
Abstract
Introduction
C
In this study, we observed the decelerated age-related accumulation of oxidative damage in ubiquinol-10-supplemented mice with accelerated senescence. Our major finding is that ubiquinol-10 induces a pathway that activates SIRT1, SIRT3, peroxisome proliferator-activated receptor γ coactivator 1α, and mitochondrial function. Activation of this pathway modulates oxidative damage in the liver and inner ear by enhancing the activity of mitochondrial antioxidant defense systems, which results in significantly delayed senescence and age-related hearing loss. These findings suggest that pharmaceutical treatments which include ubiquinol-10 supplementation to activate sirtuin pathways could prevent mitochondrial decay associated with aging and decelerate age-related diseases by increasing oxidative stress resistance.
Senescence-accelerated mouse (SAM) strains provide a unique model system for studying the aging process in higher organisms. SAM strains include the accelerated senescence-prone senescence-accelerated mouse prone (SAMP) strains that have markedly shorter life spans and exhibit early signs of aging (64). SAMP1, a strain in the SAMP series, exhibits accelerated progression of many age-associated degenerative diseases such as senile amyloidosis, impaired immune response, hyperinflation of the lungs, and AHL (31, 65). AHL is a universal feature of mammalian aging and is the most common sensory disorder in the elderly (62). SAMP1 mice show age-related hearing impairment, which is closely related to chronological age, and also manifestations of accelerated senescence (51, 52). These SAMP strains also showed mitochondrial dysfunction and a higher oxidative stress status (11). Therefore, as a model of presbycusis, SAMP1 mice should prove useful. Here we examined whether dietary supplementation with ubiquinol-10 could exert beneficial effects against accelerated senescence and age-related diseases in SAMP1 mice and elucidate the mechanism of the anti-aging effect of ubiquinol-10.
Aging and age-related diseases in higher organisms is a complex process that is likely controlled by a combination of many different genetic, environmental, nutritional, and pathologic factors. While the specific role of oxidative stress in aging and development of age-related diseases is an area of active investigation, the exact mechanisms that may define this complex relationship are unclear (69). A better understanding of this relationship may help identify useful biomarkers of oxidative stress, which can be objectively measured as indicators of normal and pathologic processes that result in age-related diseases. Cell aging is thought to be due to cumulative cellular and genomic damage that results in permanent cell-cycle arrest, apoptosis, or senescence (49). A major source of cellular damage is reactive oxygen species (ROS), which are mainly generated at complexes I and III of the cellular RC (2). ROS generation was estimated to represent 0.1%–2% of total oxygen consumption (15, 35), and an early study suggested that its generation rate is thought to increase with age due to the accumulation of damaged mitochondria in a vicious cycle in which ROS-induced mitochondrial impairment results in increased ROS production, which, in turn, leads to further mitochondrial damage in aging (10, 14). However, recent research revealed that mitochondria ROS generation does not need to increase with age to cause aging (3). Nutrient intake and numerous genetic mutations affect the rate of aging with a concomitant alteration of mitochondrial metabolism and ROS accumulation, suggesting that mitochondrial homeostasis can be regulated during the aging process. However, recent research revealed that besides the harmful effects of ROS, ROS play an important physiological role as “redox messengers” in intracellular signaling and regulation by modulating fundamental cell death and cell survival processes, including apoptosis and autophagy (28). In addition, since many exceptions and contradictory findings to the ROS accumulation theory have been reported recently (6, 18), further research to understand the role of oxidative damage and mitochondria dynamics in the aging process is needed.
Metabolic pathways are co-ordinated through reversible acetylation of metabolic enzymes in response to nutrient availability and calorie intake (59). The sirtuin family has emerged as key regulators of the nutrient-sensitive metabolic regulatory circuit. Sir2 plays a direct role in the anti-aging effects of calorie restriction (CR) in yeast (21), while overexpression of SIRT1, the mammalian Sir2 homolog, was reported to protect mice from age-related phenotypes such as type 2 diabetes (2), cancer (19), and Alzheimer's disease (13). However, there are inconsistent results as to whether SIRT1 activity extends the lifespan of Caenorhabditis elegans (19). SIRT3 regulates the global acetylation landscape of mitochondrial proteins, and SIRT3-initiated metabolic adaptations enhance mitochondrial management of ROS. SIRT3 increases the activity of antioxidants such as superoxide dismutase 2 (SOD2) and reduced glutathione (GSH), and promotes ROS scavenging (44, 66). A recent study on AHL in mice clearly showed that ROS production could diminish the effects of sirtuins (62). Mice sustain cumulative oxidative damage to hair cells and spiral ganglia neurons of the inner ear cochlea, resulting in hearing loss in aged mice. CR induced SIRT3 protein expression in mice, so it is likely that this sirtuin mediates ROS management, mitochondrial integrity, and sensory function, at least in those neurons which govern hearing.
