
Abstract
Vicinal disulfide bridges, in which a disulfide bond is formed between adjacent cysteine residues, constitute an unusual but expanding class of potential allosteric disulfides. Although vicinal disulfide rings (VDRs) are relatively uncommon, they have proven to be functionally critical in almost all proteins in which they have been discovered. However, it has proved difficult to test whether these sterically constrained disulfides participate in functionally important redox transformations. We demonstrate that chemical replacement of VDRs with dicarba or diselenide bridges can be used to assess whether VDRs function as allosteric disulfides. Our approach leads to the hypothesis that not all VDRs participate in functionally important redox reactions. Antioxid. Redox Signal. 19, 1976–1980.
Introduction
A novel but growing class of potential allosteric disulfides are vicinal disulfide bonds, in which a bridge is formed between adjacent cysteine residues. Although the formation of a vicinal disulfide ring (VDR) is relatively uncommon, it is functionally important in those proteins that possess it (2). An unusual feature of VDRs is that the steric constraints imposed by formation of an eight-membered ring typically yields a high dihedral strain energy for the disulfide bridge, forcing the backbone amide bond between the two covalently linked cysteine side chains to be non-planar (8). As a result, it has been proposed that VDRs might function as redox-activated conformational switches that enable proteins to switch between active and inactive forms depending on the redox state of the VDR (2). However, due to the difficulty of controlling the redox state of cysteine residues in biological systems, this hypothesis has been experimentally validated for only a limited number of VDRs.
Innovation
This study revealed that the functional importance of some vicinal disulfide rings (VDRs) is not due to their participation in redox transformations. Nevertheless, some VDRs do serve as allosteric disulfides and the chemical approaches outlined here provide a convenient means of distinguishing between these possibilities. The introduction of dicarba linkages is likely to be restricted to small proteins amenable to solid-phase peptide synthesis or expressed protein ligation (7). In contrast, various strategies have been developed for facile incorporation of Sec into synthetic and recombinant proteins (6); this approach should therefore find broad utility for testing the redox chemistry of VDRs and other putative allosteric disulfides.
We sought to develop a general chemical strategy for testing whether VDRs can serve as redox switches. κ-hexatoxin-Hv1c (Hv1c), a 37-residue cysteine-rich peptide found in venom of the lethal Australian funnel-web spider Hadronyche versuta (8), was used as a model system. Hv1c is the prototypic member of a family of excitatory neurotoxic peptides that are promising bioinsecticide leads due to their selective activity on insect ion channels (4).
Hv1c contains four disulfides, three of which form an inhibitor cystine knot motif that is commonly found in spider toxins (Fig. 1A). The remaining two Cys residues form a VDR between residues Cys13 and Cys14 that is essential for the toxin's insecticidal activity and its potent block of insect large-conductance calcium-activated potassium (BKCa) channels (8). Mutation of Cys13 and Cys14 to Ser or Ala virtually abolishes activity (5, 8). However, the VDR is not structurally important as replacement of Cys13 and Cys14 with isosteric Ser residues does not perturb the toxin's conformation (8). Thus, the VDR in Hv1c must have an important functional role.
