
Abstract
Introduction
I
Different cell types show broadly variable sensitivity to ischemia. However, for each cell type, the vulnerability to cell death induced by ischemia can be opposed by ischemic preconditioning, a phenomenon whereby a sublethal ischemic stress gives protection against a later, longer lasting ischemic injury. Preconditioning occurs in two phases: an early phase that rapidly evolves and fades away within few hours and a longer lasting late period, requiring transcriptional activation (84).
Among many determinants dictating cell response to hypoxia and ischemia, an ever increasing number of studies have concurred to highlight a fundamental role of microRNAs (miRNAs). miRNAs are small noncoding RNA sequences, 20–25 nucleotide-long, that regulate gene expression mainly via suppression of specific target mRNAs. For a summary of miRNA biogenesis, see the Editorial of this Forum; for extensive reviews, see Refs. (18, 72, 156).
miRNAs originate from longer precursor RNAs, called primary miRNAs, that are generally transcribed by RNA polymerase II (77). Like mRNAs, miRNA expression is regulated by transcription factors and many of them exhibit a tissue-specific distribution or can be induced upon specific stimuli such as hypoxia or starvation. The sequential action of DROSHA/DGCR8 and DICER ribonucleases III yields mature miRNA sequences, which are loaded on the effector complex called RNA-induced silencing complex (RISC). Within the RISC, the mature miRNA associates with target mRNAs and, in most circumstances, acts as a negative regulator of gene expression.
The biological role of miRNA is particularly pervasive, regulating virtually all aspects of cell biology, and heart and blood vessels are no exceptions. Indeed, several reviews extensively illustrate miRNA regulation of cardiogenesis, angiogenesis, endothelial and myocyte growth, contractility, and cardiac rhythm (6, 41, 46, 114, 128, 129). Not surprisingly, the expression of specific miRNAs is altered in many cardiovascular diseases and experiments of gain- and loss of function in mice have shown that miRNA deregulation is a condition necessary and sufficient for multiple diseases. A huge amount of data have been accumulating linking miRNAs, such as let-7 family, miR-1, miR-133a/b, miR-19a/b, miR-150, miR-195, miR-199, miR-221, miR-23a/b, miR-29a/b, miR-30 family, and miR-320, to ischemic disease clinical data [for recent comprehensive reviews, see Refs. (41, 46, 56, 64, 75, 81, 128)].
Another element of interest is constituted by indications that miRNAs may represent excellent prognostic/diagnostic biomarkers. Indeed, mounting evidence accumulating in the last few years shows that miRNAs are present not only in tissues, but also in various biological fluids including blood, urine, and saliva (36, 135). Remarkably, miRNAs circulate in the peripheral blood in a highly stable, cell-free form that is protected from endogenous RNase activity and can be detected in both plasma and serum.
As previously pointed out, hypoxia constitutes a crucial pathogenetic element of ischemic cardiovascular diseases. This review will be focused on the regulation and role of hypoxia-induced miRNAs (hypoxamiRs). In particular, we will describe those miRNAs that not only are directly regulated by hypoxia, but also regulate cell responses to decreased oxygen tension. We will first review miRNAs involved in more general events such as hypoxia-regulated transcription, miRNA biogenesis, and angiogenesis. Next, we will describe hypoxamiR implication in some of the most common ischemic cardiovascular diseases. HypoxamiR deregulation, functions, and transcriptional regulation are summarized in Table 1.
3-NPA, nitropropionic acid; 5-AzaC, 5-azacytidine; AO, atherosclerosis obliterans; BCNU, 1,3-bis(2 chloroethyl)-1-nitrosourea; BM, bone marrow; CI, cerebral ischemia; CM, cardiomyocytes; CNE, nasopharyngeal carcinoma cells; CoCs, colon cancer cells; DFOM, deferoxamine mesylate; EC, endothelial cells; EGCG, epigallocatechin-3-gallate; EFF, effector (transcription factor/signaling pathway); EP, endothelial pericytes; EPC CAD, endothelial precursor cells from CAD patients; HF, heart failure; FOXO3A, forkhead box O3; hUCB-MSCs, human umbilical cord blood-derived multipotent stem cells; hypoxamiR, hypoxia-induced miRNA; IP, ischemic preconditioning; I/R, ischemia–reperfusion; IWH, ischemic wound healing; MI, myocardial infarction; Mm, mus musculus; MØ, macrophages; PH, pulmonary hypertension; PmSC, preconditioned mesenchymal stem cells; RC, renal cells; SC, stem cells; SMs, small molecules; THR, transplanted heart recovery; WH, wound healing.
Hypoxia-Inducible Factor 1: An miRNA-Mediated Modulator
Hypoxia-inducible factor 1 alpha (HIF1A) is the first identified member of a family of oxygen-sensitive transcription factors that allows aerobic organisms and tissues to adapt to hypoxia (94, 124). This is mostly accomplished through the transcriptional activation of more than 200 genes, constituting a significant portion of the hypoxia-induced transcription. Moreover, stimuli other than hypoxia, such as nitric oxide and reactive oxygen species, can also activate HIF1A. Being a master transcription factor of the hypoxic response, HIF1A is implicated in virtually all ischemic diseases (101, 108).
Under normal oxygen tension, the hydroxylation of HIF1 at two specific proline residues, by the prolyl-4-hydroxylase domain-containing enzymes (PHD), targets HIF1A for polyubiquitination and proteosomal degradation by the von Hippel–Lindau tumor suppressor protein (62, 95). Under hypoxic conditions, PHDs are inhibited, saving HIF1A from degradation and allowing it to accumulate and translocate to the nucleus. Once in the nucleus, HIF1A dimerizes with HIF1 beta (HIF1B) and binds to specific hypoxia-responsive element sequences of target gene promoters. The transcriptional activity of the HIF1 heterodimer is also regulated by an asparaginyl hydroxylase, named factor inhibiting HIF1 (FIH-1) (92).
While the regulation of protein stability by the ubiquitin-proteasome pathway plays a fundamental role, other mechanisms of HIF1A regulation are present as well. Indeed, HIF1A mRNA levels dramatically increase in response to hypoxia or ischemia in heart, skeletal muscle, kidney, brain, and lung (7, 11, 76, 144). Albeit this may be primarily due to increased mRNA transcription, decreased mRNA degradation may play a role as well. In this respect, miRNAs seem to play an important role, by binding to the 3′-untranslated region (UTR) of HIF1A mRNAs and either blocking its translation or inducing its degradation (Fig. 1). For instance, miR-199a is downregulated in cardiac myocytes upon hypoxia and this reduction is required for the rapid upregulation of its target, HIF1A (117). The override of miR-199a decrease during hypoxia inhibits HIF1A expression and HIF1A-mediated stabilization of p53. As a consequence, apoptosis is attenuated as well. On the other hand, the blockade of miR-199a during normoxia results in the upregulation of HIF1A, both directly and indirectly. Indeed, miR-199a also targets sirtuin-1 that, in turn, negatively regulates PHD2, required for the de-stabilization of HIF1A. The relevance of this molecular circuitry is highlighted by the fact that, miR-199a inhibition reproduces hypoxia preconditioning in vitro (117).
