
Abstract
Introduction
T
In this review, we will give an overview on the current knowledge of ascorbic acid and SVCTs in the PNS, from cellular and molecular aspects to clinical studies.
Ascorbic Acid Function in Myelination In Vitro
In 1986, first reports on the effect of ascorbic acid on isolated SCs and their capacity to myelinate axons in vitro were published (2, 37). Baron-van Evercooren et al. (2) showed that ascorbic acid treatment induced the formation of an ECM in SC cultures. This ECM contained collagen IV, fibronectin, entactin, and heparan sulfate proteoglycan. Olsen and Bunge (37) first published the achievement of in vitro myelination of neuronal axons. They showed that long-term culture and addition of ascorbic acid to the culture medium were necessary for myelination of Salamander DRG cultures. They also showed that ascorbic acid promoted the formation of an ECM.
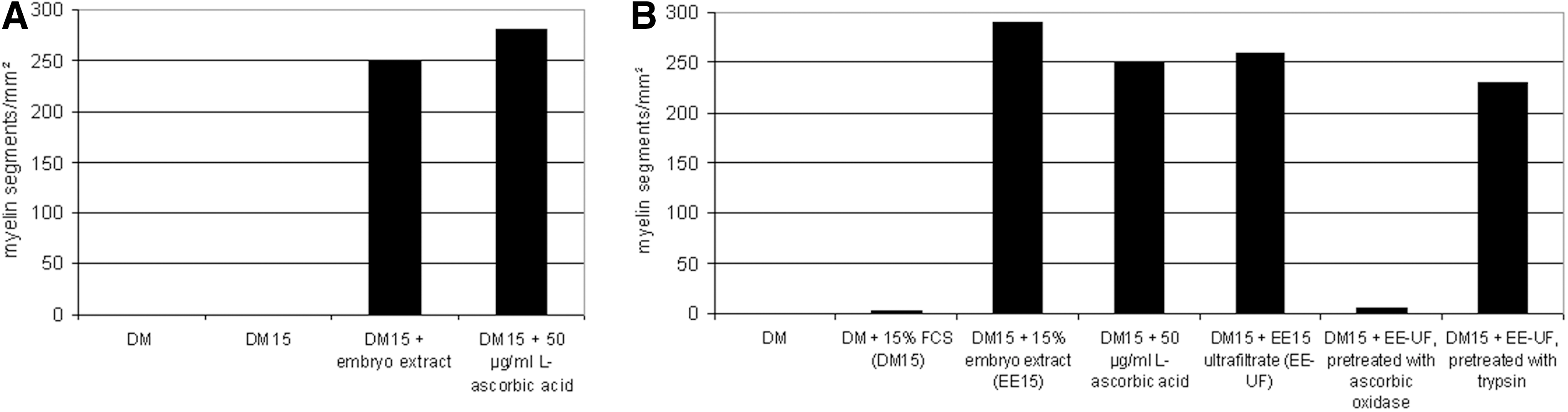
Following these observations, Eldridge et al. conducted a detailed study on the necessary components of media to promote in vitro myelination in rat SC/DRG co-cultures (14). They found that the addition of ascorbic acid was equally effective as chick embryo extract for in vitro myelination (Fig. 1A). Chick embryo extract was used at that time as an additive to allow for in vitro myelination. Ultrafiltration did not abolish the pro-myelinating effect of embryo extract, indicating that the active component was not macromolecular. Treating embryo extract with ascorbate oxidase before the addition to the medium diminished the pro-myelinating effect almost completely, while treatment with trypsin did not (Fig. 1B). This provided further evidence that the active, pro-myelinating component in embryo extract was, in fact, ascorbic acid. It should be noted, however, that the addition of fetal bovine serum was also necessary; so, apart from ascorbic acid, another serum component was also necessary for in vitro myelination. Moreover, ascorbic acid was shown to induce the formation of a collagen- and laminin-containing ECM in SC and SC/DRG co-cultures (14). In a second study, Eldridge et al. analyzed the effects that the ascorbic-acid-induced ECM had on myelination in SC/DRG co-cultures (13). They could show that the exogenous addition of an ECM was able to promote myelination to a similar degree as ascorbic acid (Fig. 2A). It was further demonstrated that the addition of laminin as an exogenous overlay allowed myelination as effectively as ascorbic acid (Fig. 2B). The authors assumed that ascorbic acid acted as a co-factor to prolyl-hydroxylase, facilitating the formation of triple-helical collagen (13). The function of ascorbic acid in formation of triple-helical collagen as co-factor of prolyl-hydroxylase has been widely accepted (3). However, it has not actually been shown that this is the precise mechanism of action of ascorbic acid in myelination in vitro.

