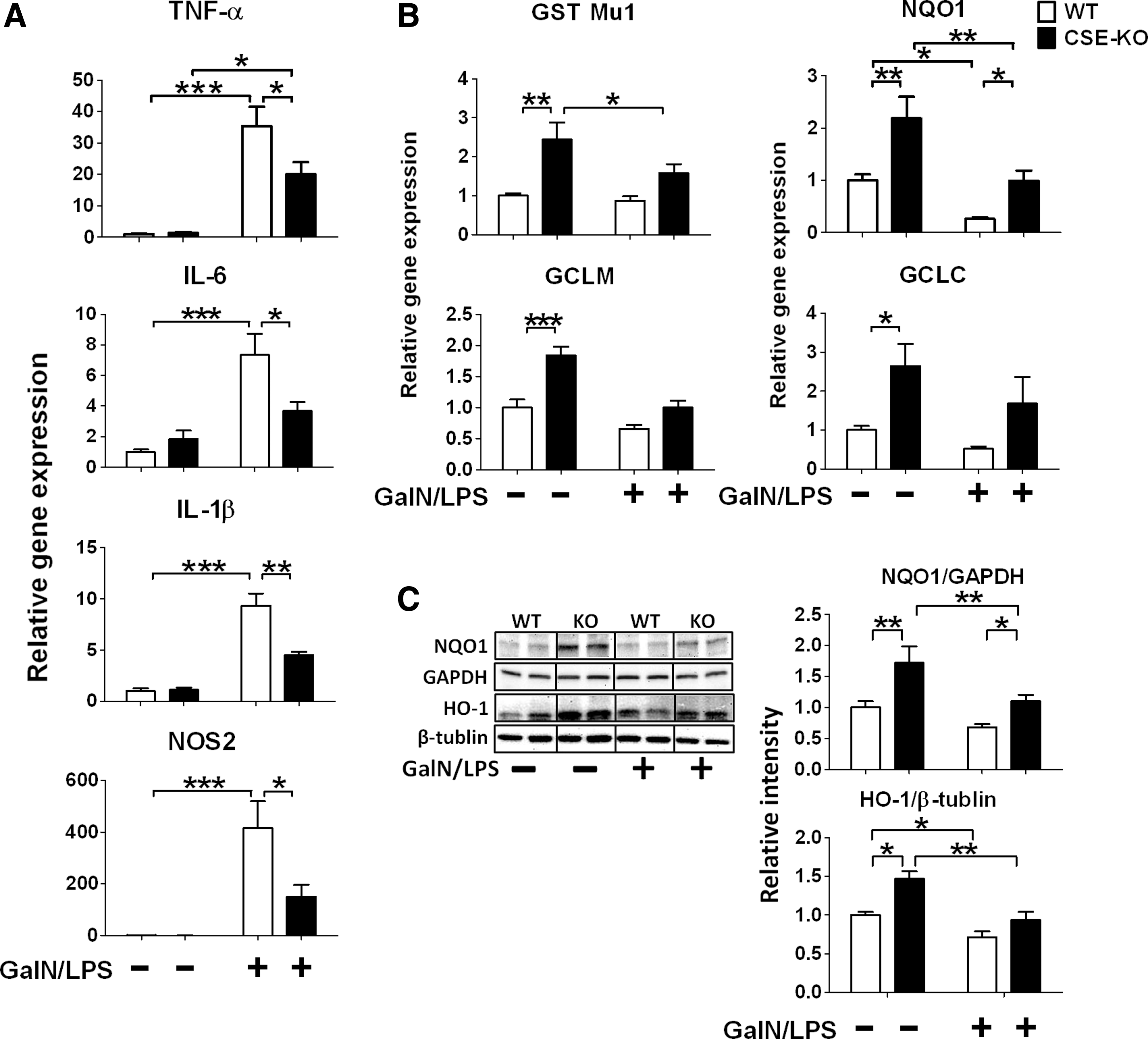
A
Using cystathionine γ-lyase (CSE)-deficient mice and primary hepatocytes, we unexpectedly revealed cytoprotective effects of CSE deficiency against inflammatory acute liver failure. Our results also showed a profound impact of CSE deficiency on sulfide metabolism that triggered several well-defined cytoprotective signaling mechanisms including augmentation of thiosulfate levels. Lastly, we confirmed the robust cytoprotective effects of sodium thiosulfate against acute liver failure suggesting an important translational potential of our findings.
Hydrogen sulfide (H2S) is an endogenously-produced gasotransmitter (30). In mammalian tissues, H2S is generated by cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE) (22), and 3-mercaptopyruvate sulfurtransferase (3MST) (21). On the other hand, H2S is serially oxidized to persulfide, sulfite (SO3 2−), thiosulfate (S2O3 2−), and sulfate (SO4 2−) in reactions catalyzed by several enzymes including sulfide:quinone oxidoreductase (SQR), sulfur dioxygenase ethylmalonic encephalopathy 1 (ETHE1), sulfurtransferase rhodanese, and sulfite oxidase (SUOX) (6, 8). H2S can exert a host of biological effects on various targets ranging from cytotoxicity to cytoprotection.
Cytoprotective effects of H2S have been attributed to its effects on several well-defined signaling mechanisms. For example, a number of studies reported that H2S activates NF-E2 p45-related factor 2 (Nrf2)-dependent signaling (7). Nrf2 is a transcription factor that regulates antioxidant genes as an adaptive response to oxidative stress or pharmacologic stimuli. Downstream targets of Nrf2 include direct antioxidant proteins (e.g., glutathione peroxidase, heme oxygenase-1 [HO-1], and NAD(P)H:quinone oxidoreductase-1 [NQO1]) and thiol metabolism-associated detoxifying enzymes (e.g., glutathione-S-transferase and glutamate-cysteine ligase) (8). It has been shown that Nrf2-dependent antioxidant proteins exert protective effects in animal models of inflammatory ALF (4, 15). Additionally B-cell lymphoma 2 (Bcl-2) activated by Nrf2 exerts anti-apoptotic effects (18).
Role of H2S in inflammatory organ injury remains incompletely defined. While acute administration of high doses of H2S donor compounds appears to be invariably toxic, lower and steady levels of H2S may be cytoprotective against systemic inflammation. Along these lines, we have recently reported that breathing low concentration of H2S prevents lethal endotoxemia in mice at least in part by increasing thiosulfate, an oxidative metabolite of H2S (28). On the other hand, inhibition of CSE by DL-propargylglycine (PAG) has been reported to exert anti-inflammatory effects in rodent models of sepsis, suggesting a pro-inflammatory effect of CSE (1). Nonetheless, role of endogenous H2S on inflammation is incompletely defined due to the limited selectivity of chemical inhibitors of H2S-producing enzymes including PAG.