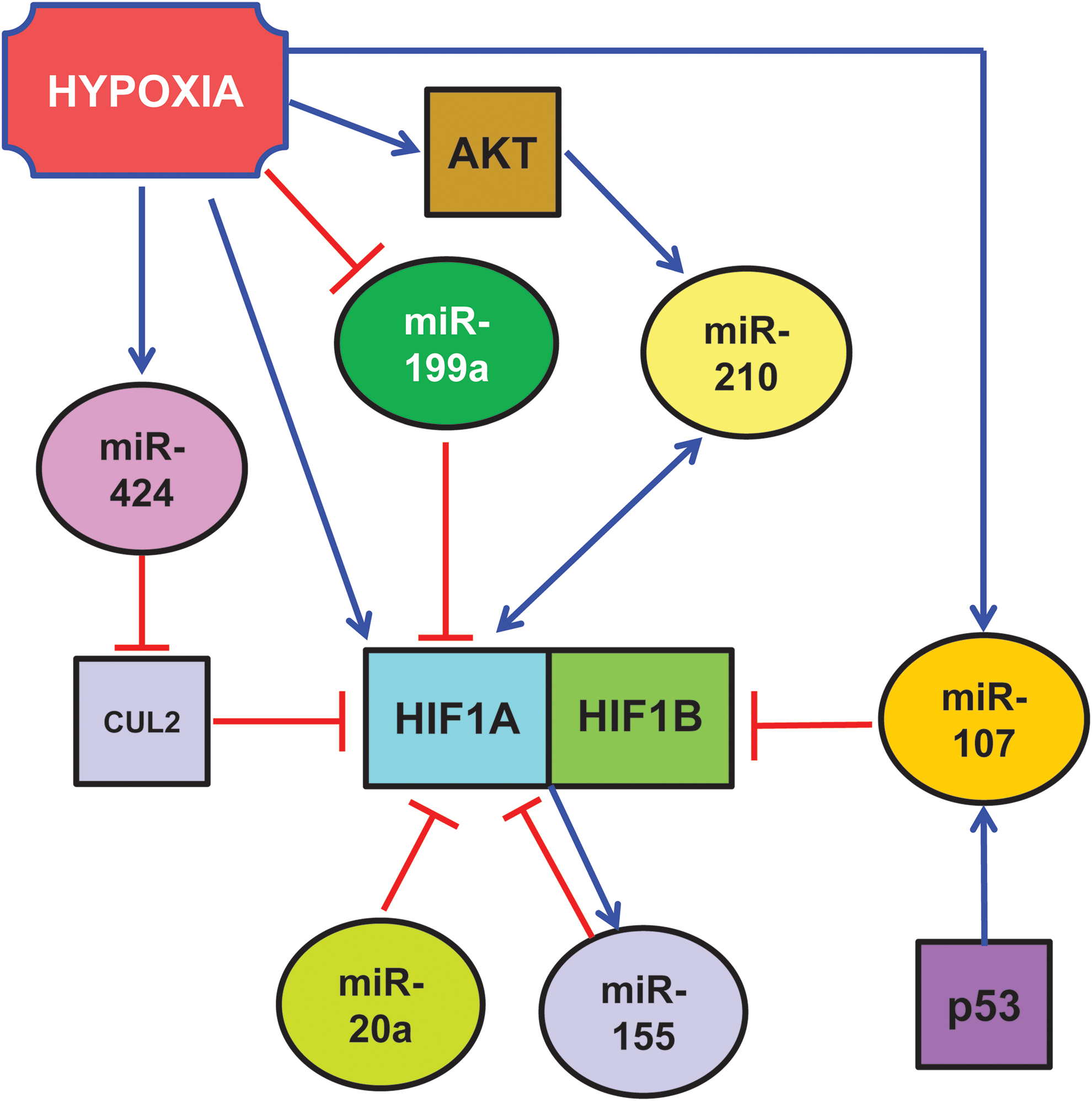
Other miRNAs also target HIF1A transcript: Taguchi et al. found that HIF1A is a target of miR-17-92 cluster and in particular of its miR-20a component (Fig. 1) (132). In support of these findings, Kang et al. showed that a pyrrolopyrazine metabolite of the antiangiogenic drug Oltipraz inhibits HIF1A via miR-199a and miR-20a induction (66). However, both Taguchi et al. and Kang et al. used cancer cells as experimental systems, and the relevance of their findings in the cardiovascular system remains to be investigated.
HIF1A can be also targeted by miRNAs indirectly, that is, modulating fundamental regulators of its activity. Indeed, argonaute-1 (AGO1) and the miRNA machinery are necessary for Drosophila adaptation to hypoxia (35).
One way for miRNAs to regulate HIF1A is by targeting its ubiquitination machinery. Ghosh et al. found that miR-424 was induced in human endothelial cells exposed to hypoxia (Fig. 1) (48). miR-424, in turn, targets cullin 2, a scaffolding protein critical to the assembly of the ubiquitin ligase system, thereby stabilizing HIF1A. Hypoxia-induced miR-424 is not HIF1A dependent, but it relies on SPI1/PU.1 that, in turn, is increased in hypoxic endothelium by RUNX-1 and C/EBPα. The relevance of this pathway in post-ischemic vascular remodeling and angiogenesis is demonstrated by the fact that miR-424 promotes angiogenesis in vitro in mice. Moreover, mu-miR-322, the mouse homolog of human miR-424, is significantly upregulated along with HIF1A in experimental models of ischemia.
Another modifier of HIF1 is miR-31, which targets the 3′-UTR of FIH, an inhibitor of HIF under normoxic conditions. Thus, by targeting an HIF-inhibitor, miR-31 stimulates HIF function (81).
Other HIF1A-binding proteins can be miRNA target as well. For instance, p53 regulates hypoxic signaling in human colon cancer through the transcriptional induction of miR-107 that, in turn, suppresses the expression of HIF1B (Fig. 1) (151). Indeed, inhibition and overexpression of endogenous miR-107 enhances and inhibits, respectively, HIF1B expression and hypoxic signaling. Further, overexpression of miR-107 in tumor cells suppresses tumor angiogenesis, tumor growth, and tumor vascular endothelial growth factor (VEGF) expression in mice. Again, the relevance of these molecular circuitries in the cardiovascular system has not been demonstrated yet.
HIF1A can induce the expression of several miRNAs that, in turn, can regulate HIF1A with a feedback loop, either positive or negative (Fig. 2). Among HIF1A-induced miRNAs, miR-210 deserves a particular attention, since miR-210 induction is a virtually constant feature of the hypoxic response in both normal and transformed cells (17, 21, 40, 44, 61, 73, 109, 138). Indeed, HIF1A binds at specific sites of miR-210 promoter, activating its expression. Specifically, Kulshreshtha et al. (73) demonstrated in cancer cell lines, using a combination of promoter reporter and chromatin immunoprecipitation assays, the role of HIF1A in the upregulation of miR-210 by hypoxia. More recently, a specific functional HRE site was identified in hypoxic cancer cells (61) and in differentiating myoblasts (30). miR-210 expression is mainly stimulated by HIF1A, but an HIF1A-independent, AKT-dependent pathway has been described as well (99).
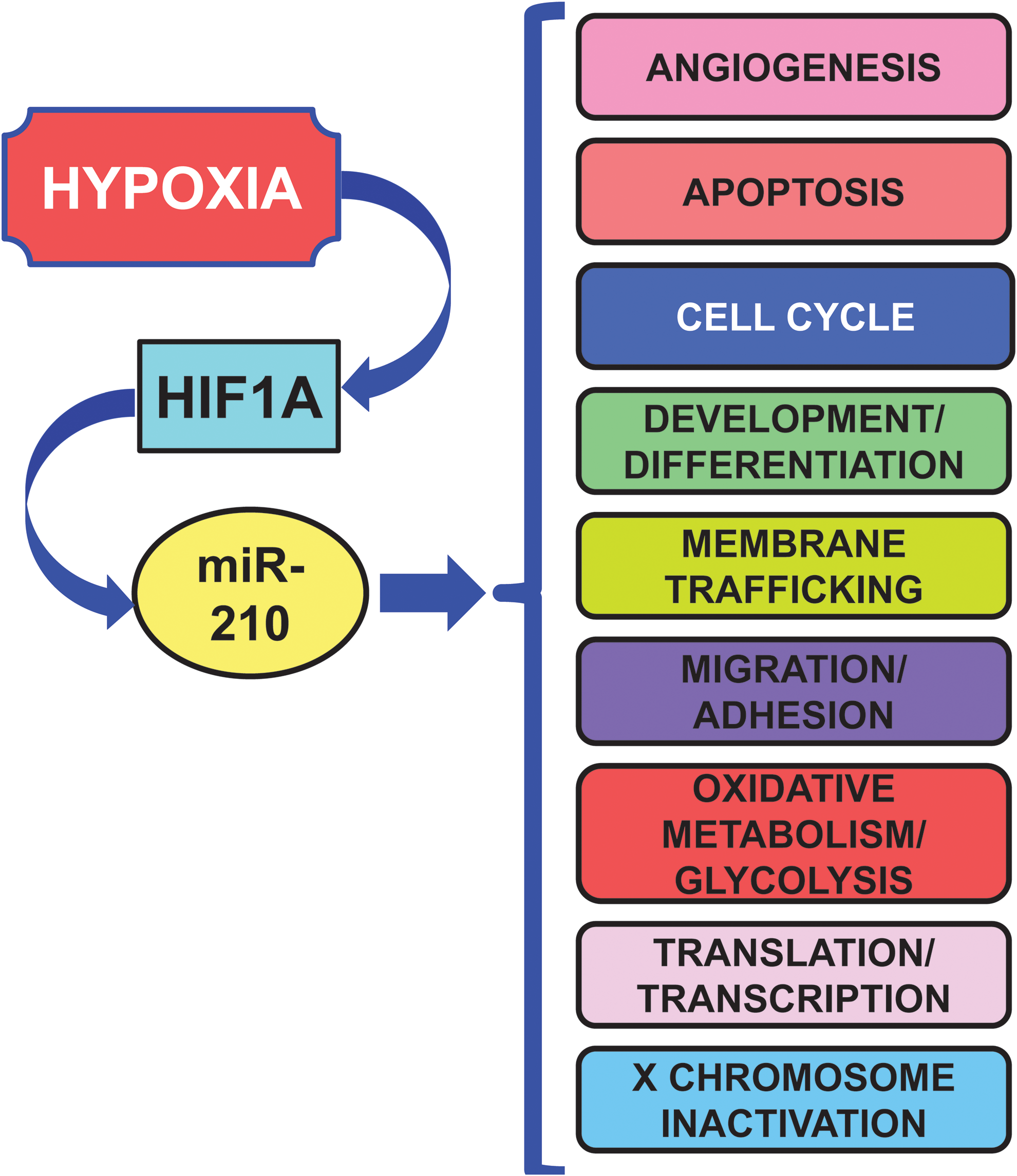
According to its consistent induction in different hypoxic situations, miR-210 regulates several key aspects of cardiovascular disease, summarized in Figure 3 and Table 2 (17, 40, 44, 61, 109, 161).
