
Abstract
Introduction
P
This study established the survival protein myocyte enhancer factor 2D (MEF2D) as a direct oxidative downstream target of neurotoxic stress and revealed that oxidation-induced inhibition of MEF2D activity may underlie the pathogenic processes of Parkinson's disease.
Autophagy is a process by which lysosome-mediated degradation of intracellular components occurs (5). There are three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Adequate autophagic activity is important for cellular homeostasis, and defects in this pathway have been identified as playing a role in a growing list of human disorders, including neurological diseases (37). For example, the dysregulation of autophagy has been implicated both in chronic neurodegenerative diseases, such as Alzheimer's disease, PD, Huntington disease, and amyotrophic lateral sclerosis (23, 26), and in acute neuronal damage, such as the cases of ischemia and trauma (27, 28). The deletion of the genes required for macroautophagy in the central nervous system causes a degenerative loss of neurons (8, 15). The CMA process involves the binding of chaperone heat-shock cognate protein 70 kDa (Hsc70) to select substrate proteins and their subsequent delivery to lysosomes, the membrane receptor lysosomal-associated membrane protein 2A (LAMP2A). Moderate oxidative stress activates CMA (13), and an increase in CMA function has been shown to restore cellular function (42). In contrast, several proteins that are known to cause familial PD, such as mutated α-synuclein and ubiquitin C-terminal hydrolase L1 (UCH-L1), disrupt the CMA process (12, 24), suggesting that reduced CMA activity may underlie PD pathogenesis (2).
The transcription factor myocyte enhancer factor 2D (MEF2D) is known to promote neuronal survival in several model systems (1, 6, 30). Several survival and death signals converge on MEF2D and regulate its activity. Recent studies showed that CMA plays a critical role in degrading non-functional MEF2D from DA neurons, and this process may be disrupted under the pathological conditions which are associated with PD (40). Furthermore, MEF2D is present in mitochondria and regulates complex I activity. The disruption of MEF2D functioning in mitochondria, in part, underlies neurotoxin-induced toxicity in PD (30). Importantly, several studies have shown that enhancing MEF2D activity in SNc DA neurons protects them from MPTP-induced toxicity (30, 32). Our current study showed that the oxidative stress caused inhibitory modifications of MEF2D in DA SN4741 cells and in the SNc areas of mice and postmortem PD brain tissues. Oxidized MEF2D is preferentially removed by CMA, and failure to remove damaged MEF2D by CMA is correlated with an increase in sensitivity to oxidative stress-induced toxicity.
Results
6-Hyroxydopamine induces oxidative modifications of MEF2D
Since our previous studies showed that MEF2D activity is inhibited by oxidative stress and is also inhibited in models of PD (6, 30), we tested whether oxidative stress may directly modulate MEF2D in a mouse midbrain DA neuronal cell line, namely, in SN4741 cells. We chose to use this cell line, because it has been widely used to study the neuronal response induced by PD-related stress signals (33). Our data showed that short-term exposure to 6-hyroxydopamine (6-OHDA), a neurotoxin used to model PD in rodents, led to a clear reduction of MEF2D levels in the cytoplasm (Fig. 1A, left panel). Short exposure to 6-OHDA did not change nuclear MEF2D levels (Supplementary Fig. S1; Supplementary Data are available online at

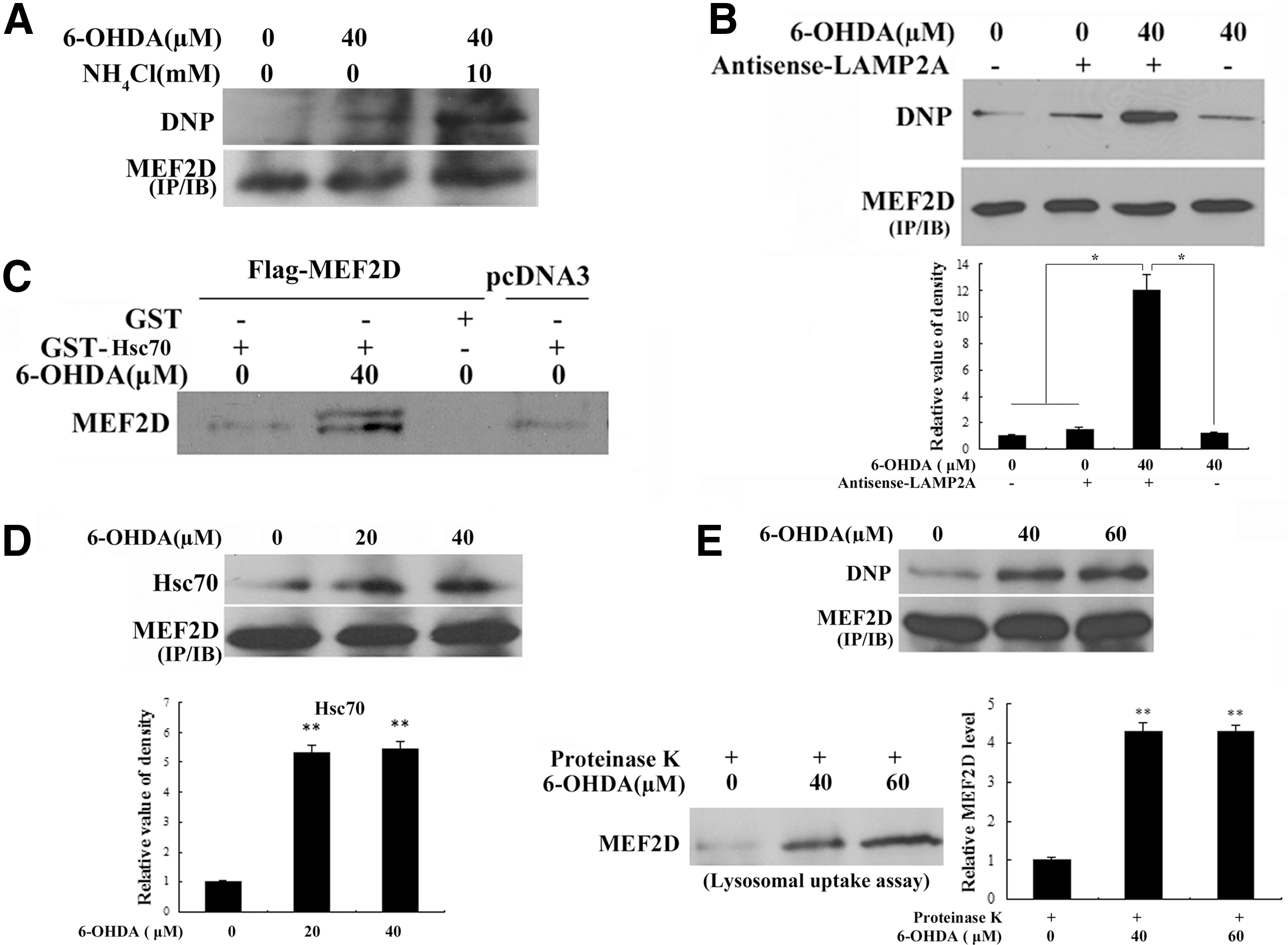
6-OHDA reduces the stability of MEF2D by CMA in DA SN4741 cells