Ascorbic Acid in Animal Models of Inherited Neuropathies
Inherited neuropathies are a large group of neuropathies that are caused by mutations in one of several known genes. Currently, more than 40 disease-causing genes for inherited neuropathies are known. The inherited sensorimotor peripheral neuropathies are pooled under the term CMT neuropathies. CMT neuropathies belong to the most common neurogenetic disorders. Prevalence rates between 1:1200 and 1:9000 have been reported, depending on the country and region studied (7, 15, 24, 44). The most common CMT neuropathy is CMT1A, which is caused by a duplication of a region on chromosome 17 containing the causative gene peripheral myelin protein 22 (PMP22) (32, 34, 50, 51). CMT1A is a severely demyelinating neuropathy leading to distal-symmetric pareses and muscle atrophies, sensory loss, and foot deformities (54). Patients with CMT1A typically develop first symptoms in the first to second decade of life. The condition is slowly progressive, often causing the need for ankle-foot-ortheses and other aids. In rare cases, patients can become wheelchair bound. Life expectancy is not shortened in CMT1A. The second to most common form of CMT is CMTX, caused by mutations in the connexin-32 gene, followed by CMT1B and CMT2A1, caused by mutations in myelin protein zero and mitofusin-2, respectively (23, 26, 55). The clinical syndrome and course of the disease are similar. In the current literature, there are no reports on oxidative stress, vitamin or other dietary deficiencies in CMT patients. Some patients with CMT develop neuropathic pain or symptomatic restless-legs-syndrome, which can be treated symptomatically. Symptomatic treatment is also available for erectile dysfunction and daytime sleepiness, which occur at higher rates in CMT patients compared with healthy people (4, 12). A causative treatment, however, has not been established to date.
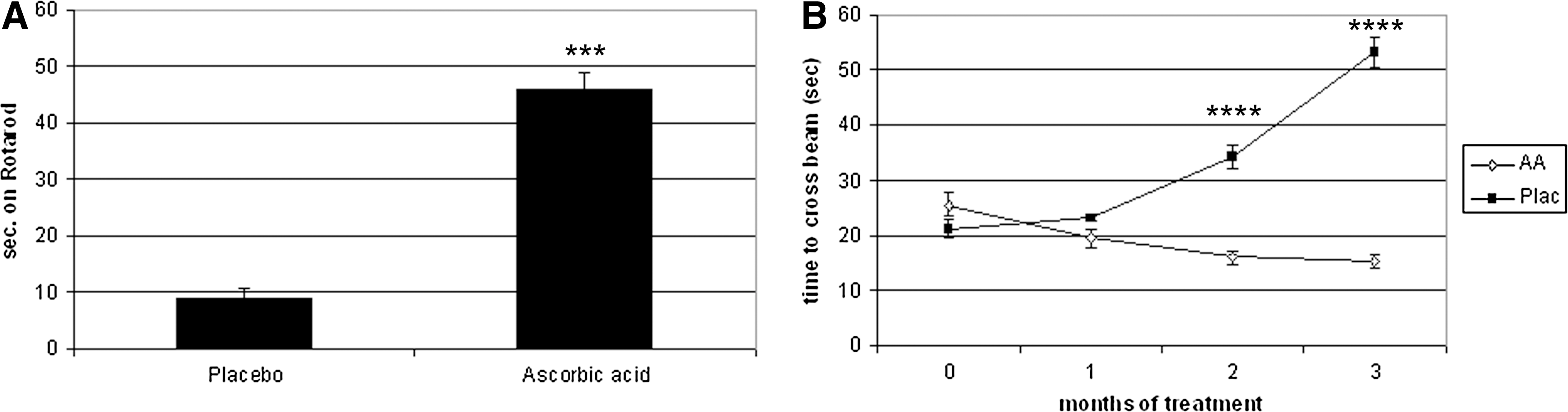
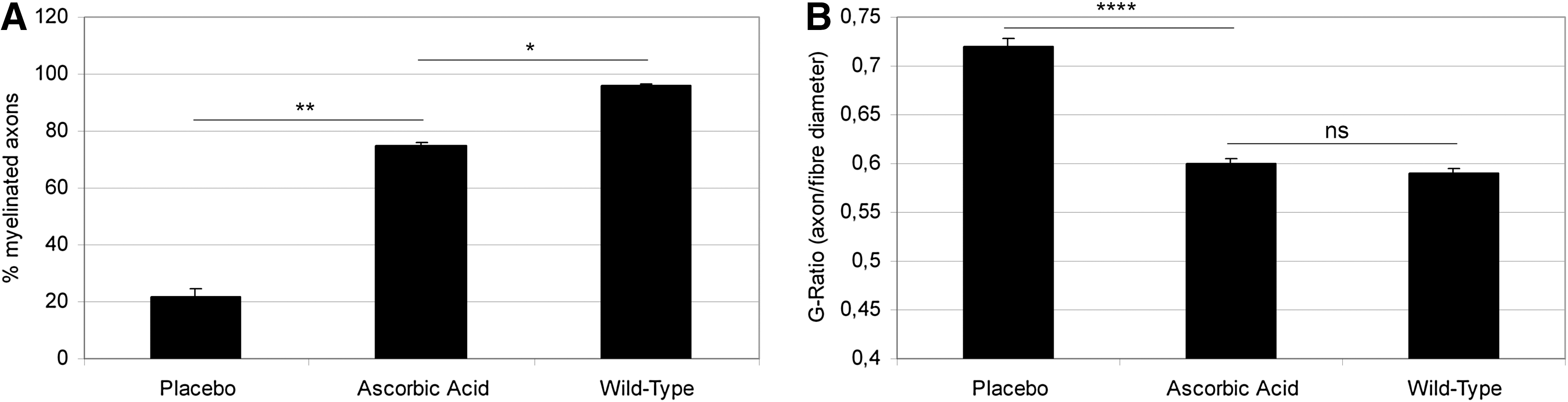