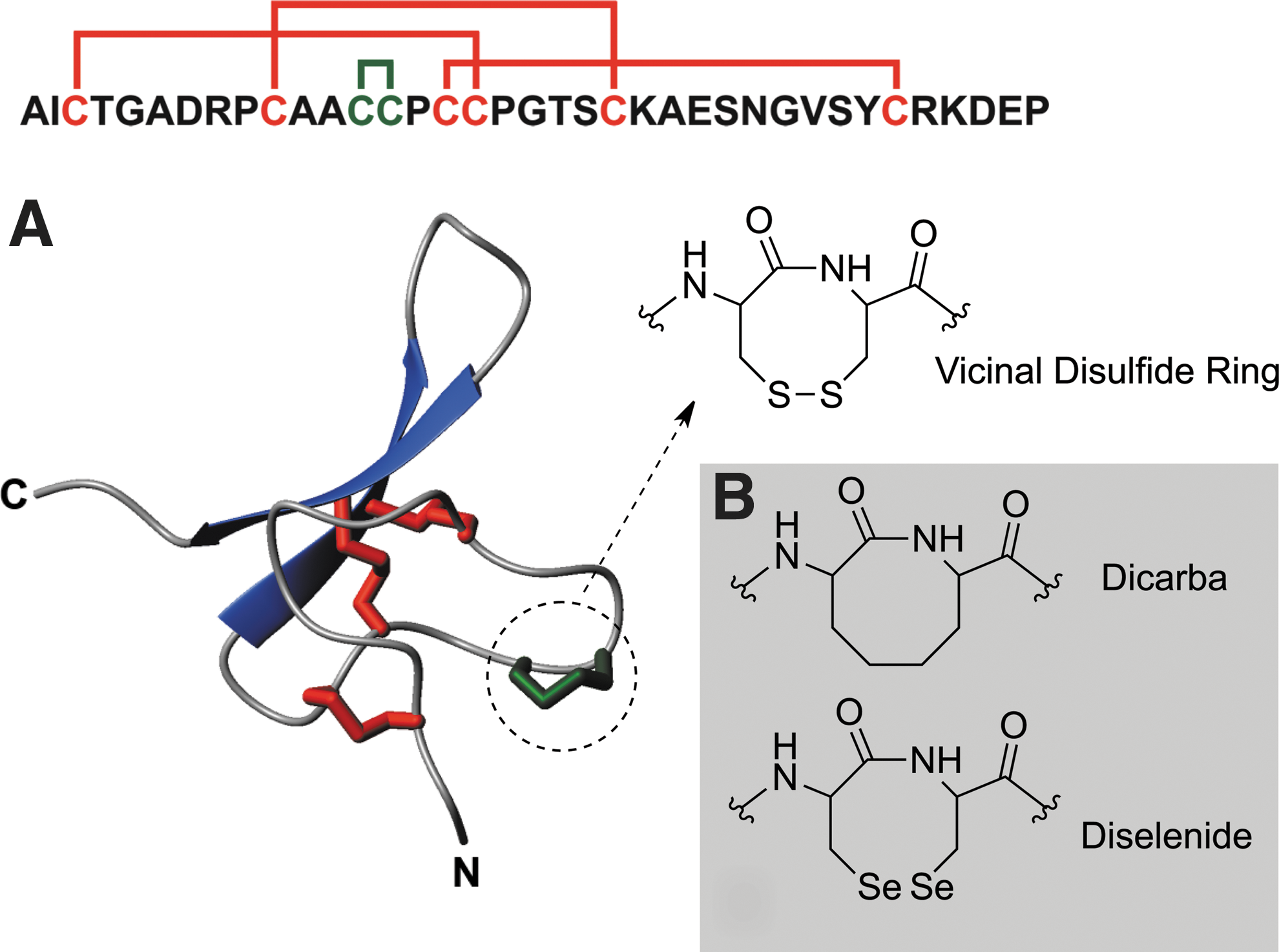
The Cys13–Cys14 peptide bond in Hv1c assumes a distorted trans conformation (ω=–145) (8) but the dihedral strain energy of 15.5 kJ·mol–1 calculated for the VDR is not particularly high, and it is in fact lower than for the Cys10–Cys22 disulfide (22 kJ·mol–1). However, the dihedral strain energy calculated for the Cys13–Cys14 VDR in Hv1c is similar to that calculated for the redox-active Cy370–Cys371 VDR in human transglutaminase 2 (14 kJ·mol–1) (3). Thus, it is unclear whether the VDR in Hv1c is redox-active or whether the VDR itself constitutes a special recognition site that is critical for receptor binding. To distinguish between these two hypotheses, we designed a series of Hv1c analogues in which the native vicinal disulfide (S-S) was replaced by more stable ethylene (dicarba; CH2-CH2) or diselenide (Se-Se) linkages (Fig. 1B). These modifications were designed to abrogate possible redox reactions of the VDR while still maintaining its shape and stereochemistry.
Disulfide bonds have been replaced with nonreducible ethylene bridges in several bioactive peptides. Despite significant differences in covalent geometry between the two connectivities, the ethylene substitution is often well tolerated, resulting in analogues with greater chemical stability. Diselenides are excellent surrogates for disulfide bonds (6), but their much lower redox potential (−380 mV for Se–Se versus −180 mV for S–S at pH 7) makes them less susceptible to reduction by oxidoreductases.