Studies have shown that the stability of MEF2 is regulated under various conditions, including stress (36). Our studies in Figure 1 revealed that levels of MEF2D decrease, as they are increasingly oxidatively modified. These observations suggest that oxidative modifications may regulate MEF2D stability. Since our previous studies had established MEF2D as a direct substrate of CMA in DA neurons (40), we investigated whether the neurotoxin-induced loss of MEF2D proteins was mediated by a lysosomal pathway. We exposed SN4741 cells to 6-OHDA with or without NH4Cl. This analysis showed that 6-OHDA reduced the level of MEF2D, and the inhibition of lysosomal function with NH4Cl significantly blocked the 6-OHDA-induced loss of MEF2D (Fig. 2A). To show that 6-OHDA reduces MEF2D levels primarily through CMA-mediated degradation, we over-expressed a mutated form of MEF2D (MEF2DΔN18), which has been shown to be resistant to degradation by CMA in SN4741 cells (40), and then exposed the resulting cells to 6-OHDA. Although 6-OHDA reduced the levels of wild-type (wt) MEF2D, it failed to significantly alter the levels of MEF2DΔN18 (Fig. 2B). Furthermore, inhibition of macroautophagy had a smaller effect on the 6-OHDA-induced loss of MEF2D (Fig. 2C). Inhibition of the proteasome through MG132 (Supplementary Fig. S2A), which did not cause a significant loss of cellular viability under our experimental conditions (Supplementary Fig. S2B), also failed to attenuate 6-OHDA-induced loss of MEF2D (Fig. 2D). These findings suggest that proteasomes do not play a major role in 6-OHDA-induced loss of MEF2D. Previous studies have shown that the caspase pathway plays a role in the regulation of MEF2D stability. However, the inhibition of caspase-3 activity by z-DEVD-fmk did not reverse the 6-OHDA-induced MEF2D reduction (Fig. 2E). Together, these results support CMA as the primary mechanism by which 6-OHDA promotes MEF2D degradation.

Neurotoxin 6-OHDA activates CMA in DA SN4741 cells
Activation of CMA is a part of the response when cells are deprived of nutrients. We examined whether 6-OHDA regulates CMA in SN4741 cells by determining the level of the rate-limiting CMA regulator LAMP2A. We treated the SN4741 cells with 6-OHDA and measured the levels of LAMP2A mRNA using quantitative real-time polymerase chain reaction (RT-PCR) and the levels of protein using immunoblotting. This analysis showed that a short exposure to 6-OHDA significantly increased the levels of LAMP2A mRNA (Fig. 3A). Consistent with this, 6-OHDA also caused an increase in the level of LAMP2A protein as assessed using immunoblotting (Fig. 3B, bottom) and immunocytochemistry (Fig. 3B, top). In addition, the reduction of LAMP2A levels using an antisense approach in the SN4741 cells significantly prevented the MEF2D degradation induced by 6-OHDA (Fig. 3C). In contrast to LAMP2A, the level of Hsc70, another key CMA regulator, did not significantly change after 6-OHDA treatment (Fig. 3D). These findings suggest that 6-OHDA may significantly enhance CMA activity via LAMP2A in the SN4741 cells. To measure CMA activity more directly, we carried out lysosomal binding and uptake assays, the gold standard in the determination of CMA activity. We treated the SN4741 cells with 6-OHDA, prepared highly purified lysosomes free of significant mitochondrial and ER contamination (Fig. 3E, bottom), and tested their binding capacity to RNase A, a known CMA substrate. This analysis indicated that 6-OHDA significantly increased the amount of RNase A associated with lysosomes (Fig. 3E, top). Similarly, 6-OHDA also increased the amount of RNase A taken up by purified lysosomes (Fig. 3F, top). The increased uptake was attributable neither to the non-specific inhibitory effects of 6-OHDA on the substrate nor to its effects on the lysosomes, because proteinase K was still able to efficiently degrade the extra-lysosomal RNase A (Fig. 3F, bottom right), and the levels of the lysosomal luminal and member proteins Cathepsin D and LAMP1 remained unchanged after 6-OHDA treatment (Fig. 3F, bottom left). Thus, 6-OHDA activates CMA in DA SN4741 cells.