To determine the role of endogenously-produced H2S on inflammatory organ injury, we examined outcomes of GalN/lipopolysaccharide (LPS)-induced ALF in CSE-deficient mice on the C57BL6 background. A combination of GalN/LPS has been widely used to induce ALF in animal models. GalN sensitizes the liver toward other stimuli in part reflecting the role of uridine-containing compounds in hepatic biotransformation. Coadministration of LPS and GalN potentiates hepatic damage, leading to hepatocyte apoptosis (20, 29). Given the protective effects of physiological levels of H2S against systemic inflammation, we hypothesized that CSE deficiency aggravates GalN/LPS-induced liver injury in mice. Here, we report that CSE deficiency unexpectedly attenuates liver inflammation and mortality in mice subjected to GalN/LPS-challenge, and prevents cell death in primary hepatocytes incubated with GalN/tumor necrosis factor (TNF)-α.
Results
CSE deficiency improved survival rate in mice after GalN/LPS challenge
Four out of eleven wild-type (WT) mice died within 24 h after challenge with GalN (300 mg/kg) and LPS (1 μg/kg). In contrast, no CSE-deficient mice died after GalN/LPS challenge (Fig. 1A, p=0.030 by log-rank test, n=11 each group). No mice of either genotype died thereafter up to 48 h after GalN/LPS challenge.

CSE deficiency attenuated liver injury after GalN/LPS challenge
Plasma alanine aminotransferase (ALT) levels at 6 h after GalN/LPS challenge were measured as a marker of liver injury. GalN/LPS challenge markedly increased ALT levels in WT mice, but not in CSE-deficient mice (Fig. 1B). We found that hepatic architecture in WT mice was disrupted with hemorrhage, necrosis, and neutrophils at 6 h after GalN/LPS challenge (Fig. 1C). In contrast, histological liver injury was markedly attenuated in CSE-deficient mice challenged with GalN/LPS (Fig. 1D).
CSE deficiency did not affect LPS-induced TNF-α production in primary peritoneal macrophage
Since GalN/LPS-induced hepatotoxicity is mediated by LPS-induced TNF-α production in macrophages (5), we examined the impact of CSE deficiency on TNF-α production in primary murine peritoneal macrophages. TNF-α level in the cell culture medium of WT primary peritoneal macrophages incubated with LPS increased in a time-dependent manner, reaching its plateau levels after 6 h (Fig. 2A). There was no difference in the TNF-α levels released by WT or CSE-deficient primary peritoneal macrophages at 6 h after incubation with LPS (Fig. 2B). These results suggest that CSE deficiency did not prevent GalN/LPS-induced ALF by inhibiting TNF-α production by macrophages.

CSE deficiency improved survival of primary hepatocytes after GalN/TNF-α challenge
Since CSE deficiency did not affect LPS-induced TNF-α production by peritoneal macrophages, we examined whether or not CSE deficiency protects hepatocytes from GalN/TNF-α. CSE deficiency improved survival of primary hepatocytes at 20 h after GalN (5 mM)/TFN-α (10 ng/ml) challenge compared with WT hepatocytes, as assessed by lactate dehydrogenase (LDH) and Cristal Violet (CV) assays (Fig. 2C, D). These observations suggest that CSE deficiency protects hepatocytes per se against cytotoxicity exerted by GalN/TNF-α.
CSE deficiency prevented systemic inflammation and upregulated antioxidant gene expression after GalN/LPS challenge in the liver
Levels of TNF-α, interleukin (IL)-6, IL-1β, and nitric oxide synthase (NOS) 2 mRNA in the liver were increased at 6 h after GalN/LPS challenge in WT mice. CSE deficiency markedly attenuated GalN/LPS-induced upregulation of inflammatory cytokines (Fig. 3A). We also examined Nrf2-dependent antioxidant expression before and after GalN/LPS challenge. CSE deficiency upregulated gene and protein expression levels of NQO1 at baseline and after GalN/LPS challenge. Additionally, CSE deficiency upregulated protein expression levels of HO-1 at baseline (Fig. 3B, C). Similarly, levels of glutamine-cysteine ligase catalytic subunit (GCLC), glutathione S-transferase Mu1 (GST Mu1), and glutamine-cysteine ligase, modifier subunit (GCLM) mRNA in the liver were upregulated at baseline and tended to be higher at 6 h after GalN/LPS challenge in CSE-deficient mice compared with WT mice (Fig. 3B). These results suggest that CSE deficiency augments antioxidant protein expression in the liver and prevents inflammatory cytokine induction after GalN/LPS challenge.
CSE deficiency increased phosphorylated Akt, Nrf2, and Bcl-2, and attenuated GalN/LPS-induced phosphorylation of JNK in the liver
To elucidate the mechanisms responsible for upregulated antioxidant protein expression induced by CSE deficiency, we examined levels of phosphorylated Akt, phosphorylated JNK, Nrf2, and Bcl-2 in the liver. CSE deficiency increased phosphorylated Akt at Thr308 and Bcl-2 at baseline and augmented Bcl-2 expression after GalN/LPS challenge in the liver. Levels of Nrf2 in the nuclear fraction of liver tissue were increased in CSE-deficient mice at baseline and decreased in WT mice at 6 h after GalN/LPS challenge. CSE deficiency attenuated the reduction of Nrf2 in the nuclear fraction of liver tissue after GalN/LPS challenge. Levels of phosphorylated JNK at Thr183 and Tyr185 were increased at 6 h after GalN/LPS challenge in the liver tissue of WT mice. In contrast, CSE deficiency markedly attenuated the increments of phosphorylated JNK (Fig. 4A). These results suggest that protective effects of CSE deficiency are associated with upregulation of Akt, Nrf2, and Bcl-2-dependent signaling and inhibition of JNK phosphorylation.