Based on the established role of ascorbic acid in myelination in vitro, ascorbic acid was an interesting candidate for a causative therapeutic approach to CMT1A. A transgenic CMT1A mouse model carrying several copies of a PMP22-containing yeast artificial chromosome was used to test ascorbic acid as a therapeutic agent in CMT1A (25, 40). Ascorbic acid was administered to mice by peroral force-feeding of 1.12 mg (∼56 g per kg body weight) in 0.2 ml water once a week. The investigators could show that ascorbic acid had a positive effect on the performance of PMP22-transgenic mice in both Rotarod and beam-walking tests (Fig. 3A, B). Furthermore, they showed extensive remyelination in the ascorbic acid-treated animals. Ascorbic acid-treated animals showed an astounding increase in myelinated axons (Fig. 4A) and a decrease of the G-Ratio, almost approaching that of wild-type animals (Fig. 4B). The authors attributed these effects of ascorbic acid treatment to a transcriptional down-regulation of PMP22, which would be expected to correct the overexpression on PMP22 to a normal level. This was shown by reverse-transcription polymerase chain reaction of dissected nerves of placebo and ascorbic acid treated mice. Furthermore, the authors developed a hypothesis on the mechanism of action of ascorbic acid in transcriptional regulation. This hypothesis stated that ascorbic acid inhibited adenylyl cyclase, leading to a decrease in cyclic adenosine-monophosphate (cAMP). cAMP is a known inducer of PMP22 transcription; so, a decrease in cAMP would be expected to reduce PMP22 expression. Further evidence for this hypothesis was provided in subsequent studies by the same group (28 –30). The C22 PMP22-transgenic mouse line used in the study by Passage et al. carries multiple copies of the PMP22 gene, causing a very severe neuropathy with a devastating phenotype much more severe than the clinical phenotype of CMT1A patients (40). Particularly, patients with CMT1A have a normal life expectancy, while the C22 mice die within less than 1 year. This impairs the translation of findings from this mouse model to the clinical situation.