Oxidative modification of MEF2D facilitates its degradation by CMA
Since oxidative modifications of MEF2D correlate with its degradation and increased CMA activity, we tested the possibility that the oxidation of MEF2D facilitates its degradation through the effects of CMA. For this study, we treated SN4741 cells with 6-OHDA and simultaneously blocked lysosomal activity using NH4Cl. We then isolated MEF2D using immunoprecipitation and blotted precipitated MEF2D for carbonyl oxidation using the OxyBlot protocol. Following the 6-OHDA treatment, the inhibition of lysosomal functioning led to a significant accumulation of MEF2D with carbonyl oxidation (Fig. 4A). Similarly, the reduction of LAMP2A levels by antisense LAMP2A also caused an accumulation of oxidized MEF2D (Fig. 4B). To test whether 6-OHDA affects the interaction of MEF2D with Hsc70, we over-expressed MEF2D in SN4741 cells, treated these cells with 6-OHDA, incubated cellular lysate with glutathione S-transferase (GST)-Hsc70, and determined the amount of MEF2D bound to GST-Hsc70. Exposure to 6-OHDA enhanced the interaction between MEF2D and GST-Hsc70 in a pull-down assay (Fig. 4C). In addition, 6-OHDA significantly increased the interaction between endogenous MEF2D and Hsc70 in a co-immunoprecipitation assay (Fig. 4D). Consistent with the earlier findings, we immunoprecipitated comparable levels of MEF2D from the control and 6-OHDA-treated lysates and showed that 6-OHDA increased MEF2D oxidization (Fig. 4E, top). Incubation of these lysates with lysosomes purified from rat livers using a lysosomal uptake assay showed that high levels of MEF2D oxidation correlated closely with its increased uptake by lysosomes (Fig. 4E, bottom). These results strongly support the fact that 6-OHDA-induced oxidization of MEF2D significantly enhances its interaction with Hsc70 and promotes its degradation by CMA.
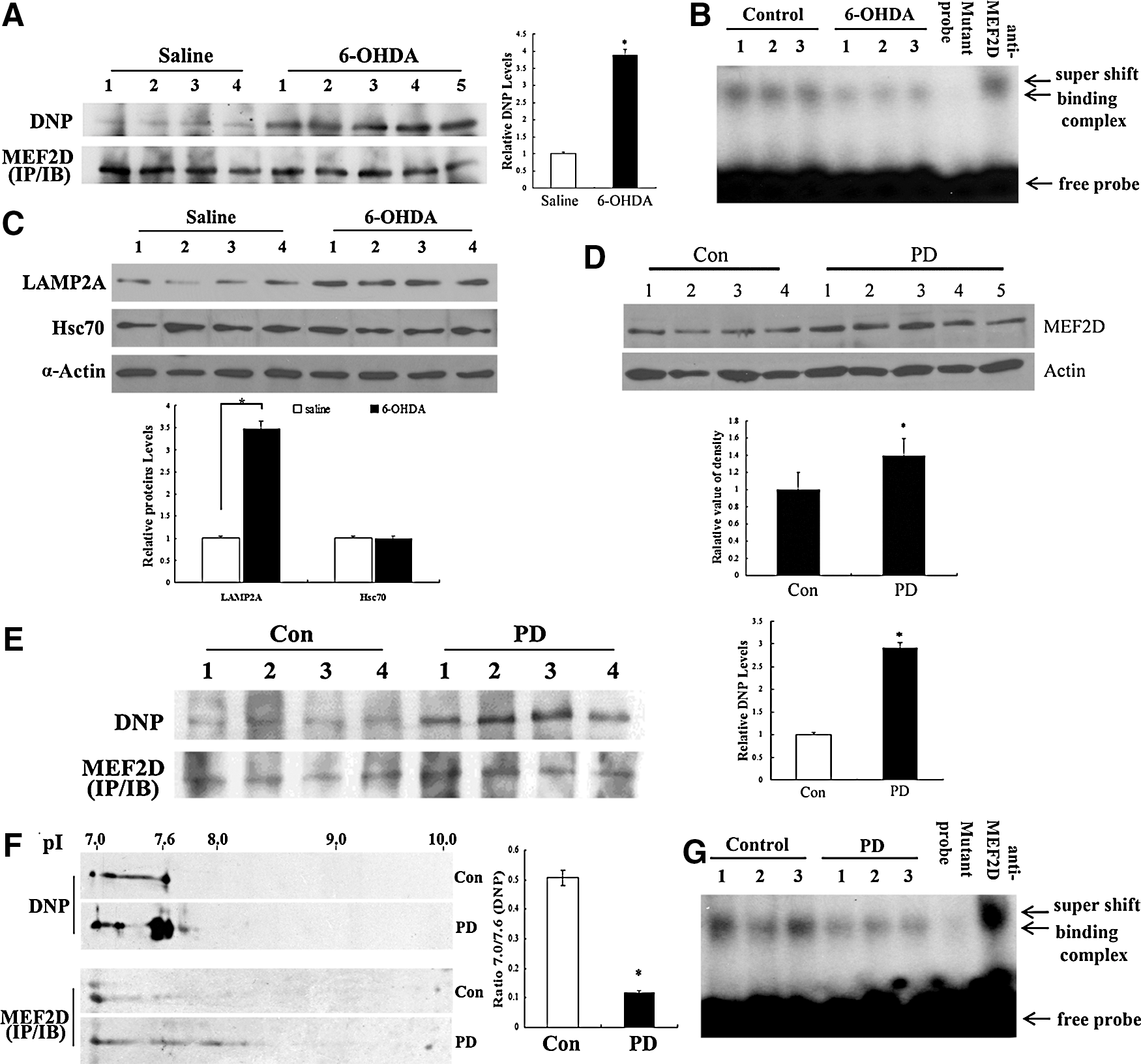
Oxidation of MEF2D is increased in the brain tissues of a rodent PD model as well as in postmortem PD patient brain samples
To corroborate the findings made in a DA neuronal cell line, we determined MEF2D oxidation in brain tissues in vivo. For this reason, we employed 6-OHDA injections in mice through a well-established rodent PD model (10, 35). After the 6-OHDA injections, we immunoprecipitated MEF2D from brain lysates and blotted for carbonyl oxidation. This analysis revealed a robust oxidation of MEF2D in vivo (Fig. 5A), which paralleled a clear reduction in