Our recent study indicated that ubiquinol-10 up-regulated genes are downstream of PPARα (54). PPARα is found mainly in oxidative tissues such as cardiac muscle, skeletal muscle, and liver, and its activation is known to be regulated by peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) (12), which controls mitochondrial biogenesis and respiration in tissues. Thus, we hypothesized that an important mediator of the metabolic effects of ubiquinol-10 may be PGC-1α activation. Since SIRT1 is known to deacetylate and activate PGC-1α, we further hypothesized that ubiquinol-10 may increase the activity of sirtuin families and PGC-1α in mice in a way similar to CR, and, in turn, reduce the oxidative damage that can promote AHL.
In this report, we show that ubiquinol-10 can prevent age-related oxidative stress and activate mitochondrial function by inducing sirtuin and Pgc-1α gene expression in SAMP1 mice, which protects against the symptoms of age-related diseases.
Results
Ubiquinol-10 reduced oxidative damage in SAMP1 mice
To investigate whether ubiquinol-10 plays a role in senescence, we conducted a study using SAMP1 mice. We supplemented the diets of young, middle, and old SAMP1 mice with ubiquinol-10, starting at 1, 7, and 13 months of age, respectively. Mice supplemented with ubiquinol-10 from a young age showed no significant changes in body weight, but a significant decrease was observed in mice that were supplemented with ubiquinol-10 beginning in middle or old age as compared with mice fed the control diet (Supplementary Fig. S1A, B; Supplementary Data are available online at
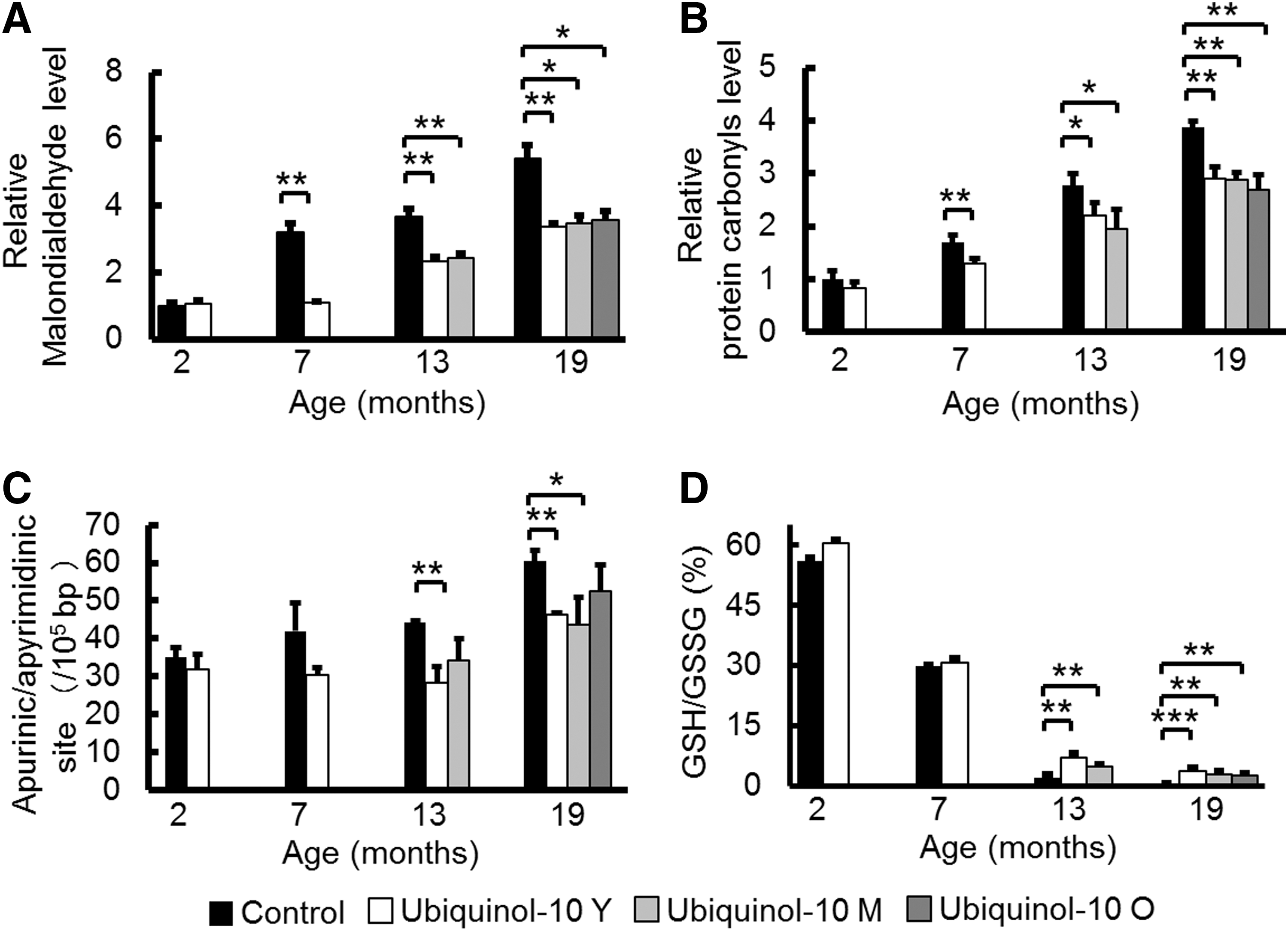