Clinical Studies on Ascorbic Acid in CMT Neuropathy
The positive therapeutic effect of ascorbic acid on PMP22-transgenic mice as a mouse model for CMT1A prompted the initiation of multiple clinical trials of ascorbic acid in CMT1A patients in many countries. The first of these trials to be finished and published was a study by Burns et al. (9) on CMT1A children in Australia. CMT1A children received either ascorbic acid (30 mg/kg bodyweight) or placebo for 1 year. An improvement in nerve conduction velocity (NCV) was the primary outcome parameter. This was not reached, although there was a slight, but insignificant increase in the verum group (9) (Fig. 5). Two other studies with doses from 1 to 3 g/day had a treatment period of 1 year and also showed no positive effects of ascorbic acid treatment (35, 52). However, a treatment period of 1 year might be too short to see the potential effects of treatment, as CMT is a slowly progressive disease. A further clinical trial with ascorbic acid at a dose of 1.5 g/d was performed with a treatment period of 2 years and a much bigger size of treatment groups (39). This study had improvements in clinical parameters such as the CMT neuropathy score as primary outcome parameters. However, it also failed to show a positive effect of ascorbic acid treatment on CMT1A neuropathy (39). Overall, five randomized clinical trials were performed on ascorbic acid treatment of CMT1A, and none was able to show a therapeutic effect. A meta-analysis shows that the overall effect of ascorbic acid treatment on the CMT neuropathy score was not significantly different from placebo (Fig. 6).


The question remains as to why ascorbic acid improved the course of CMT neuropathy in mice but not in humans. Apart from the shortcomings of the mouse model (see previous section, last paragraph), there may be other reasons for this discrepancy.
First, one has to consider that CMT1A is a slowly progressive disease; so, even a study period of 2 years may be too short to see a potential effect of any treatment. Studies might have to be conducted over 5 or more years, which would cause great practical and financial difficulties in study design. Furthermore, since CMT1A is a slowly progressive disease, clinical outcome parameters that are very sensitive to change in the course of the disease are necessary (38, 43). Hence, the multitude of ascorbic acid trials has led to increasing efforts in the CMT community to design and validate better outcome parameters that can be used for therapy trials (45).
Second, in clinical and mouse studies, no measurements of ascorbic acid levels in the peripheral nerves themselves were done. In studies on humans, this would be difficult to do, as nerve biopsies can cause undesirable effects such as wound infections, numbness, or neuropathic pain. Due to this difficulty, there are no data on the actual ascorbic acid concentrations in the nerve proper in ascorbic acid studies. Ascorbic acid concentrations were measured in the blood of patients in some studies and showed significant increases (9, 35, 39). It has been shown that the peripheral nerve, such as the brain, is an ascorbic acid-privileged tissue in that it contains much higher concentrations of ascorbic acid than blood (19). Supplementation of ascorbic acid in CMT patients may not have been successful in raising the concentrations in peripheral nerves even further. In this regard, a combination of ascorbic acid treatment with up-regulators of ascorbic acid transporters on the one hand and studies of gene variants modifying ascorbic acid transport in patients on the other hand would be interesting research avenues for the future.