MEF2D DNA-binding activity (Fig. 5B). Consistent with our cellular studies, 6-OHDA also caused an increase in the levels of LAMP2A but not in the levels of Hsc70 in the mouse brain (Fig. 5C). This correlated with a decrease in MEF2D levels in the SNc (Supplementary Fig. S3). To strengthen these findings, we tested the level of MEF2D carbonyl oxidation in postmortem PD brain tissues. We first assessed the total level of MEF2D and showed that, consistent with our previous report (40), MEF2D proteins were increased in postmortem PD brains compared with the controls (Fig. 5D). We immunoprecipitated MEF2D from control and PD postmortem brain lysates and blotted for carbonyl formation. This analysis showed that the level of MEF2D carbonyl formation in PD brains was significantly higher than that of the controls (Fig. 5E). Consistent with increased oxidative modifications, two-dimensional (2D) gel electrophoresis analysis revealed an obvious shift in the pI of MEF2D that was immunoprecipitated from PD postmortem brains and compared with the controls (Fig. 5F). The ability of MEF2D in PD brain tissues to bind to DNA was markedly reduced (Fig. 5G). Overall, these studies indicate that MEF2D is oxidized in vivo in the context of PD.
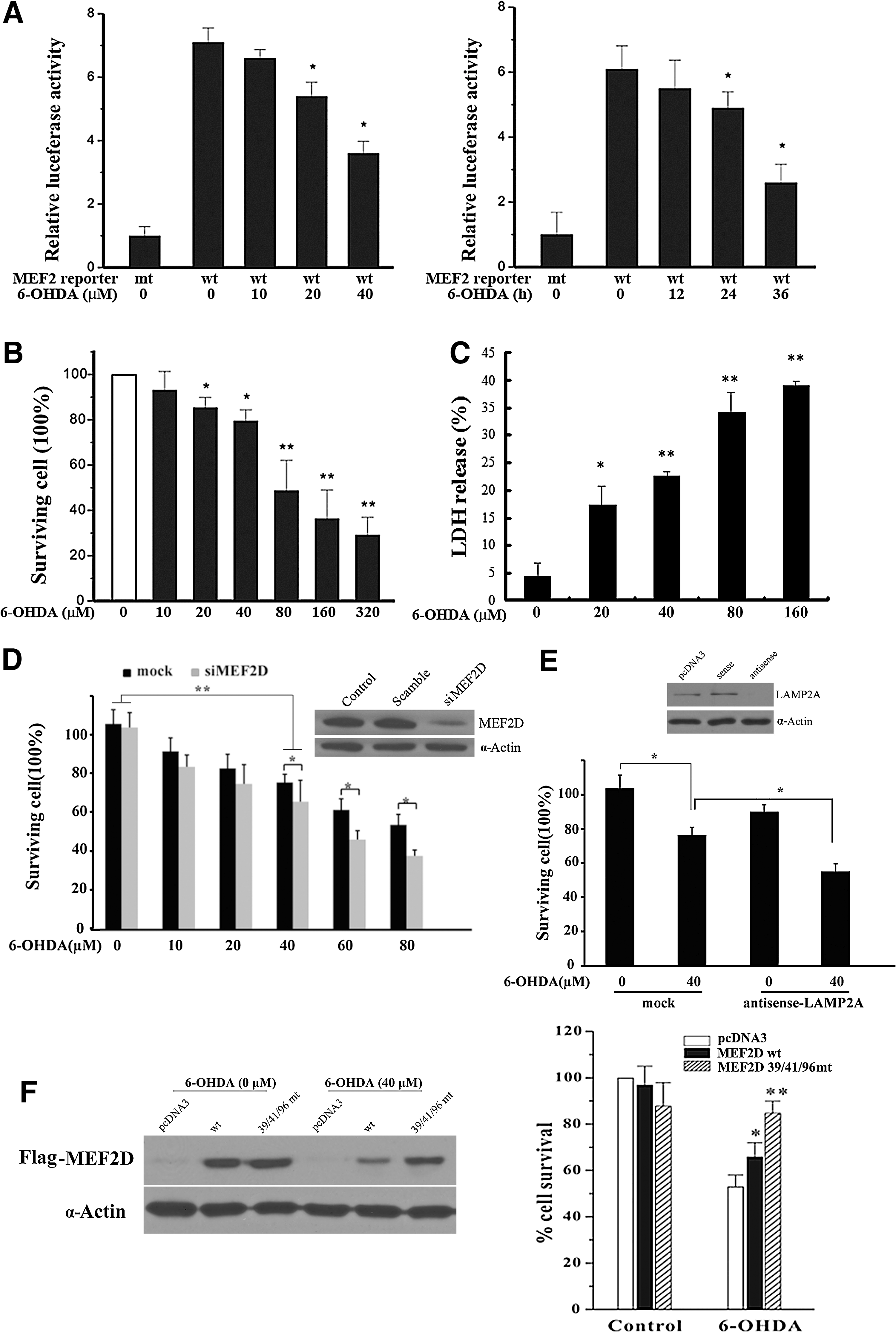
6-OHDA inhibits MEF2-mediated transactivation and survival in DA neuronal SN4741 cells
MEF2 proteins are known to promote neuronal survival in several experimental paradigms (6, 36). A recent study indicated that the inhibition of MEF2 function underlies the pathogenic process in PD (40). We studied whether 6-OHDA may affect MEF2D-mediated survival. First, we showed that the exposure of SN4741 cells to 6-OHDA led to a dose- and time-dependent inhibition of MEF2 transactivation activity (Fig. 6A). Exposure to 6-OHDA caused a dose- and time-dependent decrease in SN4741 viability, determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Fig. 6B and Supplementary Fig. S4), and a release of lactate dehydrogenase (LDH) (Fig. 6C). Since the data from the MTT and LDH assays correlate well with each other, we used MTT analysis for subsequent studies. We showed that a reduction in the levels of either MEF2D or CMA regulator LAMP2A sensitized the cells to 6-OHDA-induced toxicity (Fig. 6D, E). Cysteine residues 39, 41, and 96 of MEF2D are located within or close to the highly conserved MADS and MEF2D domains. We generated an MEF2D mutant with cysteine residues 39, 41, and 96 changed to alanine (39/41/96 mt). Compared with MEF2D wt, mutation of these cysteine residues rendered MEF2D much more resistant to 6-OHDA-induced degradation (Fig. 6F, left). Moreover, the over-expression of this MEF2D mutant significantly protected the SN4741 cells against 6-OHDA-induced neurotoxicity (Fig. 6F, right). Thus, oxidative modifications of MEF2D underlie oxidative stress-induced loss of neuronal viability.
Discussion
The transcription factor MEF2D promotes neuronal survival in different experimental paradigms (18, 21, 32). Our current study showed that oxidative stress readily oxidizes MEF2D at cysteine residues and also carbonylates the protein. This establishes a direct link between oxidative stress and neuronal survival factor MEF2D. The finding that MEF2D is highly oxidized in postmortem PD brain tissues compared with the controls suggests that oxidation-mediated inhibition of MEF2D may underlie the pathogenic process in PD. Various stress signals have been shown to inhibit MEF2D activity in neurons. The signaling mechanisms of stress-induced inhibition of MEF2 include inhibitory phosphorylation by Cdk5 and GSK3β (38). In addition, stress signals reduce the level of MEF2. For example, caspases activated by excitotoxicity directly cleave MEF2D (36). Unlike MEF2A, which is readily ubiquitinated and degraded by the proteasome (29), MEF2D is found to be degraded mainly by CMA (19). CMA-mediated regulation of MEF2D is disrupted by genetic risk factors that are associated with PD, such as α-synuclein. Neurotoxins were reported to interrupt mitochondrial MEF2D functioning in models of PD (30). High levels of α-synuclein, neurotoxic insult, and excitotoxicity are known to cause oxidative stress. Therefore, it is likely that the oxidation of MEF2D may mediate, at least in part, the toxic effects of those diverse stress conditions.
Oxidative stress has been proposed as a key unifying mechanism mediating multiple pathological stresses for triggering or exacerbating the pathological processes in PD (9). Since the impairment of MEF2D functioning underlies the loss of SNc DA neurons (30, 40), the establishment of the inhibitory oxidative modification of survival factor MEF2D reveals a significant mechanism underlying oxidative stress-induced toxicity in PD. Since high MEF2D activity protects SNc DA neurons from toxic stress in rodent models of PD (30, 41), our findings provide further evidence for enhancing MEF2D function as an effective therapeutic strategy for PD.