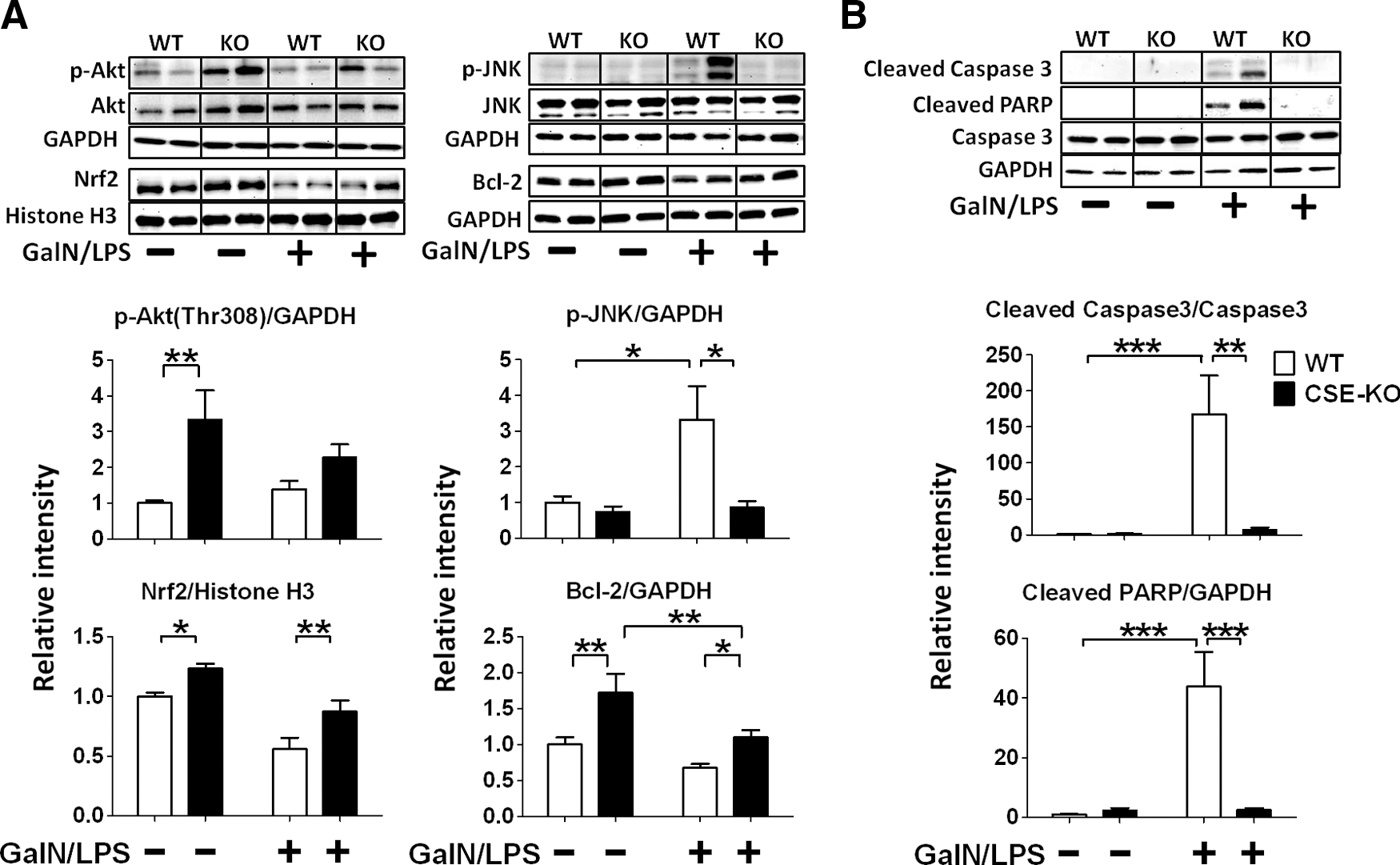
CSE deficiency attenuated GalN/LPS-induced activation of caspase 3 and poly (ADP-ribose) polymerase in the liver
Levels of cleaved caspase 3 and cleaved poly (ADP-ribose) polymerase (PARP) were increased at 6 h after GalN/LPS challenge in the liver tissue of WT mice. In contrast, CSE deficiency markedly attenuated increments of cleaved caspase 3 and cleaved PARP, suggesting anti-apoptotic effects of CSE deficiency (Fig. 4B).
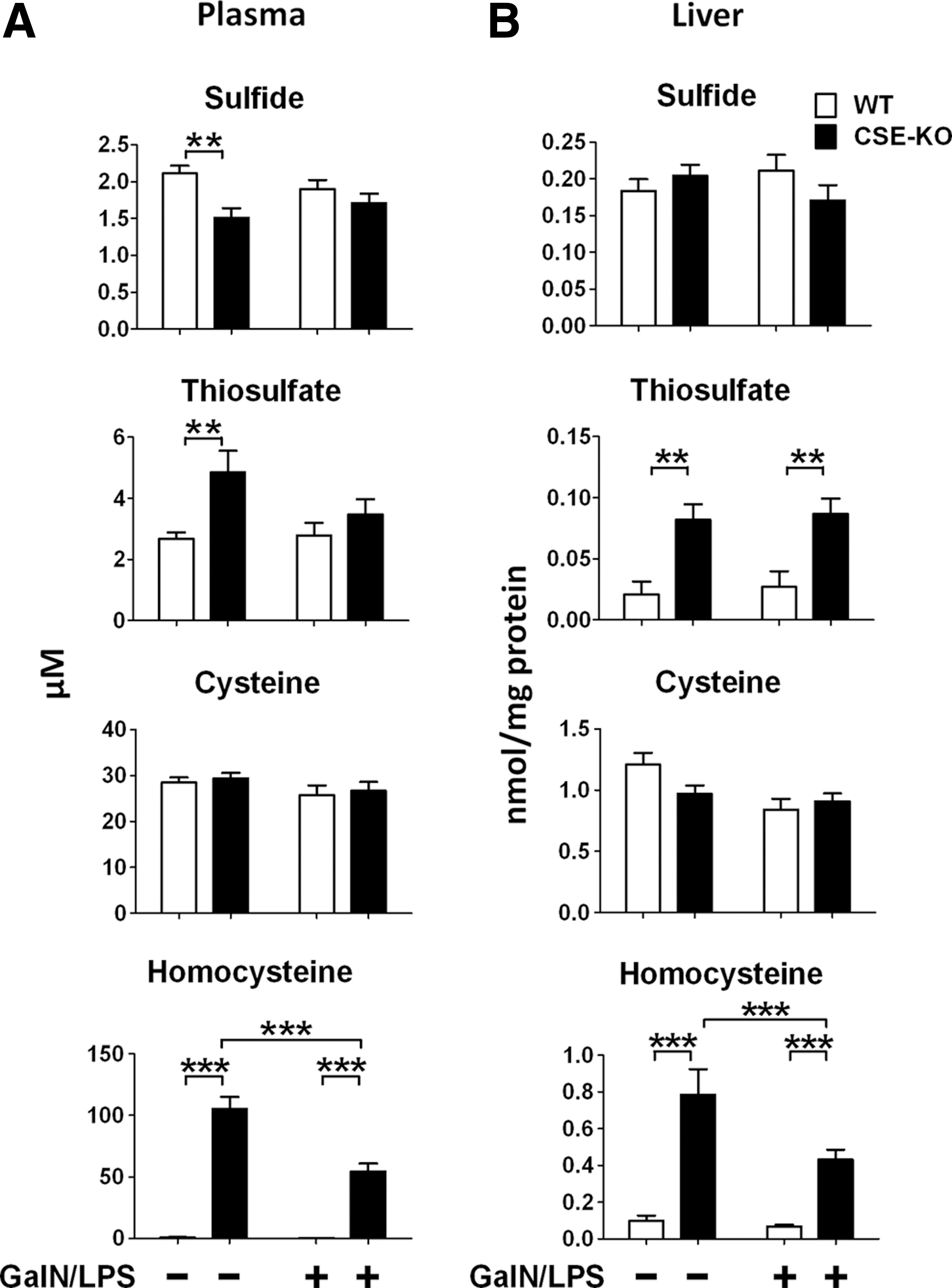
CSE deficiency affected sulfide metabolites levels in plasma and liver tissues
To elucidate the impact of CSE deficiency and/or GalN/LPS on sulfide metabolism, we measured plasma and liver concentrations of sulfide, thiosulfate, cysteine, and homocysteine (Hcy) with high-performance liquid chromatography (HPLC) after derivatization with monobromobimane (MBB) as sulfide dibimane (SDB), thiosulfate bimane (TSB), cysteine bimane (CB), and homocysteine bimane (HB), respectively (21, 28). As previously reported, plasma free sulfide levels were lower in CSE-deficient than in WT mice at baseline. On the other hand, CSE-deficient mice had similar levels of plasma sulfide after GalN/LPS challenge with WT mice. Liver sulfide levels did not differ between CSE-deficient and WT mice without or with GalN/LPS challenge (Fig. 5A, B). Interestingly, CSE deficiency increased plasma thiosulfate levels at baseline and liver thiosulfate levels at baseline and after GalN/LPS challenge (Fig. 5A, B). Cysteine levels were not affected by CSE deficiency in both plasma and liver without or with GalN/LPS challenge (Fig. 5A, B). Hcy was markedly increased by CSE deficiency in both plasma and liver with or without GalN/LPS challenge (Fig. 5A, B).