Third, schemes of ascorbic acid administration were different in the clinical trials compared with the animal trial. In the animal trial, ascorbic acid was given orally once a week at a dose of ∼56 mg per kg of body weight (conferring to ∼4 g in a 70 kg adult patient) (40). In all clinical trials, ascorbic acid was also given orally, but once or twice daily, not weekly. Since ascorbic acid transporters may be down-regulated when ascorbic acid concentrations are constantly increased, this difference in administration may be relevant (1, 33).
Expression and Function of the SVCT in the PNS
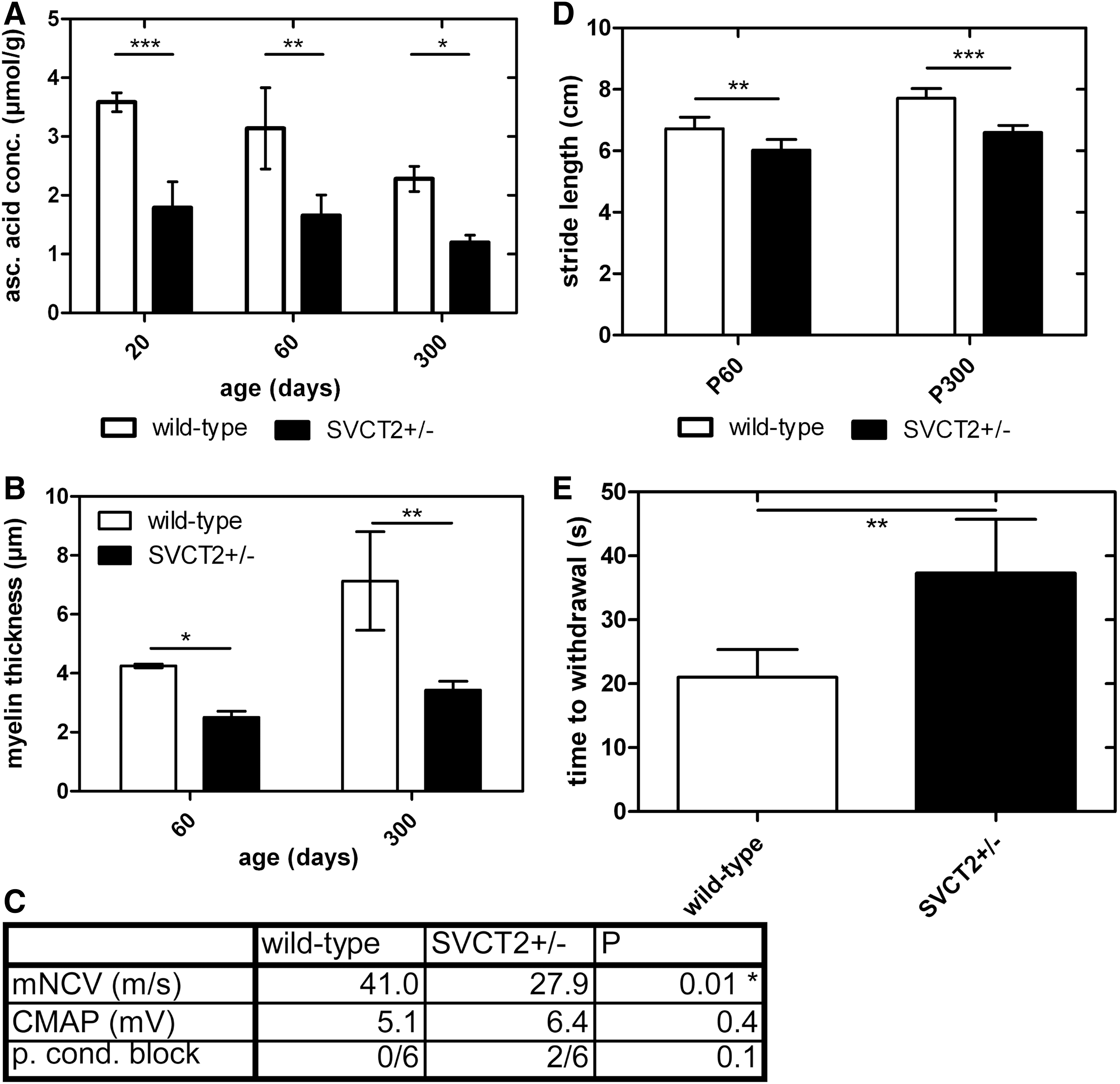
The genes for the SVCT (slc23a1 and 2 for SVCT1 and 2, respectively) were cloned in 2000 (20, 47, 53). Since then, a growing body of evidence has been developed showing that SVCT1 and 2 are the major ascorbic acid transporters in human and murine tissues (42). SVCT2 has been shown to be expressed and functionally active in brain, neuroendocrine tissue, lung, and other organs (5, 21, 46); while SVCT1 is largely expressed in epithelia controlling whole-body homeostasis of ascorbic acid (6, 27, 33). In the brain, several studies showed expression and uptake activity of SVCT2 in both neurons and glial cells (10, 11, 16, 22, 31, 36, 41, 46). In SCs, the glial cells of the PNS, SVCT2 was also shown to be the major transport pathway of ascorbic acid (18). It was demonstrated to be expressed in cultured rat SCs in vitro as well as in murine and rat SCs and peripheral nerve axons in vivo (Fig. 7A, B). Radioactive uptake assays showed uptake of labeled ascorbic acid into SC cultures with enzyme kinetics, dependent on sodium and bivalent cations, and blocked by the SVCT inhibitor phloretin (Fig. 7C–F). Furthermore, ascorbic acid uptake was inhibited by siRNA against SVCT2, but not by siRNA against SVCT1 (Fig. 7G). This study demonstrated that SVCT2 was the main transporter for ascorbic acid in SCs in vitro. In a subsequent study, the function of SVCT2 in mice was investigated in vivo. Here, the authors showed a significant decrease in ascorbic acid concentrations in heterozygous SVCT2 knockout