Results and Discussion
We prepared native and diselenide Hv1c by Boc-mediated solid-phase peptide synthesis (SPPS) and then oxidatively folded the peptides as described (6). The diselenide toxin was built by incorporating Sec residues at positions 13 and 14 during peptide assembly on resin (Fig. 2), with the VDR formed upon HF cleavage of the peptide from the solid support.

For synthesis of dicarba Hv1c, the eight-membered lactam ring was constructed prior to peptide assembly on solid support (Fig. 2). The cyclic dipeptide
The bioactivity of native Hv1c and the two VDR analogues was compared by measuring their ability to block BKCa currents (I K(Ca)) in cockroach neurons at the IC50 (3 nM) previously determined for block of peak I K(Ca) current by native Hv1c (6). All toxins caused similar inhibition of I K(Ca), namely 49%±4%, 60%±4%, and 57%±4% for native, diselenide, and dicarba Hv1c respectively (Fig. 3A); one-way ANOVA followed by a Bonferroni's post-hoc test revealed no significant differences in the extent of current block (p>0.1; n=4). We also tested the insecticidal activity of the toxins by injection into blowflies. The diselenide analogue was slightly more potent than the native toxin, whereas dicarba Hv1c was only slightly less potent (Fig. 3B). We conclude that the diselenide and dicarba variants of Hv1c have similar insecticidal activity and produce the same degree of BKCa channel inhibition as the native toxin.
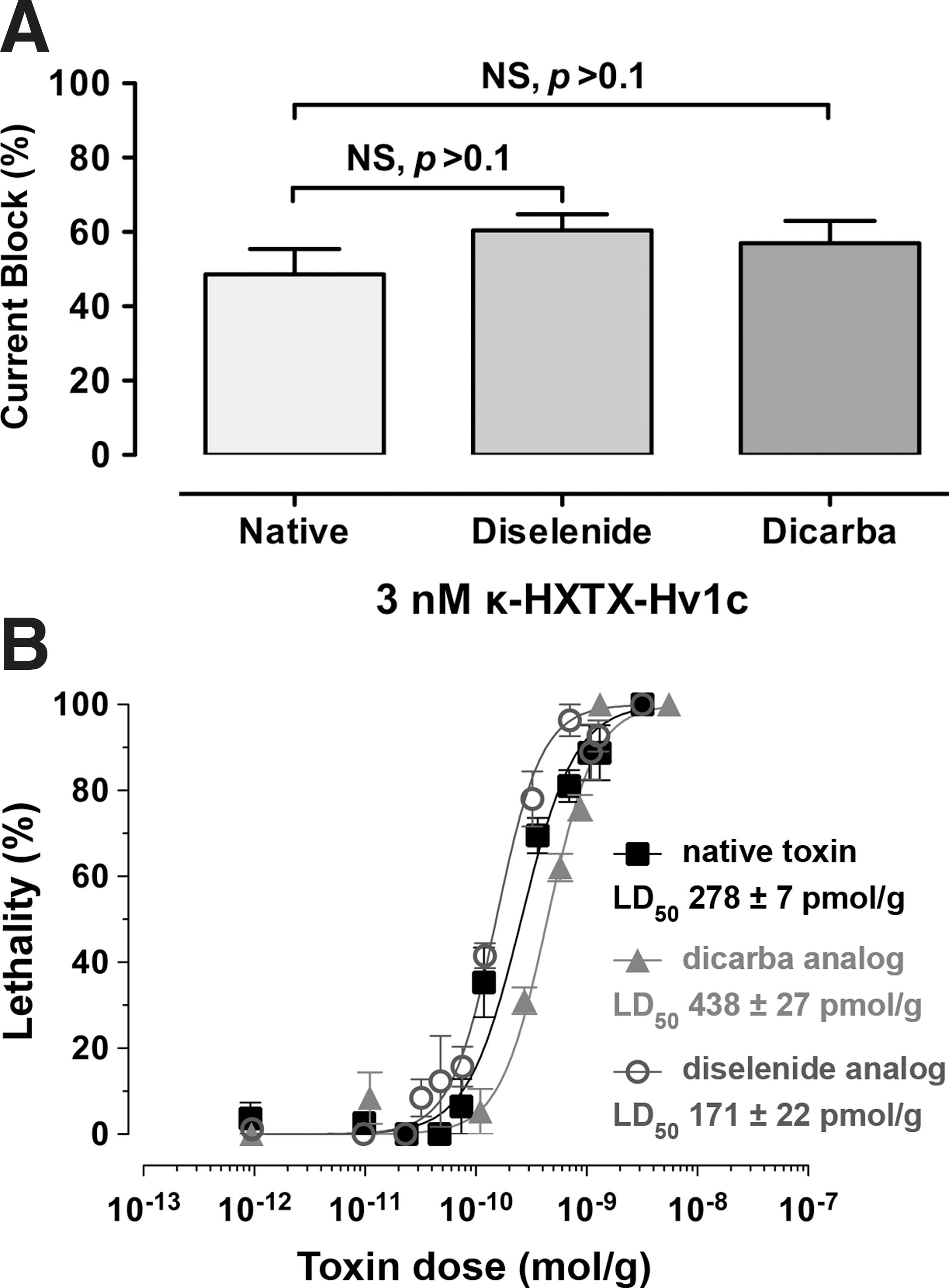