CSE deficiency upregulated SQR and 3MST
To elucidate the mechanisms responsible for the altered sulfide metabolism at baseline and after GalN/LPS challenge in CSE-deficient mice, we examined protein levels of rhodanese, ETHE1, SUOX, SQR, 3MST, CBS, and CSE in the liver at baseline and 6 h after GalN/LPS challenge. Rhodanese, ETHE1, and SUOX were not altered by CSE deficiency at baseline and after GalN/LPS (Supplementary Fig. S1; Supplementary Data are available online at

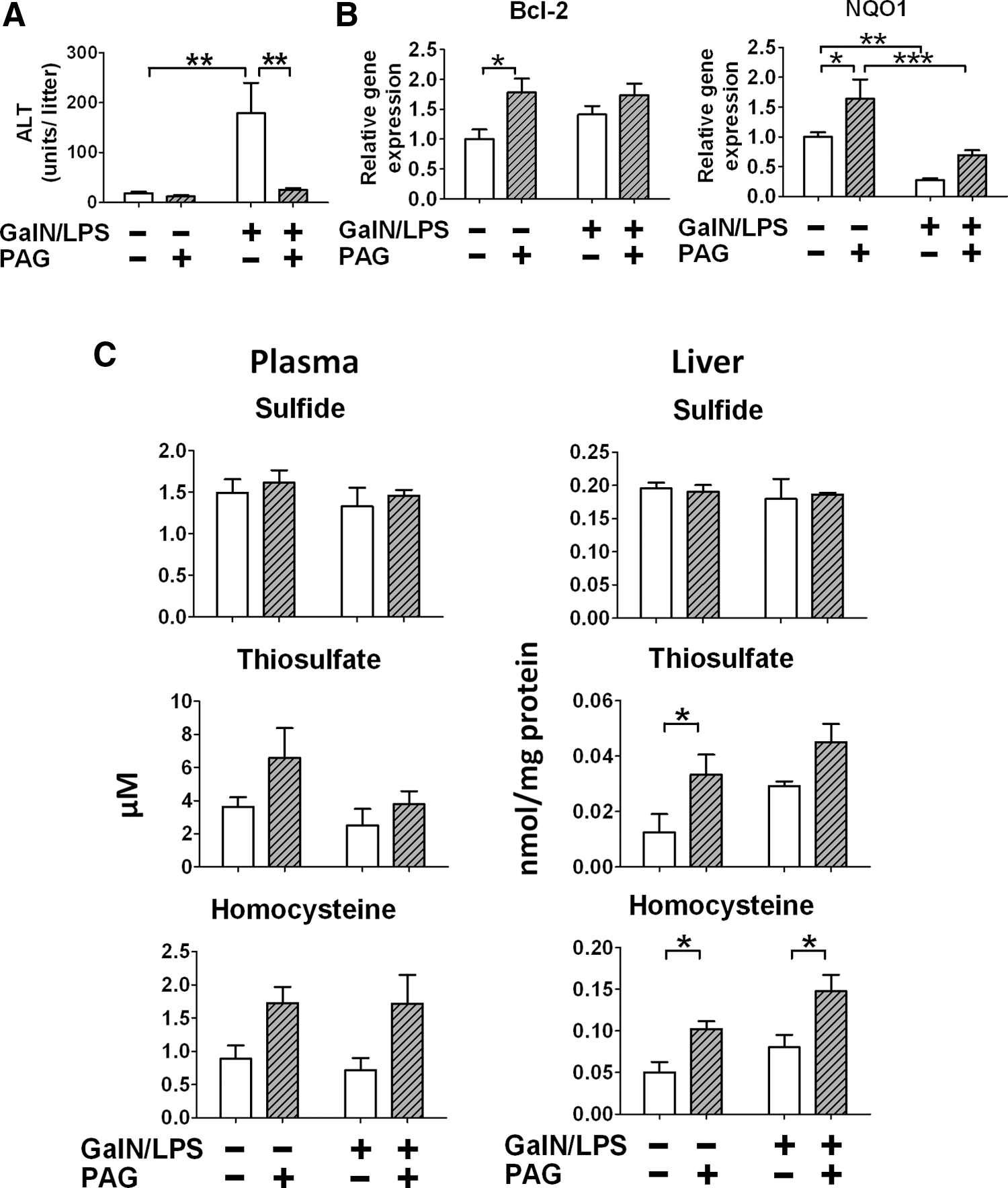
PAG attenuated GalN/LPS-induced liver injury, up-regulated antiapoptotic and antioxidant gene expression, and altered sulfide metabolism in plasma and liver tissues
To determine whether or not the protective phenotype of CSE-deficient mice is caused by compensatory mechanisms developed by the life-long deficiency of CSE, effects of acute pharmacological inhibition of CSE on GalN/LPS-induced acute hepatitis were examined. Administration of PAG, a moderately selective CSE inhibitor, markedly prevented GalN/LPS-induced increase of plasma ALT level in WT mice (Fig. 7A). Levels of Bcl-2 and NQO1 mRNA in the liver were increased by administration of PAG without GalN/LPS challenge and tended to be higher at 6 h after GalN/LPS challenge after administration of PAG (Fig. 7B). Administration of PAG did not alter sulfide levels in plasma and liver. In contrast, liver thiosulfate levels markedly increased at 6 h after administration of PAG. Similarly, liver Hcy levels markedly increased at baseline and 6 h after GalN/LPS challenge after administration of PAG. These observations suggest that acute inhibition of CSE activity, and congenital deficiency of CSE, prevents GalN/LPS-induced liver injury possibly via altering sulfide metabolism and upregulating antioxidant defense.