mice (SVCT2+/−) (Fig. 8A) (19). SVCT2+/− mice also showed thinner myelin sheaths, reduced NCV, and deficits in motor and sensory tests (Fig. 8B–E). This provides in vivo evidence for the pro-myelinating effect of ascorbic acid assumed from in vitro studies on SC/neuron co-cultures (see section: Ascorbic Acid Function in Myelination In Vitro). Furthermore, the expression of ECM components such as collagens and laminins was studied in peripheral nerves of SVCT2+/− and wild-type mice. A decrease in extracellular collagen V and XXVIII as well as laminin 2, which are collagen and laminin forms specifically expressed in the PNS, was shown. This led to the hypothesis that the mechanism of action of ascorbic acid in myelination may be the induction of an ECM, which, in turn, is necessary for normal myelination. Ascorbic acid is a co-factor of prolyl-hydroxylase, which is a critical enzyme in the formation of triple-helical collagen. Hence, it was thought from in vitro studies that ascorbic acid was necessary for proline hydroxylation to allow for correct formation of a collagen-containing ECM (13). However, in our study, the concentration of hydroxy-proline in peripheral nerves of SVCT2+/− mice was not reduced, but the expression of collagen genes was impaired, indicating that ascorbic acid may regulate collagen formation at the transcriptional, not the post-translational level (19). Previous reports have shown that ascorbic acid affects the transcription of collagen genes in fibroblasts (17, 48, 49). It may be possible that collagen formation is regulated by ascorbic acid on different pathways depending on cell and tissue types. In the PNS and certain other tissues, regulation might work through collagen gene transcription; while in other tissues, it might work through proline hydroxylation. Another hypothesis is that the effects on proline hydroxylation and collagen gene expression are dependent on the concentration of ascorbic acid. In our study, we have a reduction of ascorbic acid concentration by 30%–50% in the PNS. This may suffice to cause transcriptional changes but not deficits in proline hydroxylation.