Since we reported that ubiquinol-10 showed its most distinct effects on gene expression in the liver, to elucidate the mechanism of decelerated aging effects, we first measured oxidative damage to lipids, protein, and DNA in SAMP1 mouse livers (54). We evaluated malondialdehyde levels as a measure of oxidative damage to lipids in the liver of control and ubiquinol-10 diet mice at 2, 7, 13, and 19 months and observed age-related increases in lipid damage in control diet mice that were suppressed by ubiquinol-10 supplementation (Fig. 1A). The same result was found for oxidative damage to proteins (e.g., protein carbonyls) (Fig. 1B) and DNA as assessed by apurinic/apyrimidinic (AP) sites (Fig. 1C). Glutathione acts as the major antioxidant in cells, with NADPH-dependent glutathione reductase regenerating GSH from oxidized glutathione (GSSG). We measured the ratio of GSH:GSSG in the livers of control and ubiquinol-10 diet SAMP1 mice at 2, 7, 13, and 19 months of age and found that age-related decreases in the ratios of GSH:GSSG could be suppressed in mice supplemented with ubiquinol-10 from a young age, as well as during middle and old age (Fig. 1D). These results showed that ubiquinol-10 reduces oxidative damage to lipids, proteins, and DNA in SAMP1 mice and that ubiquinol-10 may promote a more reductive environment for liver mitochondria.
To confirm the accumulation of CoQ in the liver, homogenate, cytosol, and mitochondrial concentrations of total coenzyme Q9 (CoQ9) (dominant CoQ form in mice) and total CoQ10 (the sum of the concentrations of the reduced and oxidized forms, respectively) were determined (23). Ubiquinol-10 supplementation in SAMP1 mice produced different changes in the liver concentrations of CoQ10 and CoQ9 from 7 months of age. Supplementation with ubiquinol-10 for 7 months significantly elevated homogenate liver, cytosol, and mitochondrial concentrations of CoQ10. However, there were no significant increases in the CoQ9 concentrations in SAMP1 mice supplemented with ubiquinol-10 (Supplementary Fig. S2).