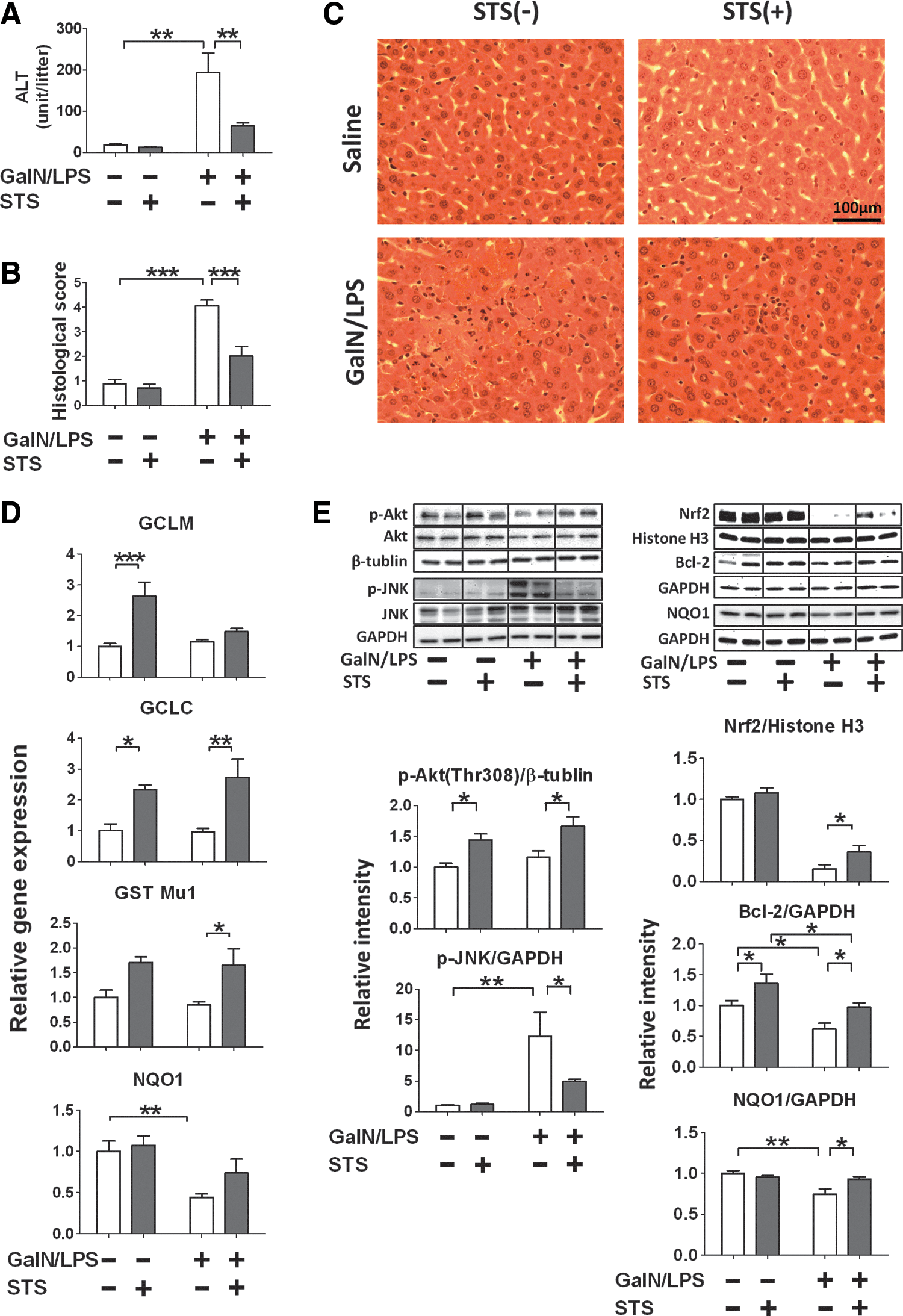
Administration of sodium thiosulfate attenuated liver injury and JNK phosphorylation and increased Akt phosphorylation, Nrf2, NQO1, and Bcl-2 after GalN/LPS
Since beneficial effects of congenital CSE deficiency and acute CSE inhibition after GalN/LPS challenge were associated with markedly increased thiosulfate levels, we hypothesized that thiosulfate prevents GalN/LPS-induced liver injury. Administration of sodium thiosulfate (STS) attenuated GalN/LPS-induced increase of plasma ALT levels and histological liver injury in WT mice (Fig. 8A–C). Administration of STS upregulated expression of several Nrf2-dependent antioxidant proteins at baseline and/or after GalN/LPS challenge (Fig. 8D). Further, beneficial effects of STS were associated with increased phosphorylation of Akt, augmented nuclear translocation of Nrf2, inhibition of JNK phosphorylation, and expression of NQO1 and Bcl-2 at 6 h after GalN/LPS challenge in the liver (Fig. 8E). As expected, administration of STS markedly increased plasma and liver thiosulfate levels at 6 h after STS administration with or without GalN/LPS challenge. Sulfide levels were higher only in plasma at 6 h after STS administration with or without GalN/LPS compared with control. Plasma and liver Hcy levels were not altered by STS administration. These observations suggest that protective effects of acute or chronic CSE deficiency on ALF are at least in part mediated by augmentation of thiosulfate levels (Supplementary Fig. S2).
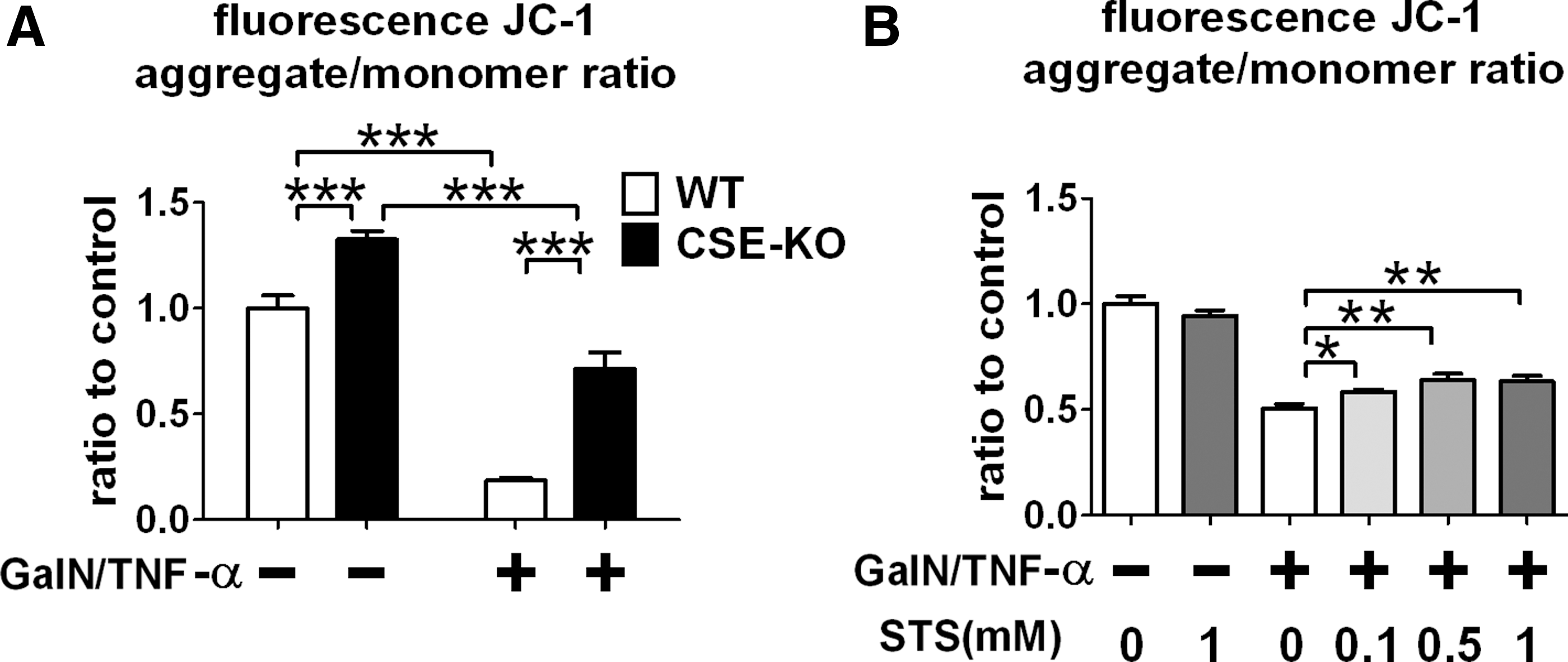
CSE deficiency or administration of STS inhibited decrease of mitochondrial transmembrane potential induced by GalN/TNF-α in primary hepatocytes
To determine the impact of CSE deficiency or thiosulfate on mitochondrial function, we examined whether or not CSE deficiency or administration of STS prevents reduction of mitochondrial transmembrane potential induced by GalN/TNF-α in primary hepatocytes. CSE deficiency inhibited reduction of mitochondrial transmembrane potential at 20 h after GalN/TNF-α challenge, as assessed by JC-1 (Fig. 9A). Similarly, administration of STS dose-dependently prevented the GalN/TNF-α-induced decrements of mitochondrial transmembrane potential compared to the control.