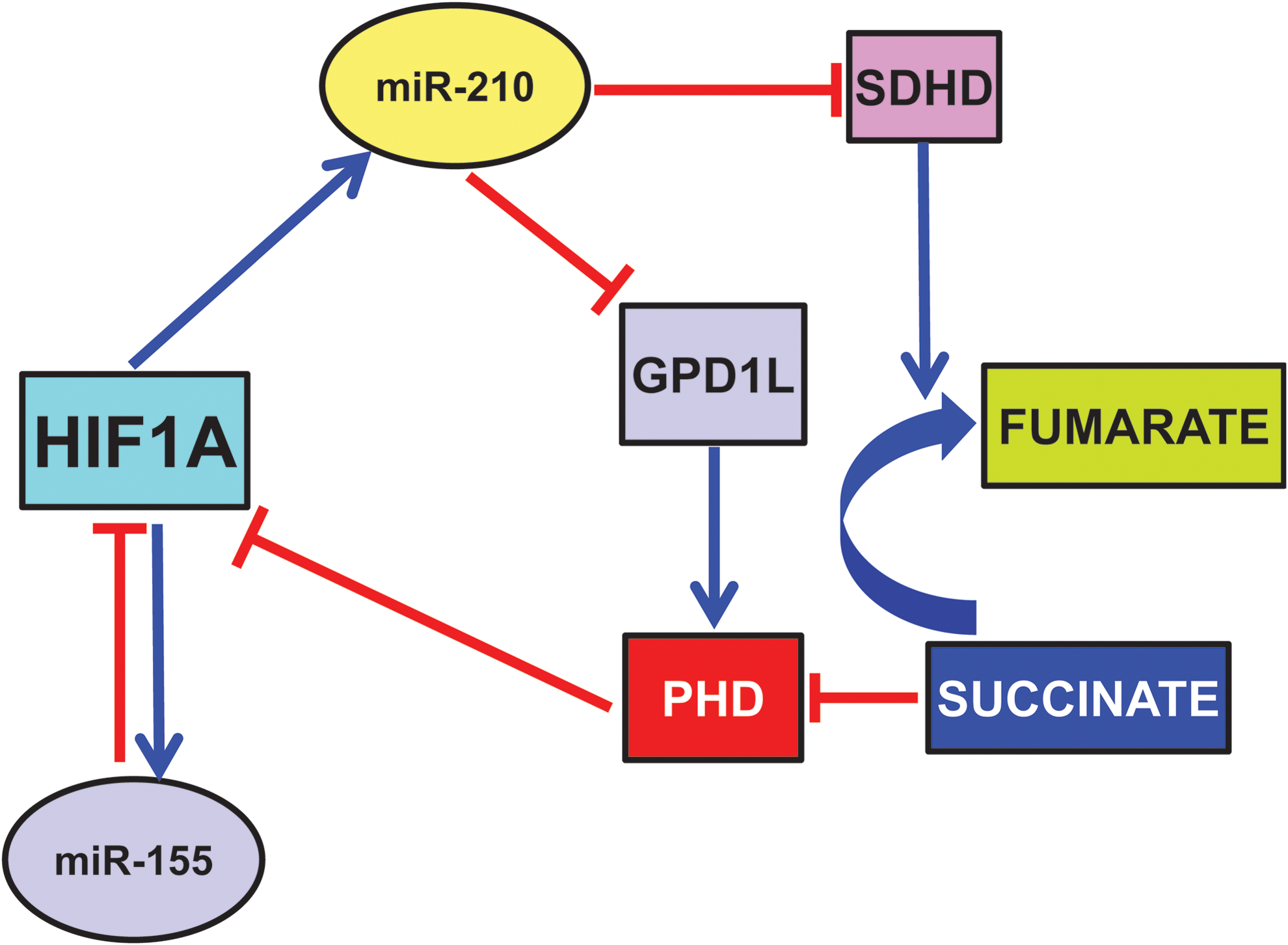
There is evidence of two mechanisms through which miR-210 triggers a positive feedback loop where HIF1A drives miR-210 expression, which further induces HIF1A protein stability. The first mechanism is via one of its direct targets, the enzyme glycerol-3-phosphate dehydrogenase 1-like (GPD1L) (42). Indeed, GPD1L increases proline hydroxylation of HIF1A, decreasing its stability. Thus, in normoxia, when HIF1A levels are low, small amounts of miR-210 result in high GPD1L protein levels and high activity of the PHDs, contributing to keep HIF1A levels at bay. When oxygen tension is lowered, decreased GPD1L protein potentiates the inhibition of PHD activity, leading to increased HIF1A stability (68).
Another mechanism is mediated by miR-210 modulation of oxidative phosphorylation (Cottrill et al., this issue). In particular, Puisségur et al. showed that the D subunit of the succinate dehydrogenase complex (SDHD) is a bona fide miR-210 target (109). Albeit more work is necessary, the current model proposes that miR-210-dependent demise in SDHD results in increased succinate accumulation, which is a by-product and natural inhibitor of PHD activity (123). However, miR-210-HIF1A positive feedback may not be present in all tissues and/or physiopathological conditions. Indeed, we found that miR-210 blockade did not affect HIF1A-pathway activation in a mouse model of acute hindlimb ischemia (158).
While the mechanism underpinning HIF activation is well understood, little is known about its resolution in cells exposed to prolonged hypoxia. Given their inhibitory nature, miRNAs are likely candidates for this role. miR-155 is an hypoxamiR directly targeting HIF1A transcript in gut epithelial cells (Fig. 2) (14). Overexpression of miR-155 decreases the HIF1A protein and transcriptional activity in hypoxia, and the blockade of endogenous miR-155 prevents the resolution of HIF1A stabilization and activity. Thus, miR-155 induction contributes to a negative feedback loop for the resolution of HIF1A activity in cells exposed to prolonged hypoxia, leading to the oscillatory behavior of HIF1A-dependent transcription.
Hypoxia Regulates the miRNA Biosynthesis Machinery
Hypoxia can modulate the miRNA biosynthesis machinery affecting miRNA levels and function, thereby eliciting broad effects (Fig. 4).

Data obtained in vitro and in vivo show that hypoxia impairs DICER expression and activity, resulting in global consequences on miRNA biogenesis (58). While most miRNAs remain unaffected, possibly due to DICER-independent maturation (31), miR-185 is decreased. This miRNA targets HIF2A, the main HIF species in endothelial cells. As a consequence, upon hypoxia HIF2A is de-repressed, increasing the expression of HIF-induced genes. Therefore, functional deficiency of DICER in hypoxia is relevant to both HIF and hypoxia-responsive/HIF target genes, especially in the vascular endothelium.
Hypoxia also induces type I collagen prolyl-4-hydroxylase [C-P4H(I)] (Fig. 4), which leads to prolyl-hydroxylation and accumulation of argonaute-2 (AGO2), a critical component of the RISC (111, 145). Hydroxylation of AGO2 is required for the association of AGO2 with heat shock protein 90, which, in turn, is necessary for the loading of miRNAs into the RISC, and translocation to stress granules. Moreover, hydroxylation of AGO2 increases miRNA levels and enhances the endonuclease activity of AGO2.