Ubiquinol-10 suppressed age-related decreases in the expression of genes and proteins related to mitochondrial function
CoQ10 is an electron carrier from enzyme complex I and complex II to complex III in mitochondria, while ubiquinol-10 may enhance the antioxidant activity of mitochondria. Therefore, we hypothesize that ubiquinol-10 supplementation might increase mitochondrial activity. To test this possibility, we extracted total RNA and proteins from the livers of 2-, 7-, 13-, and 19-month-old mice and examined age-related changes in sirtuin family members and Pgc-1α gene and protein expression levels by real-time polymerase chain reaction (PCR) and western blot, respectively. We found an age-related decrease in the expression of Sirt1, Sirt3, and Pgc-1α mRNA, and ubiquinol-10 supplementation in mice from a young, middle, and old age suppressed these age-related decreases (Fig. 2A). To confirm the results from our previous study (54), we measured Pparα mRNA expression and found similar increases in expression. In addition, western blot analyses using antibodies to SIRT1, PGC-1α, SIRT3, estrogen-related receptor alpha (ERRα), and SOD2 showed that the levels of these mitochondria-related proteins also decreased with age, and these decreases were significantly reduced for young and middle age groups after ubiquinol-10 supplementation (Fig. 2B, C). Thus, these data provide strong biochemical evidence that ubiquinol-10 suppresses age-related decreases in the expression of genes and proteins related to mitochondrial functions.

Supplementation of ubiquinol-10 decelerates senescence and suppresses AHL progression
SAMP1 mice exhibit accelerated progression of many age-associated degenerative diseases, including AHL. To investigate whether ubiquinol-10 plays a role in AHL, we analyzed the auditory brainstem response (ABR) in 2-, 7-, 13-, and 19-month-old mice. For control mice, we observed an age-related increase in the ABR hearing thresholds at high (32 kHz), middle (16 kHz), and low (8 kHz) frequencies (Fig. 3A–C), while mice supplemented with ubiquinol-10 from a young age had hearing thresholds at high frequencies that were significantly lower at 7 and 13 months (Fig. 3A). For middle (16 kHz) and low (8 kHz) frequencies, age-associated hearing losses were suppressed by ubiquinol-10 supplementation (Fig. 3B, C). These results demonstrate that ubiquinol-10 plays an essential role in AHL in SAMP1 mice.

We confirmed that aging resulted in increased ABR hearing thresholds at high (32 kHz) frequencies in 7-month-old SAMP1 mice fed a control diet (Fig. 3A), indicating that these mice displayed hearing loss. Ubiquinol-10 suppressed the exacerbation of AHL in mice fed a supplemented diet. Since ubiquinol-10 was shown to regulate mitochondrial metabolic activity in the liver, we hypothesize that ubiquinol-10 may similarly regulate mitochondrial metabolic activity in the cochleae. By determining the expression of mitochondria-related genes and proteins in cochleae, we could show that, in agreement with the ABR test and liver results, Sirt3 and Pgc-1α mRNA and protein expression was significantly increased after ubiquinol-10 supplementation (Fig. 3D, E). These results provide strong biochemical evidence that ubiquinol-10 regulates mitochondrial metabolic activity by increasing SIRT1, PGC-1α, and SIRT3 levels in the cochleae of SAMP1 mice.
Ubiquinol-10 decelerated the decline in mitochondrial function associated with aging
PGC-1α is known to be an important mediator of the metabolic effects of SIRT1. SIRT1 deacetylates and activates PGC-1α (48), while SIRT3 deacetylates and activates isocitrate dehydrogenase 2 (IDH2) (20) and SOD2 (44, 66). We observed that ubiquinol-10 supplementation suppressed the age-related decrease in Sirt1 and Sirt3 levels in the liver (Fig. 2B). Based on these findings, we examined the acetylation levels of PGC-1α, SOD2, and IDH2. PGC-1α, SOD2, and IDH2 were immunoprecipitated with their respective antibodies and acetylation levels of PGC-1α, SOD2, and IDH2 were detected with an anti-acetyl lysine antibody. We found that acetylation levels of PGC-1α, SOD2, and IDH2 increased in old mice (19 months), and ubiquinol-10 supplementation suppressed this increase (Fig. 4A–C). Thus, our findings provide evidence that ubiquinol-10 activates PGC-1α, SOD2, and IDH2 after SIRT1-dependent deacetylation and activation of SIRT3.