Since replacement of the vicinal disulfide bond in Hv1c with more redox-stable diselenide and ethylene linkages does not affect toxin activity, we can eliminate the possibility that the biological function of the toxin depends on the ability of the VDR to participate in redox transformations. It also eliminates other potential functions of the VDR such as metal binding. Thus, the VDR in Hv1c must serve some other function; the structure-activity relationship data (5) suggest that the ring structure forms part of the toxin pharmacophore and makes a direct, noncovalent interaction with its molecular target. However, the VDR does not simply serve as a nonspecific, hydrophobic protein–protein interaction epitope since replacement of Cys13 and Cys14 with either Ala or Val residues results in a mutant toxin with >400-fold lower activity (5). Rather, the increased potency of the diselenide analogue relative to native Hv1c in blocking KCa currents (IC50 of 1.5 nM vs. 3.5 nM for native toxin) (6) and in killing flies (Fig. 3) suggests that the VDR participates in a precisely configured hydrophobic interaction.
Notes
Synthesis of the dipeptide building block 3
The dialkylglycine precursor
NMR and MS data for dipeptide building block 3
1H-NMR (CDCl3, 600 MHz): δ 0.75–0.95 (m, 1H, CH2), 1.41 (s, 9H, (CH3)3), 1.25–1.55 (m, 2H, CH2), 1.65–1.69 (m, 1H, CH2), 1.72–1.77 (m, 1H, CH2), 1.81–1.85 (m, 1H, CH2), 1.90–1.95 (m, 1H, CH2), 2.06–2.09 (m, 1H, CH2), 3.73 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 4.53–4.61 (m, 2H, CH2), 4.72 (m, 1H, CH), 4.75 (dd, 1H, J=3.2, 12.6 Hz, CH), 6.00 (d, 1H, J=7.4 Hz, NH), 6.35 (d, 1H, J=2.0 Hz, Ar), 6.37 (d, 1H, J=8.4 Hz, Ar), and 7.27 (d, 1H, J=8.8 Hz, Ar) ppm. 13C-NMR (CDCl3, 150 MHz): δ 22.9, 24.3, 28.3, 30.8, 37.9, 40.9, 51.8, 55.0, 55.2, 57.2, 80.1, 98.0, 103.9, 117.6, 130.2, 155.5, 157.6, 159.9, 172.4, and 173.8 ppm. HR-MS: calcd for [M+H]+: 437.2210, found 437.2289.