However, hypoxia regulation seems complex with both positive and negative elements. By deep sequencing of small RNA libraries derived from primary endothelial cells subjected to normoxia or hypoxia, Chen et al. found that Let-7, miR-103, and miR-107 are highly induced (Fig. 4) (27). Quite surprisingly, however, miR-210 is not reported as significantly induced as previously reported by many authors (17, 21, 40, 44, 61, 73, 109, 138). Let-7, miR-103, and miR-107 miRNAs are induced by HIF1A and target the RISC component AGO1. Targeting of AGO1 results in the translational de-repression of VEGF mRNA and stimulation of angiogenesis. Indeed, data from tumor xenografts and human cancer specimens indicate that AGO1-mediated increase of VEGF may be associated with tumor angiogenesis and poor prognosis. As described in the previous section, Yamakuchi et al. reported that miR-107 also targets HIF1B (151) and HIF1B targeting by miR-107 limits endothelial progenitor cell differentiation (96). However, this does not seem to be the case in mature endothelial cells or the inhibitory effect on HIF1B is overcome by the pro-angiogenic effect of AGO1 inhibition. Another element of apparent contradiction is that AGO1 silencing impairs Drosophila adaptation to hypoxia (35). However, differences in the experimental system and in the degree of AGO1 suppression may account for these apparently conflicting results.
HypoxamiR and Angiogenesis
Angiogenesis is a physiological process involving the growth of new blood vessels from pre-existing vessels. The regulation of angiogenesis by hypoxia is an important component of homeostatic mechanisms that link vascular oxygen supply to metabolic demand.
Several studies using quantitative polymerase chain reaction (21, 40) or RNA-sequencing (138) identified miR-210 as the prominent hypoxamiR in endothelial cells (Fig. 5). miR-210 overexpression in normoxic endothelial cells stimulates the formation of capillary-like structures, and VEGF-driven cell migration, whereas miR-210 blockade inhibits these events (40, 80, 87, 147). In keeping with these findings, anti-angiogenic effect of WSS25 sulfated polysaccharide is, at least in part, mediated by miR-210 down-modulation (147). Further, directed endothelial differentiation of embryonic stem cells is associated with miR-210 increased expression (65).
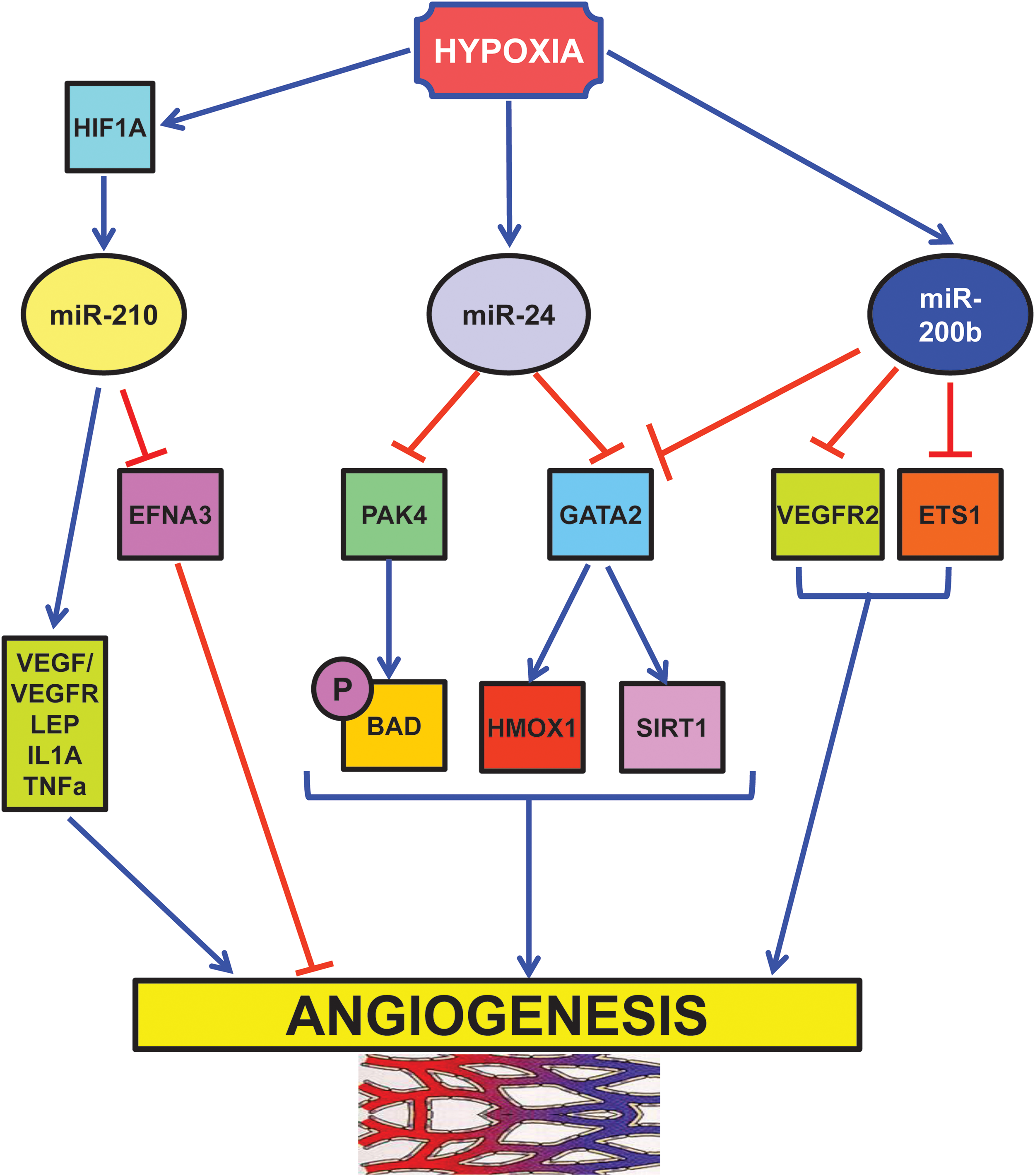
One critical direct target of miR-210 pro-angiogenic function is ephrin-A3 (EFNA3). Fasanaro et al. showed that the override of EFNA3 down-modulation either by miR-210 expression or by hypoxia prevents both tubulogenesis and chemotaxis stimulation (40). However, miR-210/EFNA3 relationship seems to be rather complex: hypoxia stimulates EFNA3 mRNA expression in endothelial cells, while miR-210 decreases EFNA3 protein levels (40). In this respect, suppression by miR-210 may represent a feedback loop to keep EFNA3 levels under control. In keeping with this model, Pulkkinen et al. found increased EFNA3 protein levels in post-ischemic mouse hippocampus (110). A possible differential expression in neuronal and endothelial compartments was not investigated.
It is also likely that miR-210 may act through other effectors as well, both direct and indirect. Accordingly, increased angiogenic factor signaling has been reported in miR-210-expressing cells (59, 80).
More in vivo evidence is needed to assess miR-210 pro-angiogenic role. Nevertheless, it is worth noting that tissue perfusion and capillary density in mouse ischemic hindlimb are significantly improved by the injection of umbilical cord blood CD34+ cells expressing miR-210 (4).
Another hypoxia-induced miRNA is miR-24 (73), which acts as a negative regulator of angiogenesis (Fig. 5) (45). Fiedler et al. found that miR-24 expression induces endothelial cell apoptosis, decreases endothelial capillary-like network formation on Matrigel, and inhibits cell sprouting from endothelial spheroids (45). These effects are mediated through direct targeting of the endothelium-enriched transcription factor GATA-binding protein 2 (GATA2) and the p21-activated kinase PAK4. These targets, in turn, impinge on BCL2-associated agonist of cell death and sirtuin-1 signaling cascades. The relevance of miR-24 modulation of angiogenesis is demonstrated by the impairment of zebrafish embryos angiogenesis upon overexpression of miR-24 or silencing of its targets. Moreover, the blockade of endothelial miR-24 limits mouse myocardial infarct size by preventing endothelial apoptosis and enhancing vascularity. These beneficial events, in turn, lead to preserved cardiac function and increased survival.
Hypoxia also negatively regulates miR-200b, a miRNA extensively studied in molecular oncology for its ability to regulate epithelial to mesenchymal transition (13). Overexpression of miR-200b in human microvascular endothelial cells inhibits and miR-200b blockade enhances angiogenesis in vitro, as evidenced by tube formation on Matrigel and cell migration (Fig. 5) (22). These events are mediated by miR-200b targeting of v-ets erythroblastosis virus E26 oncogene homolog 1 (ETS1) a crucial angiogenesis-related transcription factor: knocking down endogenous ETS1 is sufficient to inhibit angiogenesis, whereas overexpression of ETS1 rescues miR-200b-dependent impairment in angiogenic response and suppression of ETS1-associated gene expression. Interestingly, Magenta et al. found that miR-200c overexpression induces endothelial cell growth arrest, apoptosis, and senescence, indicating that the whole miR-200-family has an anti-angiogenic activity (91).