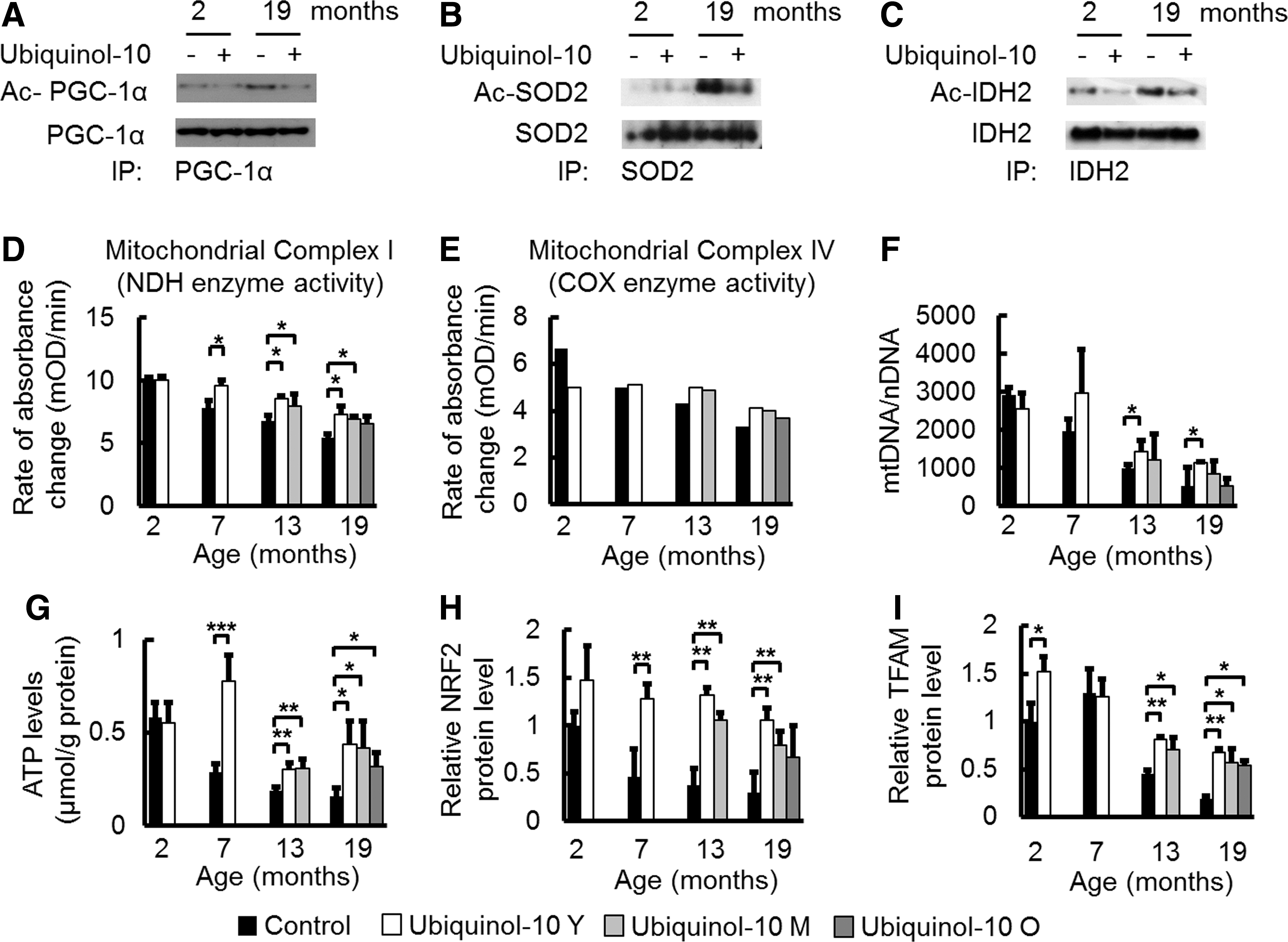
The primary function of mitochondria is production of ATP via oxidative phosphorylation, which is conducted by the four RC complexes (complexes I–IV) and ATP synthase (complex V), all of which are located in the inner mitochondrial membrane. To provide evidence that ubiquinol-10 increases mitochondria function in SAMP1 mice, we determined the effect of ubiquinol-10 on the activity of complexes I and IV (cytochrome-c oxidase, EC 1.9.3.1) in the liver, as the activities of these complexes were found to be decreased with age (36). We found that both complex I and IV activity decreased with age, and compared with control animals, decreased complex I activity was significantly suppressed in mice supplemented with ubiquinol-10 during young and middle age (Fig. 4D, E). In several studies, an increase in mtDNA damage and mutations resulted in decreased amounts of mitochondria. As such, we investigated the effect of ubiquinol-10 on mtDNA copy number during aging. Ubiquinol-10 supplementation significantly suppressed a decrease in mtDNA copy number when ubiquinol-10 supplementation started at a young age (Fig. 4F). In addition, age-related decreases in liver ATP levels in control diet mice were suppressed by ubiquinol-10 supplementation started from young, middle, and old age (Fig. 4G). We observed an effect of ubiquinol-10 on PGC-1α downstream targets and mitochondrial biogenesis-related proteins such as nuclear factor erythroid 2-related factor 2 (NRF2) and mitochondrial transcription factor A (TFAM). Western blot analyses showed that the levels of these proteins also decreased with age, but these decreases were significantly reduced for young and middle age groups after ubiquinol-10 supplementation (Fig. 4H, I). Thus, ubiquinol-10 promoted deacetylation of PGC-1α, SOD2, and IDH2 via SIRT1 and SIRT3 and increased the function and amount of mitochondria via activation of PGC-1α.