Discussion
We recently reported that breathing low concentration of H2S (80 ppm) restored LPS-induced reduction of plasma sulfide levels and prevented LPS-induced mortality and hepatic inflammation in mice. To further elucidate the role of physiological levels of H2S on inflammatory liver injury, in the current study, we examined the impact of CSE deficiency on GalN/LPS-induced ALF model in mice. Given the anti-inflammatory effects of H2S and robust expression of CSE in the liver, we hypothesized that CSE deficiency would worsen GalN/LPS-induced liver injury. To our surprise, congenital CSE deficiency markedly attenuated inflammation and apoptosis, protected hepatic architecture, and improved survival of mice after GalN/LPS challenge. We confirmed the salutary effects of CSE deficiency on the GalN/LPS-induced liver injury by demonstrating the protective effects of PAG on liver function after GalN/LPS challenge in WT mice. These observations suggest that beneficial effects of CSE deficiency are not due to unknown compensatory changes associated with life-long deficiency of CSE, but to the lack of CSE activity itself.
Galactosamine is metabolized in the liver leading to depletion of uridine nucleotides, hepatic transcriptional blockade, and hepatic necrosis. Addition of small doses of LPS and resultant production of TNF-α by immune cells significantly potentiates the hepatotoxicity of GalN. To determine cells responsible for the protective effects of CSE deficiency, we examined primary peritoneal macrophages and hepatocytes harvested from WT mice and CSE-deficient mice. While CSE deficiency did not affect LPS-induced TNF-α production by primary peritoneal macrophages, CSE-deficient primary hepatocytes were protected from GalN/TNF-α-induced cell death. These results suggest that CSE deficiency in hepatocytes prevents ALF leading to the improved survival of mice.
To elucidate the mechanisms responsible for the hepatoprotection observed in CSE-deficient mice against ALF, we measured levels of sulfide metabolites in plasma and liver homogenates. We confirmed that free plasma sulfide levels are lower in CSE-deficient mice than in WT mice at baseline, as previously reported (16, 33). However, plasma sulfide levels did not differ between two genotypes after GalN/LPS challenge. Free sulfide levels in liver tissue homogenates were similar in the two genotypes without or with GalN/LPS challenge. The maintained sulfide levels in CSE-deficient mice liver may be due to the compensatory upregulation of 3MST in the liver. H2S may also be produced from Hcy and cysteine catalyzed by CBS (19). It is important to note that CSE-deficient mice used in the current studies were backcrossed to a C57BL/6J genetic background using Max-Bax marker-assisted accelerated backcrossing (Charles River Laboratories) (14). This breeding strategy yielded congenic mice that carried the mutation (i.e., CSE deficiency) and were otherwise greater than 99.9% genetically identical to inbred C57BL/6J mice. In contrast, CSE-deficient mice used in the previous studies were on a mixed C57BL/6J x 129SvEv background (34). Less marked reduction of free sulfide levels observed in the current study compared with previous studies are likely due to the difference in the genetic background of CSE-deficient mice between these studies.
While CSE deficiency did not alter liver sulfide levels, Hcy and thiosulfate levels were markedly increased in CSE-deficient mice compared with WT mice with or without GalN/LPS challenge. Although the hyperhomocysteinemia (HHcy) in CSE-deficient mice observed in the current study is consistent with previous reports (16), this is the first report of increase of thiosulfate levels induced by CSE deficiency. Increased thiosulfate levels in CSE-deficient mice may be due to the markedly upregulated SQR expression levels in the mutants, which may accelerate conversion of persulfide to thiosulfate. In addition, H2S can be oxidized to glutathione persulfide, sulfite, sulfate, or thiosulfate without being catalyzed by SQR (9). Of note, administration of PAG also increased liver levels of Hcy and thiosulfate in WT mice suggesting that the altered sulfide metabolisms are results of inhibition of CSE activity itself. Based on these observations, we hypothesized that increased Hcy and (or) thiosulfate levels contribute to the protective effects of CSE deficiency against GalN/LPS-induced ALF.
The beneficial effects of CSE deficiency against ALF were associated with translocation of Nrf2 in the nucleus and upregulation of Nrf2-dependent signaling including antioxidant proteins NQO-1 and HO-1, and anti-apoptotic protein Bcl-2. These observations appear to be in conflict with a number of publications that suggest that protective effects of H2S are mediated by upregulating Nrf2-dependent signaling via sulfhydration (7). However, it has been reported that expression of Nrf2 depends on stress levels; low oxidative stress could increase the expression of Nrf2, whereas high oxidative stress might decrease Nrf2 and Nrf2-dependent antioxidant genes (6, 31). It was recently reported that hydrogen peroxide activates Nrf2 by inactivation of Kelch-like ECH-associated protein 1 (Keap1) via formation of an intramolecular disulfide bridge releasing inhibition of Keap1 on Nrf2 (7). The same authors also showed that Nrf2 upregulates SQR. Since HHcy is known to cause oxidative stress (35), it is conceivable that chronically elevated Hcy levels in CSE-deficient mice produced “low” oxidative stress levels activating Nrf2-dependeng signaling.
Although the life-long HHcy may have contributed to the hepatoprotective phenotype of CSE-deficient mice, administration of PAG, at the dose that prevented ALF, only transiently and modestly increased Hcy levels. Therefore, the beneficial effects of transient CSE inhibition by PAG cannot be attributed to the cytoprotective signaling induced by the persistent oxidative stress associated with chronic HHcy (Supplementary Fig. S3). On the other hand, both CSE deficiency and PAG increased liver thiosulfate levels. To determine whether or not elevated thiosulfate levels per se confer beneficial effects after GalN/LPS challenge, we examined effects of administration of STS. We found that STS markedly prevented liver injury, enhanced Akt phosphorylation and Bcl-2 expression, and induced several Nrf2-dependent antioxidant proteins. Similar to CSE deficiency, administration of STS markedly inhibited GalN/LPS-induced phosphorylation of JNK. JNK phosphorylation is induced by extracellular stresses and inflammatory cytokines (27) and counteracts the antiapoptotic effects of Bcl-2 (12, 24). The observations that CSE deficiency and STS administration prevented GalN/TNF-α-induced reduction of mitochondrial transmembrane potential further support the anti-apoptotic effects of STS. While administration of STS increased sulfide levels in plasma and thiosulfate levels in plasma and liver with or without GalN/LPS challenge, liver levels of sulfide and Hcy were not affected by STS. These observations suggest that increased thiosulfate levels per se conferred cytoprotective effects in the liver (Fig. 10).