HypoxamiR and Heart Diseases
Myocardial infarction and hypoxamiR
Myocardial infarction (MI) is the irreversible damage of myocardial tissue caused by sustained hypoxia and ischemia. In most cases, MI occurs when a coronary artery becomes occluded upon the rupture of an atherosclerotic plaque, thereby leading to coronary thrombosis. When a vessel becomes completely occluded, the heart area perfused by that vessel will become ischemic and hypoxic causing myocardial cell death. Indeed, myocardial oxygenation measurement in infarcted rats by paramagnetic resonance oxymetry indicates that pO2 in the infarction site decreases from ∼20 to ∼3.0 mmHg (20). The damaged tissue is initially composed of a necrotic core surrounded by a border zone that, over time, can either recover to normal function or become irreversibly damaged (86).
Given its prominent role as master hypoxamiR, miR-210 is one of the better characterized hypoxamiRs in MI (Fig. 6). In cardiomyocytes exposed to hypoxia in vitro, miR-210 is robustly upregulated through both HIF-dependent and -independent pathways (59, 99). miR-210 expression upregulates several angiogenic factors, inhibits caspase activity, and prevents cell apoptosis. Further, downregulation of miR-210 increases reactive oxygen species levels after hypoxia-reoxygenation. These functions are attributable to miR-210 ability to modulate mitochondrial metabolism and reactive oxygen species generation by the electron transport chain (Fig. 3) (Cottrill et al., this issue) and by the repression of pro-apoptotic genes, such as PTPN1. In keeping with in vitro data, miR-210 is also induced following MI in humans (12). To test the relevance of miR-210 induction, Hu et al. injected a minicircle vector carrying miRNA-210 precursor in the border zone of a mouse model of MI (59). The protective role of miR-210 is highlighted both by echocardiography showing a significant improvement of left ventricular fractional shortening and by histological analysis, displaying decreased cellular apoptosis and increased neovascularization.
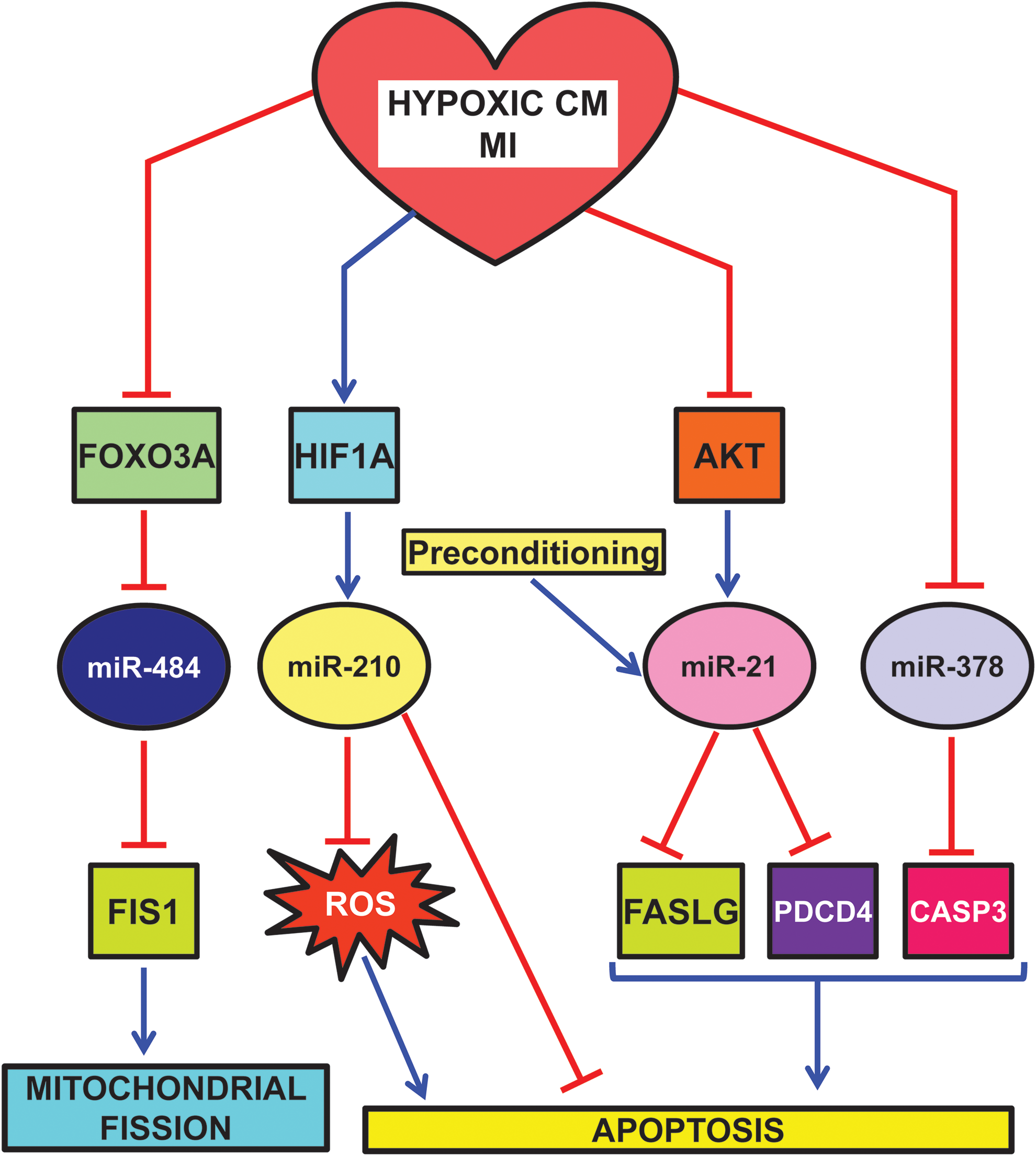
miR-210 involvement does seem to be limited to acute MI. Indeed, miR-210 is also induced in left ventricle biopsies of diabetic patients affected by post-ischemic heart failure (52).
miR-210 is not the only hypoxamiR modulating mitochondrial function and morphology (109). Indeed, mitochondria constantly undergo fusion and fission, two necessary processes for the maintenance of organelle fidelity. Wang et al. found that miR-484 can target mitochondrial fission protein FIS1, and inhibit FIS1-mediated fission and apoptosis in cardiomyocytes (Fig. 6) (142). During anoxia, miR-484 is downregulated and, as a consequence, FIS1 is de-repressed. Interestingly, miR-484 expression is regulated by forkhead box O3 (FOXO3A) transcription factor. Indeed, FOXO3A transgenic or knock-out mice exhibit, respectively, a high or low level of miR-484 and a reduced or enhanced mitochondrial fission, apoptosis, and MI damage.
Another miRNA playing an important role in MI is miR-21. miR-21 is positively regulated via an AKT-dependent pathway, which is down-modulated by sustained hypoxia (Fig. 6) (122). Accordingly, hypoxia-induced downregulation of miR-21 and upregulation of its target Fas ligand (FASLG) were reversed by activated AKT. More interestingly, the anti-apoptotic function of AKT, at least in part, requires miR-21, which is sufficient for inhibition of caspase-8 activity and mitochondrial damage. In keeping with these findings, overexpression of miR-21 in a transgenic mouse heart results in suppression of ischemia-induced upregulation of FASLG expression, a smaller infarct size, and less severe heart failure. Another target that may contribute to miR-21 anti-apoptotic role is programmed cell death 4 (PDCD4) (29).
In keeping with these data, Cheng et al. also found that miR-21 is also induced in heart after ischemic preconditioning (29). Accordingly, cardiac protection afforded by preconditioning against I/R injury is inhibited by knockdown of cardiac miR-21.
An additional miRNA down-modulated by both MI and hypoxia is miR-378 (Fig. 6) (39). In H9c2 cardiomyocyte cell line, miR-378 expression enhances cell viability, reduces lactate dehydrogenase release, and inhibits both apoptosis and necrosis. Conversely, miR-378 blockade aggravates hypoxia-induced apoptosis and cell injury. One interesting direct target is caspase-3, a key apoptosis executioner. However, more experiments are needed to ascertain the functional relevance of caspase-3 inhibition by miR-378.
Stem cells for MI therapy and hypoxamiR
miRNA induction by hypoxia also have repercussions in regenerative medicine for ischemic cardiac diseases. Preclinical studies in experimental animal models and some pioneering clinical trials have produced encouraging results that allow to envision angiogenic and myogenic repair of the ischemia-damaged heart by stem cell therapy (5). Despite these encouraging results, numerous problems still need to be solved. One of the most outstanding is massive death of donor cells in the ischemic myocardium after engraftment, since it greatly lowers the efficacy of the procedure.