Overall, these studies show that SVCT2 is the main transporter for ascorbic acid into the PNS, and that ascorbic acid and SVCT2 have a function in myelination in vivo. The mechanism of action of ascorbic acid in the PNS in vivo appears to be the formation of a collagen- and laminin-containing ECM, which had also been shown in vitro (13, 14). This mechanism of action might also be another possible reason that clinical trials with ascorbic acid did not show benefits in CMT neuropathy, at least not in adults. In progressed CMT neuropathy, there is mostly no lack of extracellular collagen. Collagenous transformation of demyelinated peripheral nerves is actually a pathological sign of inherited as well as acquired peripheral neuropathy (8). Hence, an increase in collagenous ECM should not necessarily be expected to have a positive effect on disease pathology or clinical progression. However, the situation may be different at an earlier time point in the disease when there is no collagenous transformation and an improved ECM might be helpful. Nonetheless, clinical studies on ascorbic acid treatment in CMT children and young patients were negative (9, 52); so either ascorbic acid is ineffective at early time points as well, or treatment would have to start even earlier. Ascorbic acid might also be an interesting candidate for therapy after a peripheral nerve lesion, as its ECM- and myelination-promoting activity might be useful in remyelination after injury. Since the role of SVCT2 in ascorbic acid transport into the PNS is now established, it will also be interesting to pharmacologically or genetically increase SVCT2 activity alone or in combination with ascorbic acid treatment.
Innovation
Ascorbic acid has been shown to have important functions in myelination of peripheral nerves in vitro and vivo. SVCT2 is the main transporter of ascorbic acid in the PNS, and deficiency of this transporter has consequences on peripheral nerve function. Clinical trials on ascorbid acid treatment of the hereditary neuropathy CMT1A have been largely negative.
Conclusion
Ascorbic acid has been shown to have an important function in myelination of the PNS. It has been known to be an effective additive for in vitro myelination in SC/DRG co-cultures and is still widely used as such. Recently, novel results were published indicating a role in PNS myelination in vivo as well. Furthermore, there is evidence from in vitro as well as in vivo studies for a function of ascorbic acid in ECM formation in the PNS. The ECM, in turn, is thought to be necessary for normal myelination.
The SVCT2 was shown to transport ascorbic acid into the PNS, particularly into SCs. SVCT1 was also shown to be expressed, but at a lower level confined to peripheral nerve axons. Transport studies in cultured SCs suggested ascorbic acid transport by SVCT2, not SVCT1. The reduction of the SVCT2 gene dosage in the SVCT2+/− mice was shown to reduce myelin sheath thickness with resulting electrophysiological and functional deficits of mice.
Some studies have indicated an effect of ascorbic acid on the expression of the myelin gene PMP22 with a potential therapeutic effect in CMT1A neuropathy. Such an effect was shown in a mouse model of CMT1A. Clinical trials using ascorbic acid as a therapeutic approach to CMT, however, have been negative. While ascorbic acid was well tolerated, there was no positive effect of treatment in children or adults with CMT1A over a treatment period of 1–2 years. While there is still debate on the reasons for the failure of ascobic acid in clinical trials, it is clear that it cannot be recommended as a treatment of CMT1A on the basis of the current data. To our knowledge, a potential effect of ascorbic acid on other demyelinating or axonal neuropathies has not been studied yet.
For future studies, it will be interesting to investigate the effects of SVCT2 up-regulation on myelination, disease course of peripheral neuropathy, and remyelination after nerve injury. In addition, more research into the mechanisms of axonal degeneration and the potential neuro-protective effect of antioxidants in the PNS is warranted. Overall, the PNS could serve as a model system to study the cellular and molecular functions of ascorbic acid in neural tissues.
Footnotes
Acknowledgments
The authors are thankful for the kind provision of SVCT2+/− mice by Prof. Nussbaum. Furthermore, they thank Anne Humberg, Johanna Nowitzki, and Elisabeth Lange for their excellent technical assistance.