Moderate levels of oxidative stress stimulated CMA activity in our model cells, which was consistent with the reported effect of oxidative stress on CMA in non-neuronal cells (13). One mechanism by which 6-OHDA may increase CMA activity is by increasing the levels of the key CMA regulator LAMP2A. LAMP2A is generally regarded as the rate-limiting factor for the CMA process (3). Therefore, its increase is expected to enable efficient activation of this pathway. Our data indicate that at least one of the mechanisms underlying increased LAMP2A in DA neuronal cells involves transcription, but it is possible that transcription-independent processes may also play a role.
In addition to increasing the levels of LAMP2A to augment overall CMA activity, oxidation also enhances the interaction between MEF2D and Hsc70, enabling the delivery of MEF2D to LAMP2A. Therefore, two factors contribute to the enhanced degradation of MEF2D by CMA after 6-OHDA. It is known that the unfolding of CMA substrates may enable Hsc70 to better access their CMA signal motif and, therefore, facilitate their degradation. However, the domain that bears the Hsc70-interacting motif has been mapped to the N-terminus of MEF2D, which is thought to be relatively exposed (40). Since the same domain is also involved in DNA binding, it is conceivable that when MEF2D is bound to DNA, this motif is not readily accessible to Hsc70. The accessibility to Hsc70 may be much higher when MEF2D is not bound to DNA. Therefore, conditions stimulating MEF2D interaction with Hsc70 would be expected to correlate with reduced MEF2D-DNA binding. Consistent with this reasoning, 6-OHDA, indeed, reduces the DNA binding activity of MEF2D.
MEF2D's N-terminus is composed of two subdomains: the MADS domain (aa2-56) and the MEF2 domain (aa57-86). Structural studies have shown that the N-terminus of the MEF2 molecule is responsible for MEF2 dimer formation, which is required for DNA binding. Our analysis showed that 6-OHDA oxidizes three cysteine residues, two located within the MADS domain and one just outside the MEF2 domain. Since 6-OHDA led to a clear reduction in MEF2D DNA binding, our data are consistent with the interpretation that the oxidation of these three cysteine residues specifically reduces the ability of MEF2D to associate with DNA. Although the reason for this reduced DNA binding remains to be determined, it is possible that the oxidation of the cysteine residues at the N-terminal turn between the DNA-contacting helix and the β-sheet of MEF2D may affect the flexibility of this region to make proper contact with DNA, thus reducing its DNA-binding capacity (22). Alternatively, cysteine oxidation at the N-terminus of MEF2D may also hinder its ability to form dimers. The DNA-free MEF2D monomer is expected to be more efficiently transported out of the nucleus to the cytoplasm for a subsequent interaction with Hsc70. The findings that MEF2D is regulated by cysteine oxidation raise the possibility that via its cysteine residues, MEF2D may act as an oxidative sensor which modulates neuronal response.
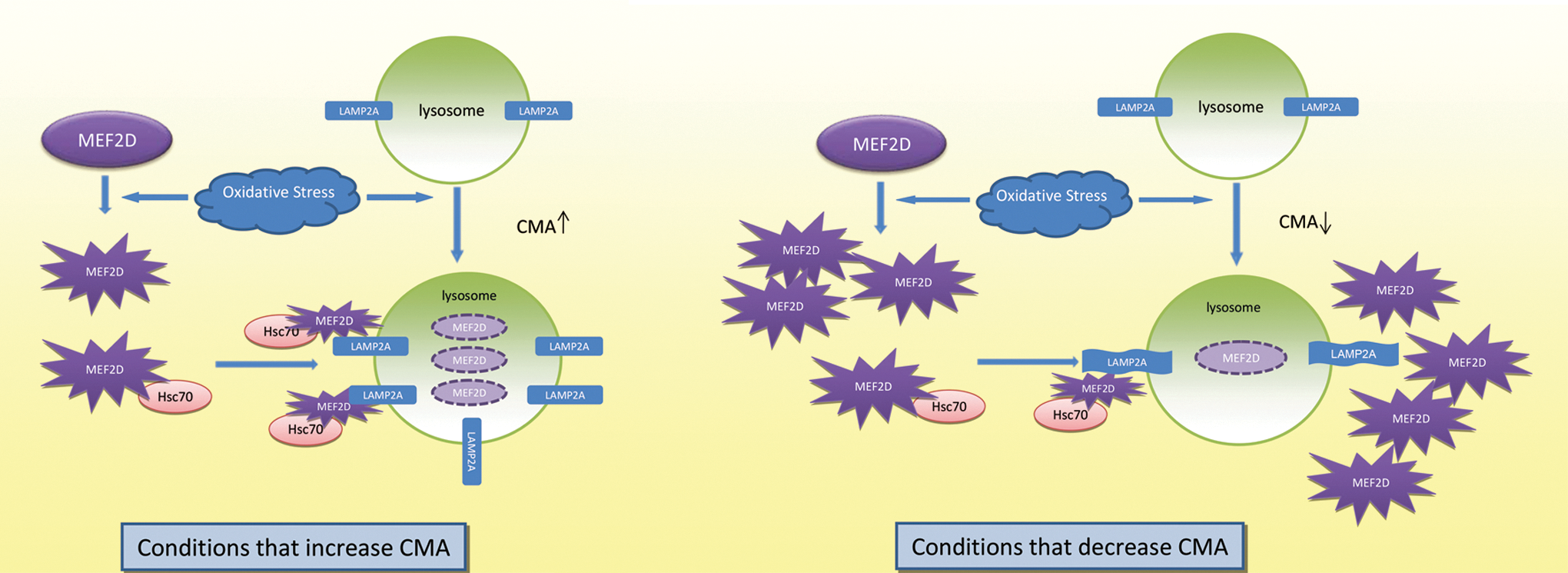
Our previous studies showed that the inhibition of lysosomal functioning leads to the accumulation of MEF2D with reduced DNA-binding activity (40). Our current studies revealed that oxidized MEF2D loses DNA-binding potential and is readily degraded by CMA. Together, these findings indicate that CMA preferentially degrades oxidized and non-functional MEF2D. Since proteolytic cleavage of MEF2D has been shown to generate a dominant negative fragment (17, 38, 39), an accumulation of non-functional MEF2D may lead to its aberrant processing and the generation of fragments that are detrimental to neuronal viability. Based on all these observations, we propose a model in which moderate oxidative stress activates CMA and removes oxidized non-functional MEF2D. This may reduce the chance of its aberrant processing and offer neuroprotection. Conditions that impair CMA processes may cause an accumulation of oxidized MEF2D. This accumulation is detrimental to both MEF2D function and neuronal viability (Fig. 7).