Kim et al. reported that miR-210 is induced by ischemic preconditioning in bone marrow-derived mesenchymal stem cells. miR-210, in turn, improves their survival under subsequent longer exposure to anoxia and following engraftment in the infarcted myocardium (69). Accordingly, high miR-210 levels in mesenchymal stem cells, either by preconditioning or by miR-210 overexpression, accelerate the functional recovery of the ischemic heart after transplantation (69). Increased survival negatively correlates with the levels of caspase-8-associated protein-2 (CASP8AP2) (Fig. 3). Interestingly, miR-210/HIF1A positive feedback loop (Fig. 2) may also play an important role in increasing cell survival (25). Moreover, in vitro co-culture experiments show miR-210 transfer from mesenchymal stem cells to cardiomyocytes, indicating that mesenchymal cells may also act as a miRNA delivery system (70).
Another promising cell type is bone marrow mononuclear cells. Xu et al. found that bone marrow cells isolated from MI patients display increased miR-210 and miR-34a levels (148). The latter appears as particularly important for this cell type: inhibition of miR-34 significantly decreases hydrogen peroxide-induced cell death in vitro, whereas its overexpression reduces the bone marrow cell survival. Significantly, ex vivo blockade of miR-34a also increases the therapeutic benefit of transplanted bone marrow cells in mice after acute MI.
It is known that hypoxia maintains cultured skeletal myoblasts in an undifferentiated state by inhibiting myogenic differentiation by Notch pathway activation (55, 93). In this respect, hypoxia, downregulates miR-1 and increases the levels of its target gene Pax7, therefore inhibiting differentiation and promoting myoblasts satellite cell self-renewal (82). The negative regulation of miR-1 is mediated by the Notch signaling pathway, in particular, by the hypoxia-induced activation of Hes1 and Hey.
Interestingly, Hes1 and miR-1 seem to regulate one another. Indeed, in normoxic condition, miR-1 overexpression promotes the differentiation of mesenchymal stem cells toward the cardiac lineage by direct targeting of Hes1 (60).
Finally, pioneering studies identified a paracrine mechanism involving the secretion of miR-132 and inhibition of its target genes. Katare et al. assessed the regenerative potential of saphenous vein-derived pericyte progenitor cells (67). Transplantation of pericytes into the peri-infarct zone of mouse hearts attenuates left ventricular dilatation and improves ejection fraction. These beneficial effects are, at least in part, due to hypoxia-induced miR-132. Indeed, in vitro studies show that miR-132 is actively secreted by pericytes; moreover, pericyte conditioned medium stimulates endothelial cell tubulogenesis and reduces myofibroblast differentiation, through inhibition of methyl-CpG-binding protein 2 (MECP2) and RAS p21 protein activator (GTPase-activating protein) 1 (RASA1), which are both validated miR-132 targets. Further, miR-132 inhibition decreases pericyte ability to improve contractility, reparative angiogenesis, and interstitial fibrosis following MI.
HypoxamiR and Stroke
Stroke is a multifactorial disease with two major causal events: obstruction of a blood vessel in the brain (cerebral ischemia) or disruption of a cerebral vessel (hemorrhagic stroke). In cerebral ischemia, the most characterized hypoxamiR is, again, miR-210.
miR-210 is upregulated in adult rat ischemic brain cortexes, along with NOTCH1 signaling molecules that are also increased (88). This may be relevant for the compensatory angiogenesis occurring after cerebral ischemia that increases the blood flow to the injured area and limits extension of the ischemic penumbra. Indeed, NOTCH signaling regulates the vascularization process that occurs in ischemic tissues. In keeping with the in vivo findings, cultured hypoxic endothelial cells also display increased miR-210 and NOTCH1-pathway components. These events are tightly linked, since miR-210 overexpression causes the upregulation of NOTCH1 signaling molecules and induces endothelial cells to migrate and form capillary-like structures on Matrigel.
miR-210 is also a potential stroke marker. Zeng et al. measured miR-210 levels in blood samples from stroke patients harvested at different times (159). Compared with healthy controls, blood miR-210 was significantly decreased in stroke patients, with a minimum at 7 and 14 days after stroke onset. This is quite surprising since miR-210 is induced both by hypoxia and ischemia in multiple cell types and tissues, including the ischemic brain (88). However, in keeping with the anti-apoptotic function of miR-210, decreased miR-210 levels may relate to the severity of damage. Indeed, miR-210 level is significantly higher in stroke patients with good outcome than in patients with poor outcome (159).
HypoxamiR Peripheral Artery Disease and Wound Healing
Peripheral artery disease commonly involves the arteries supplying the leg and is mostly caused by atherosclerosis (105). Restriction of blood flow due to arterial stenosis or occlusion often leads to muscle pain on walking (intermittent claudication). Any further reduction in blood flow causes ischemic pain at rest, which affects the foot. Ulceration and gangrene may then supervene and can result in loss of the limb, if not treated. Although many patients with claudication remain stable, some progress to critical limb ischemia and need revascularization. Unfortunately, those who benefit from successful revascularization suffer from high rate of recurrent symptoms or revision surgery and many still require progressive amputations.
As for almost all ischemic diseases, miR-210 role has been investigated in peripheral artery disease and ischemic wounds. Indeed, we found that miR-210 is induced in a mouse model of hindlimb ischemia (158). Accordingly, Li et al. propose miR-210 as serum biomarker for atherosclerosis obliterans (78).
In a mouse model of ischemic wound healing, in vivo imaging shows significant HIF1A stabilization in ischemic wounds (9) (Fig. 7). HIF1A induces miR-210 expression that, in turn, silences its target E2F transcription factor 3 (E2F3), which is markedly downregulated in the wound-edge tissue of ischemic wounds. E2F3 transcription factor has a well established pro-proliferative effect. Indeed, knock down of E2F3 limits keratinocyte (HaCaTs) proliferation; accordingly, miR-210 overexpression and inhibition experiments demonstrate a key role of miR-210 in limiting keratinocyte proliferation. Interestingly, miR-210 does not seem to have the same role in all cell types. Indeed, miR-210 overexpression has no significant effect on endothelial cell proliferation (40).
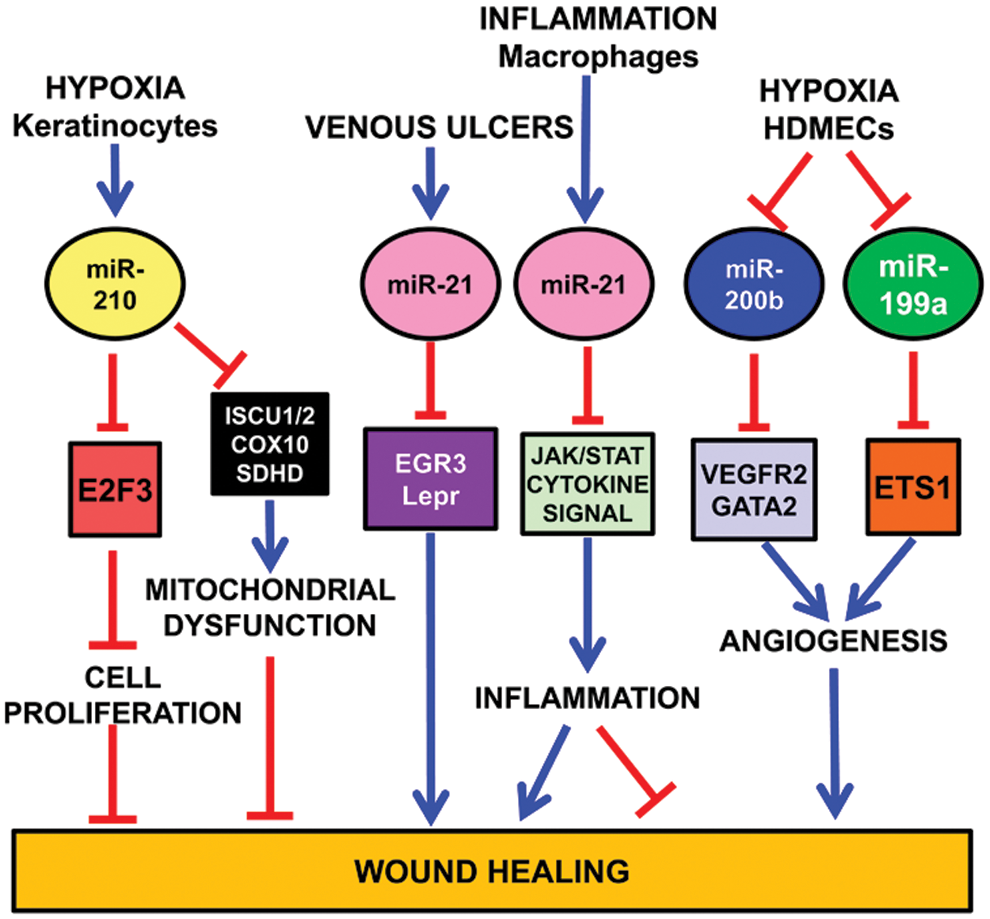
As previously discussed, miR-210 targets ISCU1/2 (21, 28), COX10 (28), and SDHD (109), slowing down mitochondrial respiration under a severe hypoxic insult. On the one hand, this response allows affected cells to survive longer. On the other hand, miR-210 also impairs functions necessary for active tissue regrowth that is required for wound healing (126) (Fig. 7).