Ubiquinol-10 suppressed age-related decreases in cAMP levels and the expression of phosphorylated CREB, AMPK, and LKB1
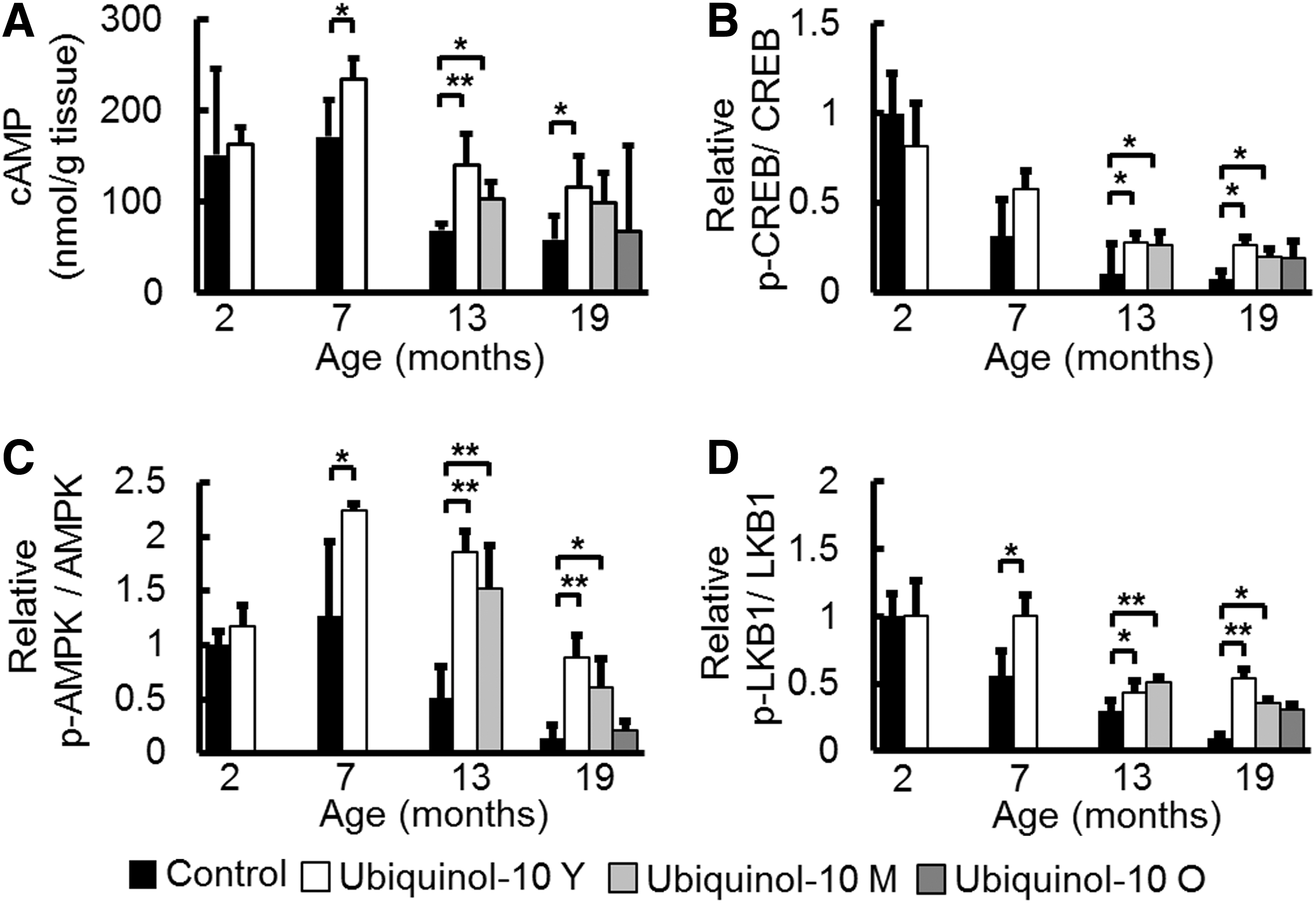
While the mechanism by which ubiquinol-10 activates SIRT1 and PGC-1α remains unclear, a recent study revealed that increased cyclic adenosine monophosphate (cAMP) levels mediated the metabolic effects of PGC-1α on mitochondrial biogenesis and function by increasing PGC-1α expression (43). We found that cAMP levels decreased with aging, and compared with control animals, ubiquinol-10 supplementation from young and middle age significantly suppressed this decrease (Fig. 5A). The cAMP response element-binding protein (CREB) is a transcription factor that is induced by a variety of growth factors and inflammatory signals and subsequently mediates the transcription of genes containing a cAMP-responsive element. A previous study showed that CREB binds to a target sequence in the PGC-1α promoter, thereby enhancing its expression in response to cAMP. We found that age-related decreases in the ratio of phosphorylated-CREB/CREB were suppressed by ubiquinol-10 supplementation (Fig. 5B). AMP-activated protein kinase (AMPK) is a trimeric complex that senses nutrient deprivation by responding to changes in the AMP/ATP (9) and ADP/ATP (73) ratios. AMPK, which is emerging as a key regulator of whole-body metabolism, was shown to increase NAD+ levels and activate SIRT1 and PGC-1α (8). We determined that phosphorylated AMPK levels were reduced by aging and that these reductions could be slowed by ubiquinol-10 (Fig. 5C). We also found that ubiquinol-10 supplementation suppressed age-related decreases in phosphorylated serine/threonine kinase B1 (LKB1), which is the critical upstream kinase required for AMPK phosphorylation (Fig. 5D). These data provide biochemical evidence that ubiquinol-10 regulates mitochondrial metabolic activity by increasing the activity of the cAMP-AMPK-Sirt1-PGC-1α pathway.