Materials and Methods
Animals
Adult male C57BL/6 mice were purchased and housed in standard enriched environment cages in a temperature-controlled room with a 12-h light/dark cycle and free access to food. The animals were given at least a week acclimation period to the facility before the administration of 6-OHDA.
Patient cases
Brain tissue was collected from the Brain Bank, the Center for Neurodegenerative Disease, Emory University, with the proper consents. Use of the deidentified archived specimens did not require IRB approval. The control and PD cases were matched with regard to age (control, n=9, 65.4±5.1 years; PD, n=9, 72.5±7.1 years), race (all mixed European descent), sex (control, 4:5 female/male; PD, 5:4 female/male), and postmortem interval (control, 7.5±3.1 h; PD, 8.5±2.8 h). They were not diagnosed with other neurodegenerative diseases, including Alzheimer disease. The IRB of Emory University approved all procedures, and all subjects provided written informed consent.
Plasmids, antibodies, and chemicals
Flag-MEF2D, Flag-MEF2DΔN18, wt, and mutated luciferase reporters were previously described (40). Antibodies were purchased from the following vendors: anti-MEF2D from BD Biosciences (610775), anti-Hsc70 (ab1427) and anti-LAMP2A (ab18528) from Abcam, LAMP1 (3243S) and Cathepsin D (2284S) from Cell Signaling Technology, and horseradish peroxidase (HRP)-streptavidin from Amersham Bioscience (RPN2195). The OxyBlot protein oxidation detection kit was purchased from Chemicon International (S7150), QuikChange site-directed mutagenesis kit from Stratagene (200522), TRIzol from Invitrogen (15596-026), TURBO DNA-free™ kit from Ambion (AM1907), SYBR Green Brilliant III Ultra-fast reagent kit from Applied Biosystems (6008223), 6-OHDA from Sigma (H8523), Biotin-iodoacetamide from Invitrogen (B1591), iodoacetamide (IAM) and α-tocopherol from Sigma (I6125), and MTT Kit I from Boehringer Mannheim (1465007). The Cytotoxicity LDH Detection Kit was purchased from Roche (04744926001).
Cell culture
SN4741, a mouse embryonic substantia nigra-derived cell line, was cultured at 33°C in DMEM (Gibco) containing 10% FBS.
Immunofluorescence
The SN4741 cells were plated on glass slides in 24-well plates. After the 6-OHDA treatment, cells were fixed with 4% formaldehyde solution, blocked with 5% goat serum in phosphate-buffered saline (PBS), incubated with anti-LAMP2A antibody (1:200) overnight, washed thrice with PBS, incubated with anti-rabbit antibody at room temperature, washed thrice with PBS, and stained with DAPI for 5 min.
Immunoprecipitation of BIAM-labeled MEF2D
The cells were treated with 6-OHDA (40 μM) for 18 h and lysed in oxygen-free lysis buffer (50 mM Bis-Tris–HCl (pH 6.5), 0.5% Triton X-100, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 150 mM NaCl, 1 mM EDTA, protease inhibitor, and 20 μM BIAM). After incubation at 37°C in darkness for 30 min, the labeling reaction was stopped by adding IAM to 5 mM. MEF2D was precipitated and immunoblotted with anti-streptavidin HRP.
Protein oxidation detection
The SN4741 cells were allowed to reach 70%–80% confluence. After treatment with 6-OHDA for 18 h, the cells were lysed in a buffer containing 50 mM DTT. The levels of oxidized MEF2D were detected using the OxyBlot protein oxidation detection kit. Briefly, the samples were denatured by adding 12% SDS to a final concentration of 6% SDS, derivatized by adding 1×DNPH solution, incubated at room temperature for 15–30 min, neutralized with neutralization solution and 2-mercaptoethanol, and immunoblotted with anti-DNP.
Electrophoretic mobility shift
An electrophoretic mobility shift assay was performed as previously described (20). The cell lysates were incubated with an MEF2-specific probe or a mutant probe. For the super shift assay, the lysates were incubated with MEF2D antibody for 15 min before incubation with a labeled probe. A commonly used A/T rich MEF2 consensus binding site was synthesized as an MEF2 probe (5′-AGCTTCGCTCTAAAAATAACCCTGATC-3′ wt); for the mutant probe, the three nucleotides in italics were mutated to GGC.