Similar to miR-210, miR-21 also is a negative player in cutaneous wound healing (126). Indeed, miR-21 is upregulated in human venous ulcers and overexpression of miR-21 inhibits epithelialization and granulation tissue formation in a rat wound model. Interestingly, miR-21 directly targets early growth response factor 3 and the leptin receptor, which may mediate miR-21 effects, at least in part (104).
Further, miR-21 is TGF-β-inducible, contributing to its growth arrest properties in keratinocytes (143). However, conflicting reports have been published as well (155).
Fibrosis is another important process that is induced by miR-21 (118). Indeed, miR-21 overexpression in wound healing in vitro assays increases ability of fibroblasts to migrate toward the wound area, whereas knocking down of miR-21 has the opposite effect (90). Concordant evidences come from different systems: in a mouse model of heart I/R, miR-21 regulates matrix metalloprotease (MMP-2) expression in cardiac fibroblasts of the infarct zone via a phosphatase and tensin homologue pathway (118). Moreover, miR-21 contributes to myocardial disease by stimulating MAP kinase signaling in fibroblasts of failing hearts (134).
Finally, miR-21 also regulates the inflammatory process (119). Indeed, miR-21 is induced by proinflammatory stimuli such as IL-6, TNF-α, and TLRs (89, 127). Moreover, miR-21 targets several genes involved in JAK/STAT and cytokine pathways (47, 126), and JAK-STAT signaling is directly implicated in cytokine-mediated inflammation. Thus, miR-21 may have an anti-inflammatory effect (16, 133) (Fig. 7).
Also miR-200 family plays an important role in wound healing. In human dermal microvascular endothelial cells (HDMECs), downregulation of miR-200b, which targets GATA2 and VEGFR2 expression, stimulates cutaneous wound angiogenesis (24) (Fig. 7). Accordingly, diabetic wounds exhibit high levels of miR-200b and complementarily low GATA2 and VEGFR2 expression. The impairment of miR-200b downregulation in the early phase of wound healing is mediated by elevated TNF-α. These data are consistent with the finding that diabetic wounds are refractory to resolution of inflammation and display a prolonged abundance of proinflammatory mediators.
Another hypoxamiR involved in wound healing is miR-199a (Fig. 7). Inhibition of endogenous miR-199a in HDMECs supports angiogenesis, while overexpression of miR-199a blocks the angiogenic response in Matrigel cultures (23). miR-199a targets Ets-1 in HMECs. Accordingly, MMP-1, one of the Ets-1 downstream mediators enabling endothelial migration (37), is negatively regulated by miR-199a. These data together indicate that the downregulation of miR-199a is involved in the induction of wound angiogenesis through derepressing the Ets-1-MMP-1 pathway.
HypoxamiR and Kidney I/R
I/R is one of the main causes of acute tubular necrosis, underpinning most of the cases of acute renal failure. Upon ischemia, apical actin cytoskeleton of tubule cell is rapidly reorganized and adhesion molecules change their localization. These events lead to impairment of cell-to-matrix and cell-to-cell adhesion structures and cell detachment, causing severe kidney dysfunction (10).
As expected, miR-210 is induced in a mouse model of kidney I/R, correlating with increased angiogenesis in the ischemic region (Fig. 8) (80). Interestingly, pathological changes on a cellular level are also detectable in more accessible specimens such as plasma: in patients with non post-renal acute kidney injury, circulating miR-210 is upregulated and predicts survival (85).
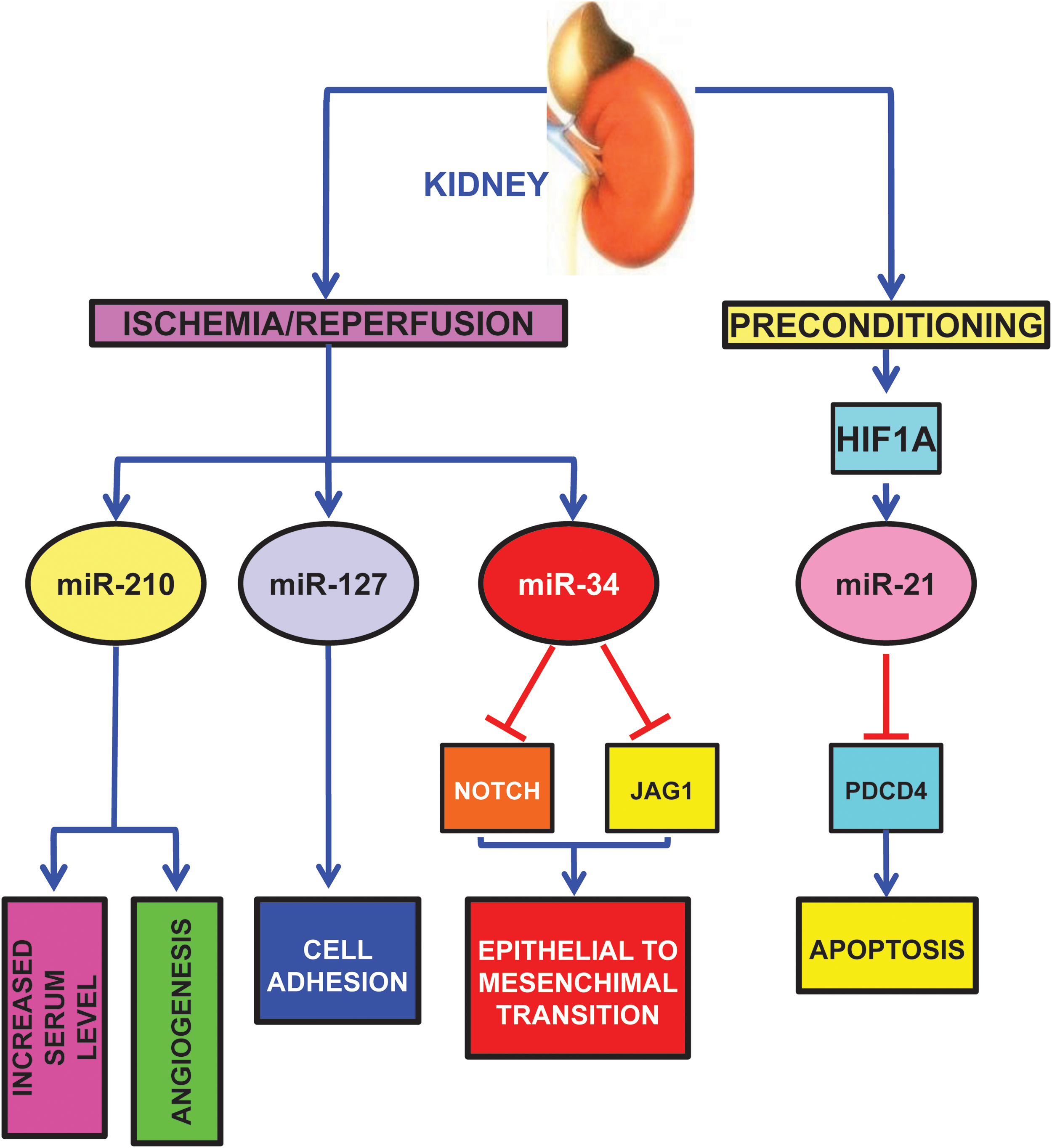
Other hypoxamiRs seem to play a role in kidney ischemia as well. miR-127 is induced in proximal tubule cells cultured in conditions mimicking I/R with an HIF1A-dependent mechanism (Fig. 8) (2).
miR-127 is involved in cell–matrix and cell–cell adhesion maintenance, since its over-expression contributes to maintain the integrity of focal adhesion complexes and tight junctions. miR-127 targets kinesin family member 3B, which regulates cell trafficking; in keeping with this finding, miR-127 inhibition promotes dextran-FITC uptake.
Hypoxia-induced renal tubular cell epithelial-mesenchymal transition is another important pathogenetic mechanism, leading to renal fibrosis. In this respect, Du et al. found that hypoxia decreases miR-34a expression (Fig. 8) (38). miR-34a, in turn, promotes epithelial-mesenchymal transition of renal tubular epithelial cells by directly targeting NOTCH1 and JAG1, and subsequently, NOTCH downstream signaling.