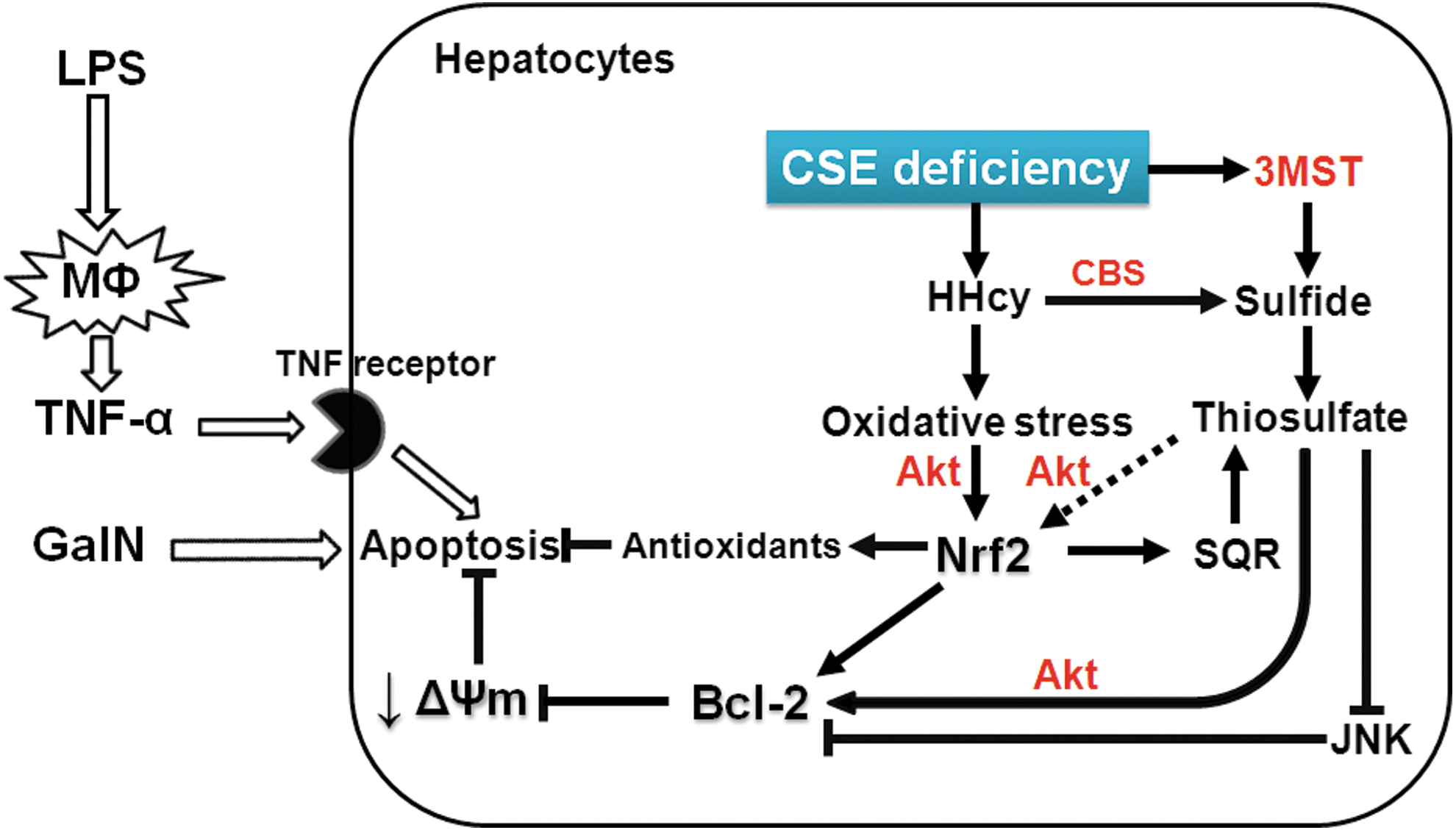
The current observations confirm our previous results that administration of STS prevented death from endotoxin shock in mice. It is of interest to note that two apparently opposing strategies, inhalation of H2S in the former (28) and CSE deficiency in the current study, confer similar hepatoprotective effects at least via increasing thiosulfate levels. Taken together, these observations highlight an important biological role of thiosulfate as a potentially cytoprotective intermediate of sulfide metabolism. Since STS is clinically approved for the treatment of cyanide toxicity and is widely available, our observations have important translational potential. Further studies are warranted on the protective effects of STS in inflammatory liver injury and sepsis.
Materials and Methods
Animals and ALF model
After approval by the Massachusetts General Hospital Subcommittee on Research Animal Care, all animal experiments were performed in accordance with the guidelines of the National Institutes of Health. Male WT mice (C57BL/6J, 8–12 week old) were purchased from the Jackson Laboratory and male CSE-deficient mice were backcrossed to a C57BL/6J genetic background using Max-Bax marker-assisted accelerated backcrossing (Charles River Laboratories). These mice were given access to food and water ad libitum in our animal facility until the time of experiments. To induce ALF model, the mice were challenged with D-Galactosamine (GalN, 300 mg/kg; Sigma-Aldrich) and LPS (1 μg/kg, Escherichia coli; Sigma) intraperitoneally. Control mice received normal saline at the same volume with GalN/LPS. To examine the effects of thiosulfate, in separate groups of mice, STS (2 g/kg; American Regent, Inc.) solution was administered intraperitoneally at 30 min before and 3 h after GalN/LPS challenge in WT mice. Similarly, to examine the effect of PAG (50 mg/kg; Sigma-Aldrich), an irreversible inhibitor of CSE, PAG was intraperitoneally administered at 30 min before GalN/LPS challenge in WT mice.
Survival analysis after GalN/LPS challenge
Survival after challenge with GalN/LPS was studied in age-matched male mice. To avoid dehydration, normal saline (1 ml) was intraperitoneally given to all mice at times 0, 6, and 24 h after GalN/LPS challenge. We determined the dose of GalN/LPS that was lethal in 40% of WT mice in our pilot studies. Ambient temperature was controlled at 25°C.
Plasma liver enzyme
Blood was drawn from inferior vena cava and centrifuged to collect the plasma. Plasma ALT level, as markers of hepatic damage, at baseline and 6 h after GalN/LPS injections were measured using an Infinity™ ALT assay reagent (Thermo Fisher Scientific, Inc.) according to the manufacturers' instruction.
Histology
Liver tissues were dissected and fixed in 4% paraformaldehyde and embedded in paraffin. Five micrometer sections were stained with hematoxylin and eosin using a standard protocol and then analyzed with a fluorescence microscopy (Nikon ECLIPSE-TE2000-S). Sections were scored by an investigator blinded as to the identity of the samples. Sinusoidal congestion, cytoplasmic vacuolization, and parenchymal necrosis were scored according to the criteria described by Suzuki et al. (2, 26).