Peptide synthesis
Native Hv1c and analogues were prepared by manual SPPS using O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) as the activation reagent and the stepwise in situ neutralization protocol for Boc chemistry. The side-chain protecting groups used were Cys(MeBzl), Thr(Bzl), Asp(OcHex), Arg(Tos), Ser(Bzl), Lys(ClZ), Glu(OcHex), Asn(Xan), and Tyr(BrZ). Coupling at positions 13 and 14 involved two successive standard coupling steps of Boc-Cys(MeBzl)-OH for native toxin; two successive standard coupling steps of Boc-Sec(MeBzl)-OH for the diselenide analogue; and one coupling step of dipeptide building block
Linear peptides were folded/oxidized by dissolving 25 mg of pure peptide in 20 ml of buffer A and adding dropwise to 250 ml of oxidation buffer (0.1 M 3-(N-morpholino)propanesulfonic acid [MOPS] pH 7.3, 0.2 M KCl, 1 mM EDTA, 2 mM reduced glutathione, and 0.5 mM oxidized glutathione). The reaction mixture was stirred at RT for 12 h and quenched by adding a few drops of TFA. The mixture was diluted two-fold with water and the native disulfide isomer was purified by RP-HPLC using a preparative C18 column (Vydac) and a linear gradient of 5%–20% buffer B over 30 min followed by an isocratic flow of 20% buffer B over 10 min at a flow rate of 8 ml/min. Peptide identity was verified by ESI MS: native ([M]+ found: 3757.4, expected: 3756.4), diselenide ([M]+ found: 3851.6, expected: 3852.3), and ethylene ([M]+ found: 3721.0, expected: 3720.6).
Linear and folded peptides were analyzed by RP-HPLC using an analytical Vydac C18 column on a Shimadzu LC-2010 system with a linear gradient of 5%–30% buffer B over 25 min at a flow rate of 1 ml/min. ESI-MS was used to monitor peptide oxidation and purity. The overall conformation of the native, diselenide, and dicarba toxins were shown to be very similar by comparison of their natural abundance 2D 1H-15N HSQC spectra acquired on a Bruker Avance 900 MHz spectrometer.
Electrophysiology
Dorsal unpaired median neurons were enzymatically and mechanically isolated from the terminal abdominal ganglia (TAG) of unsexed adult American cockroaches (Periplaneta americana) as previously described (4). TAGs were dissected from cockroaches and placed in normal insect saline (NIS) containing (in mM) 180 NaCl, 3.1 KCl, 10 HEPES, and 20 D-glucose. TAGs were then incubated in 1 mg/ml collagenase (type IA) at 29°C for 40 min, washed twice in NIS, then triturated through a fire-polished Pasteur pipette to dissociate individual neurons. Cells were then distributed onto 12 mm diameter coverslips precoated with 2 mg/ml concanavalin A (type IV) and topped up to a volume of ∼1 ml with NIS supplemented with 5 mM CaCl2, 4 mM MgCl2, 5% fetal bovine serum, 1% penicillin, and 1% streptomycin (NIS+). Cells were maintained in NIS+ at 25°C or 30°C and 100% humidity and used within 30 h.
Patch clamp electrophysiology was performed at ambient temperature (20°C–23°C) as described (4). External control, toxin, and I K(Ca) blocking solutions were applied to cells directly via a perfusion needle (Automate Scientific, San Francisco, CA) at ∼50 μl/min. Large-conductance BKCa channel currents (I K(Ca)) cannot be recorded in isolation from delayed-rectifier KV channel currents [I K(DR)] because there are no selective blockers of insect I K(DR). Therefore, to isolate I BK(Ca), the selective BKCa blocker iberiotoxin (30 nM) and CdCl2 (1 mM) were perfused at the conclusion of current recordings to eliminate I K(Ca), thus allowing offline subtraction of the remaining I K(DR). Toxins (3 nM) were perfused for a period of 10 min or until equilibrium was achieved prior to addition of CdCl2 and iberiotoxin to block BKCa channels.
Insecticidal activity
Insecticidal activity was quantified using sheep blowflies (Lucilia cuprina, average mass of 15–25 mg, 10 flies per dose of toxin or 20–30 flies for control). Toxins were dissolved in insect saline and injected into the ventrolateral thoracic region using a maximum volume of 2 μL per fly. Mortality was measured after 24 h. LD50 values are the mean of three experiments.
Footnotes
Acknowledgments
This work was supported by Discovery Grant DP1095728 from the Australian Research Council. We thank Dr. Geoff Brown (Queensland Department of Agriculture, Fisheries and Forestry) for supplying blowflies.