Finally, in keeping with its function in ischemic heart preconditioning, miR-21 also plays a protective role in kidney delayed preconditioning (Fig. 8) (150). Indeed, ischemic preconditioning induces miR-21 with an HIF1A dependent mechanism and its blockade significantly exacerbates subsequent ischemia-reperfusion injury and tubular cell apoptosis in the mouse kidney. Accordingly, inhibition of miR-21 also results in the upregulation of its pro-apoptotic target PDCD4. Significantly, in the absence of ischemic preconditioning, blockade of miR-21 alone does not affect ischemia-reperfusion injury in the mouse kidney.
HypoxamiR and Pulmonary Hypertension
Pulmonary hypertension (PH) is a complex vascular disease with diverse etiologies, including hypoxia (115). Regardless the cause, PH induces a pathological dysregulation in the pulmonary vasculature and a maladaptive increase in pulmonary arterial pressure. PH involves numerous intersecting signaling pathways leading to deregulated vascular tone, cellular proliferation, and thrombosis.
Parikh et al. used a systems biology approach to integrate network analysis of genome-wide mRNA expression data, algorithmic prediction of miRNA targets and established concepts of biological pathways involved in PH, along with in vitro and in vivo experimentation (103). They identified miR-21 as the most compelling of a select group of miRNA predicted to control the expression of a convergent set of pathways in PH. Indeed, miR-21 is upregulated by both hypoxia and bone morphogenetic protein receptor 2 (BMPR2) signaling, two established PH triggers. In turn, miR-21 targets a fundamental vascular effector, Ras homolog family member B (RHOB), and induces molecular changes in pulmonary endothelial cells triggering vasodilation and decreased angiogenesis. Further, miR-21 is upregulated in the lung PH patients and in rodent models of PH. Significantly, in one of these models, the genetic absence of miR-21 causes accelerated disease progression.
However, it is worth noting that miR-21 expression pattern and functions are not entirely consistent throughout the literature. For instance, Caruso et al. report the downregulation, rather than upregulation, of miR-21 in both human and rodent PH lungs (19). Moreover, the administration of miR-21 complementary oligonucleotides in vivo, decreases pulmonary vascular remodeling in hypoxic rodents (153), in contradiction with the data obtained using genetically deleted miR-21 mice (103). Further, some studies have indicated that miR-21 can drive a pro-proliferative state in hypoxic human pulmonary artery smooth muscle cells in culture (121).
Clinical differences, and in particular the BMPR2 genotype, and technological differences, may explain these results, at least in part. However, albeit the reasons for the observed discrepancies are not fully clarified, it is evident that miR-21 carries a central function in controlling the progression of PH.
Finally, it is not surprising that miR-210 is induced in the lung of mice exposed to chronic hypoxia (51) and in human pulmonary artery smooth muscle and endothelial cells exposed to low oxygen tension in vitro (21, 51).
Potential Clinical Applications
Therapeutic perspectives
Understanding the pathogenetic meaning of hypoxamiR alterations represents a new frontier for both molecular biologists and clinicians. An expanded understanding of hypoxamiR function in gene regulatory networks associated with cardiovascular diseases will allow the identification of novel molecular mechanisms of disease and the development of innovative therapeutic strategies. This appears as particularly important considering that miRNAs are “druggable” targets. A phase IIa study has been recently published describing the use of Miravirsen, a chemically modified complementary locked nucleic acid (LNA) that binds to miR-122, disabling the binding and growth of the Hepatitis C virus (63). Interestingly, the drug was very well tolerated, suggesting that this may be a feature of this new class of LNA drugs. While much more work is needed, several promising preclinical studies indicate that this new therapeutic modality may be a viable approach also for cardiovascular diseases (114, 137).
In addition to miRNA inhibitors, there is also the possibility to mimic or re-express miRNAs by using synthetic RNA duplexes designed to mimic the endogenous functions of the miRNA of interest, with modifications for stability and cellular uptake. However, this approach has revealed to be technically challenging and is still in the preclinical stage. Moreover, even more so than for miRNA inhibitors, precursor delivery to the appropriate cell type or tissue is an important aspect of effective miRNA mimicry, to prevent unwanted side effects.
Another way to increase the level of a miRNA is by the use of viral vectors, such as of adeno-associated viruses (AAVs). These vectors display not only tissue specific expression, if opportune promoters are used, but also tropism toward different organs, according to AAV serotype (106). However, all the caveats and limitations that apply to the use of viral vectors for gene therapy should be kept in mind (53).
It is also possible to modulate miRNAs using small molecules (Table 1). One example is represented by Atorvastatin, a statin that inhibits endothelial senescence and enhances SIRT1 in HUVECs stimulated by oxidative stress (102). SIRT1 is a modulator of vascular endothelial cell homoeostasis and is a direct target of miR-34a (107). Interestingly, it has been shown that miR-34a negatively regulates SIRT1 expression in endothelial progenitor cells from coronary artery disease patients; atorvastatin can inhibit miR-34a and prevent SIRT1 decrease, providing an additional mechanism to the beneficial effects of atorvastatin on endothelial function in coronary artery disease (131).
miR-200 family is also modulated by small molecules. Indeed, oxidative stress-inducing drug 1,3-bis(2 chloroethyl)-1-nitrosourea (BCNU) upregulates miR-200c, and to a lower extent, miR-200a and b. These miRNAs, in turn, induce endothelial cell senescence and apoptosis (91). Accordingly, 5-azacytidine (5-AzaC) induces miR-200c and cellular senescence of human umbilical cord blood-derived multipotent stem cells (130).
Moreover, the administration of nitropropionic acid (3-NPA), an irreversible inhibitor of succinate dehydrogenase used to induce ischemic tolerance, down-modulates miR-199a in rat brains (149).
Finally, HIF is a promising target for a variety of diseases and an ever increasing number of small molecule agonists and inhibitors of HIF has been developed, some of which are progressing through preclinical and early clinical development (146). While no specific studies in this sense exist, it is reasonable to anticipate that pharmacological activation or inhibition of HIF will also lead to the up- or down-modulation of HIF-dependent hypoxamiR, respectively.
Diagnostic/prognostic perspectives
A new frontier has been opened by the recent discovery that miRNAs are present in the bloodstream and in other bodily fluids, in a remarkably stable form (36, 135). Because of their stability and their tissue- and disease-specific expression and the possibility to measure them with high specificity and sensitivity, miRNAs are emerging diagnostic biomarkers. Indeed, several miRNAs, including some hypoxamiR, display the potential to act as biomarkers for a wide range of cardiovascular diseases, including MI, coronary artery disease, hypertension, heart failure, viral myocarditis, and type-2 diabetes mellitus. For instance, several studies indicated an increase of circulating miR-1, miR-133a, miR-133b, and miR-499-5p following acute MI in both humans and mice (1, 3, 34, 50, 74, 140). Moreover, it has also been reported that circulating miR-208 is enhanced in some, but not all patients with acute MI (1, 32, 34, 100, 140).
Concluding Remarks
In 2007, the pioneering work of Mircea Ivan et al. identified the first group of hypoxia-induced miRNAs (73). Thereafter, evidence rapidly accumulated implicating hypoxamiR in many cardiovascular diseases.
Certain hypoxamiR seem to have a particularly pervasive role, such as miR-210. Indeed, miR-210 is modulated in virtually all ischemic diseases tested so far. We described studies implicating miR-210 in MI, stroke, peripheral ischemia, kidney I/R, PH, and wound healing. Moreover, miR-210 is also induced in the tissue around the necrotic area in human osteonecrosis (152) and in human atherosclerotic plaques (116), a finding implicating miR-210 in all thromboembolic diseases. Intriguingly behavioral and nutritional habits may also contribute to the modulation of miR-210. The HUNT-Study identified increased miR-210 levels in the serum of healthy subjects with low aerobic fitness, measured as maximal oxygen uptake, a good indicator of cardiovascular health (15). Moreover, in vitro experiments show that the green tea catechin epigallocatechin gallate can induce miR-210 with a HIF1A-dependent mechanism (141), suggesting that nutritional habits may influence miR-210 levels. However, several other hypoxamiR have been shown to be modulated in cardiovascular diseases, representing both adaptive and maladaptive mechanisms.
In conclusion, the described studies have convincingly demonstrated the robust and complex nature of hypoxamiR in the control of the hypoxic and ischemic responses. Further investigation of their regulation, their targets, and their physiological and/or pathogenic effects in cardiovascular diseases are anticipated in the coming years.
Footnotes
Acknowledgments
F.M. and S.G. are supported by Ministero della Salute and Associazione Italiana per la Ricerca sul Cancro (Grant AIRC IG-11436). Dr. Biagina Maimone, IRCCS Policlinico San Donato, Milan, is acknowledged for her critical reading of the article.