TNF-α induced by primary peritoneal macrophages
Peritoneal macrophages were prepared from unstimulated WT and CSE-deficient mice. Cells were plated in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 100 U/ml penicillin and streptomycin, and 2 mM glutamine. After overnight incubation, WT macrophages were treated with LPS (0.5 μg/ml) for different durations, and TNF-α levels in the medium were measured by ELISA kit (R&D System) according to the manufacturer's instruction. TNF-α levels were compared between WT and CSE-deficient macrophages after incubation with LPS for 6 h since TNF-α production reached a plateau after 6 h in WT macrophages.
Hepatocyte isolation and measurements of viability
Hepatocytes were isolated from mice using a modified two-step collagenase perfusion method (22). Briefly, after the cannulation of the portal vein, liver was perfused with Krebs-Ringer bicarbonate (KRB) solution containing EDTA, and then followed by KRB solution containing collagenase (Worthington) and CaCl2. Isolated cells were filtered through 100 and 70 μm nylon cell strainer (BD Falcon). Hepatocytes were purified by centrifugation (600 rpm×10 min) in 50% Percoll (GE Healthcare Bioscience) solution. The Trypan Blue method was applied to confirm cellular viability (>85%). The viable primary hepatocytes were suspended in High Glucose DMEM with 10% heat-inactivated fetal bovine serum. To measure hepatocyte viability after GalN (5 mM)/TNF (10 ng/ml) challenge, we used LDH assay (LDH Cytotoxicity Detection Kit; Roche) and CV assay, as described previously (17).
Measurements of gene expression
Total RNA was extracted from the liver tissues of mice 6 h after saline or GalN/LPS challenge using the RNAspin Mini kit (GE Healthcare Bioscience) and cDNA was synthesized using MMLV-RT (Promega). TNF-α, IL-6, IL-1β, NOS2, NQO1, GCLC, GST Mu1, GCLM, CSE, CBS, 3MST, SQR, NOX2, p67phox, and 18S ribosomal RNA transcript levels were measured by real-time PCR using a Realplex 2 system (Eppendorf North America). The primer sequences used were as follows: NOX2 (5′-AAGGAGTGCCCAGTACCAAAG-3′, 5′-CCGAACCAACCTCTCACAAAGG-3′), p67phox (5′-GTCG GCTGTTCCGTCCAAAT-3′, 5′-GAAAAGTTGTCTTGGTG AACCAC-3′) and Bcl-2 (5′-AGCGTCAACAGGGAGATGT CAC-3′, 5′-GGGCCATATAGTTCCACAAAGGC-3′), and the others were reported previously (10, 28). Changes in the relative gene expression normalized to levels of 18S rRNA were determined using the relative CT method. The mean value of samples from control WT was set as 1.
Protein levels
Liver tissue was dissected and frozen 6 h after saline or GalN/LPS injections. Nuclear and cytoplasm fractions were prepared by using a Nuclear Extraction Kit (Active Motif) according to the manufacturer's protocol. Protein levels in liver homogenates were determined using standard immunoblot techniques using primary antibodies (1:10,000; Cell Signaling Technology, Inc., unless otherwise noted) against Nrf2 (1:200; Santa Cruz Biotechnology), SQR (1:500; Abcam), ETHE1 (1:10,000; GeneTex), rhodanese (1:10,000; GeneTex), phosphorylated Akt at Thr308 (1:2000; Cell Signaling Technology, Inc.), Akt (1:5000; Cell Signaling Technology, Inc.), phosphorylated JNK at Thr183 and Tyr185 (1:1000; Cell Signaling Technology, Inc.), JNK (1:5000; Cell Signaling Technology, Inc.), Bcl-2 (1:1000; Cell Signaling Technology, Inc.), NQO1 (1:500; Cell Signaling Technology, Inc.), 3MST (1:1000; Sigma), CBS (1:2000; Santa Cruz Biotechnology), caspase 3, cleaved PARP, GAPDH, Histon H3, SUOX, and β-tublin. Bound antibody was detected with a horseradish peroxidase-linked antibody directed against rabbit IgG (1:10,000; Cell Signaling Technology, Inc.) and was visualized using chemiluminescence with ECL Advance kit (GE Healthcare Bioscience).
Sulfide, thiosulfate, cysteine, and homocysteine concentration in plasma and liver
Concentrations of free sulfide, thiosulfate, cysteine, and homocysteine in plasma and liver at 6 h after GalN/LPS challenge were measured by HPLC after derivatization with excess MBB as stable products SDB, TSB, CB, and HB, respectively, as previously described (21, 23, 32). Briefly, at 6 h after saline or GalN/LPS injections in WT and CSE-deficient mice, blood was drawn from retro-orbital vein and a small piece of the right lobe of liver was dissected. Blood was centrifuged to collect the plasma, and liver tissue was homogenized. Thirty microliters of plasma and liver supernatant were added to 70 μl of 100 mM Tris-HCl buffer (pH 9.5, 0.1 mM diethylenetriamine pentaacetic acid [DTPA]), followed by addition of 50 μl of 200 mM 5-sulfosalicylic acid after 30 min, and the mixture was centrifuged and supernatant was analyzed by HPLC with a fluorescence detector as described previously (28). We also examined sulfide, homocysteine, and thiosulfate levels in plasma and liver at 6 h after PAG or STS administration using the same method (Supplementary Fig. S3).
Measurement of mitochondrial transmembrane potential in primary hepatocytes
To measure mitochondrial transmembrane potential after GalN (5 mM)/TNF (10 ng/ml) challenge in primary hepatocytes, we used the 5,5,6,6,-tetrachloro-1,1,3,3,-tetraethylbenzimidazole carbocyanide iodine (JC-1) mitochondrial membrane potential assay kit (Cayman Chemical Company), according to the manufacturer's instruction. After incubated with GalN/TNF-α, isolated primary hepatocytes were stained by JC-1 for 15 min at 37 C°. To examine the effects of thiosulfate, STS (0.1–1 mM) was added to the culture medium at 1 h before GalN/TNF-α challenge in WT hepatocytes. JC-1 forms aggregates in healthy cells with high mitochondrial transmembrane potential. On the other hand, in apoptotic or unhealthy cells with low mitochondrial transmembrane potential, JC-1 remains as monomer. Fluorescence of JC-1 (aggregates; excitation 560 nm, emission 595 nm) and (monomer; excitation 485 nm, emission 535 nm) were detected with a fluorescence plate reader. We used the ratio of fluorescent intensity of aggregates to that of monomers as an indicator of mitochondrial transmembrane potential (3, 11, 25).
Statistics
All data are presented as mean±SE. Data were analyzed by ANOVA using Sigmastat 3.01a (Systat Software, Inc.) and Prism 5 software package (GraphPad Software). Newman–Keuls multiple comparison post hoc test or Bonferroni post hoc test were respectively performed for one-way ANOVA or two-way ANOVA. Kaplan–Meier survival analysis was performed using Log-rank test. p-Values smaller than 0.05 were considered significant.
Footnotes
Acknowledgment
This work was supported by National Institutes of Health grant HL101930 to F. Ichinose.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.