Isolation of lysosomes
The cells and liver tissues were washed with PBS and homogenized in an extraction buffer (Lysosome Isolation Kit; Sigma). After separation by density gradient centrifugation, the lysosomes were isolated from the light mitochondrial–lysosomal fraction as previously described (40).
Binding and uptake by the lysosomes
The lysosomal binding and uptake assays were carried out as previously described (40). Briefly, for the binding assay, the isolated lysosomes were treated with 100 μM chymostatin for 10 min at 0°C and were incubated with RNase A for 20 min at 37°C in an MOPS buffer. After incubation, the lysosomes were washed four times with PBS, and the bound RNase A was blotted. For the uptake assay, proteinase K was added after the incubation of lysosomes and substrates for 10 min at 0°C.
Measurement of cytotoxicity
Neurotoxicity was assessed using a tetrazolium salt MTT assay and an LDH assay. The MTT assay was performed according to the specifications of the manufacturer. Briefly, 10 μl of 5 mg/ml MTT labeling reagent were added to SN4741 cells that were cultured in a 96-well plate in 100 μl of medium, and the plate was incubated for 4 h in a humidified incubator at 37°C. After incubation, the absorbance of the samples was measured at a wavelength of 570 nm with an excitation of 655 nm. The LDH release assays were tested using an LDH detection kit. Medium from individual wells was collected and incubated with a reaction mixture at 25°C for 30 min and then, a stop solution was added, followed by the detection of absorbance at 490 nm (Molecular Devices).
Luciferase reporter gene assay
MEF2 luciferase reporter assays were performed as previously described (6). SN4741 cells were transfected with MEF2 luciferase reporter genes (wt MEF2 binding site: TCGACGGGCTATTTTTAGGGCC; mutated binding site: TCGACGGGCGATTTTTCGGGCCG) using Lipofectamine 2000 (Invitrogen). Cellular extracts were assayed for both luciferase and β-Gal activities. The relative fold of luciferase activity was calculated based on the efficiency of transfection.
GST pull-down assay
The SN4741 cells were washed in PBS, lysed in a lysis buffer (20 mM Tris pH 7.4, 40 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 10 mM NaF, 10 mM sodium pyrophosphate, 10 mM sodium β-glycerophosphate, and 1% Triton X-100), and centrifuged. The supernatant was incubated with purified GST fusion proteins on Sepharose beads (GE) for 2 h at 4°C.
Quantitative RT-PCR
Total RNA was isolated using TRIzol and further purified using a TURBO DNA-free kit to remove potential genomic DNA contamination. Reverse transcription was performed using the RETROscript First-Strand Synthesis kit. Quantitative PCR was performed using a Stratagene Mx 3000P System with the SYBR Green Brilliant III Ultra-fast reagent kit. Primer sequences were as follows (13): LAMP2A forward: 5′-GTCTCAAGCGCCATCATACT-3′; reverse: 5′-TCCAAGGAGTCTGTCTTAAGTAGC-3′. Actin forward: 5′-AAGGACTCCTATAGTGGGTGACGA-3′; reverse: 5′-ATCTTCTCCATGTCGTCCCAGTTG-3′.
6-OHDA mouse model of PD
The 6-OHDA mouse model of PD was created as previously described (11, 25, 34). All procedures were approved by the IACUC of Emory University. Mice were randomly assigned to two groups. The group of PD model mice (n=9) received injections of 6-OHDA (3 μg in 2 μl) into the left SN of midbrain at the following coordinates: AP, −3.16; ML, −3.85; and DV, −2.64 mm. The group of control animals (n=9) received only saline at the same coordinates.
2D gel electrophoresis
The lysates from the human brain tissues (150 μg) were resuspended in a rehydration buffer [8 M urea, 4% CHAPS, 50 mM dithiothreitol, 0.5% immobilized pH gradient buffer (Amersham Biosciences), and traces of bromophenol blue] at a final concentration of 2–4 mg/ml. The samples were then loaded onto a 7–10 linear pH gradient Immobiline DryStrip (Amersham Biosciences) and separated using isoelectric focusing in an Ettan IPGphor system (Amersham Biosciences) for 15,000 V·h. The first-dimension gels were subsequently separated on 10% Tris-HCl/SDS-polyacrylamide gel electrophoresis (PAGE) gels, and proteins were detected using western blotting. We used 2D SDS-PAGE standards (Bio-Rad) to calibrate pI and molecular weights (16).
Statistics
All statistical analyses of data were performed with SPSS 15.0. The quantitative data are presented as the mean±SEM, and the differences among the control and various treatment groups were compared using one-way ANOVA. Student's t-tests were used to compare the different treatment conditions. A value of p<0.05 was considered significant.
Footnotes
Acknowledgments
The authors thank the Brain Bank at the Center for Neurodegenerative Disease at Emory University School of Medicine for providing brain tissues and Eric Dammer and Jeremy H. Herskowitz for helpful discussions and the editing of this article. This work was partially supported by FMMU Research Foundation (Q.Y.), Chinese National 973 Projects Grant 2011CB510000 (Q.Y.), NIH grants ES015317 (Z.M.), AG023695 (Z.M.), NS079858 (Z.M.), and ES016731 (Z.M.), and the Michael J. Fox Foundation (Z.M.).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.