Ubiquinol-10 increased the deacetylation status of PGC-1α by SIRT1 activation and stimulated AMPK phosphorylation in HepG2 cells
To confirm that ubiquinol-10 can activate SIRT1, PGC-1α, and AMPK signaling pathways, we examined its effect on these protein levels in human hepatoma HepG2 cells. SIRT1, PGC-1α, and phosphorylated AMPK were increased in the ubiquinol-10-treated HepG2 cells in a dose-dependent manner (Fig. 6A), with the expression of SIRT1, PGC-1α, and phosphorylated AMPK increasing with a low ubiquinol-10 dose (≤3 μM). The PGC-1α acetylation levels were decreased by ubiquinol-10, and the SIRT1 inhibitor nicotinamide reversed the effect of SIRT1 (Fig. 6B). Based on these observations, we next sought to determine whether SIRT1 was capable of directly interacting with PGC-1α. As shown in Figure 6C, a SIRT1 antibody co-immunoprecipitated with PGC-1α. Ubiquinol-10 increased the levels of co-immunoprecipitated PGC-1α, while nicotinamide reversed this effect (Fig. 6C). Since ubiquinol-10 may activate AMPK by increasing cAMP production, we next treated HepG2 cells with ubiquinol-10 in the presence of the adenylyl cyclase (AC) inhibitor MDL-12,330A (Fig. 6D). MDL-12,330A inhibited the ability of ubiquinol-10 to increase the phosphorylation of both AMPK and the AMPK substrate acetyl-CoA carboxylase (ACC), which is a marker of AMPK activity. These findings indicate that cAMP-AMPK signaling activation is essential for ubiquinol-10 to exert its effects.

Ubiquinol-10 increased mitochondrial metabolic activity
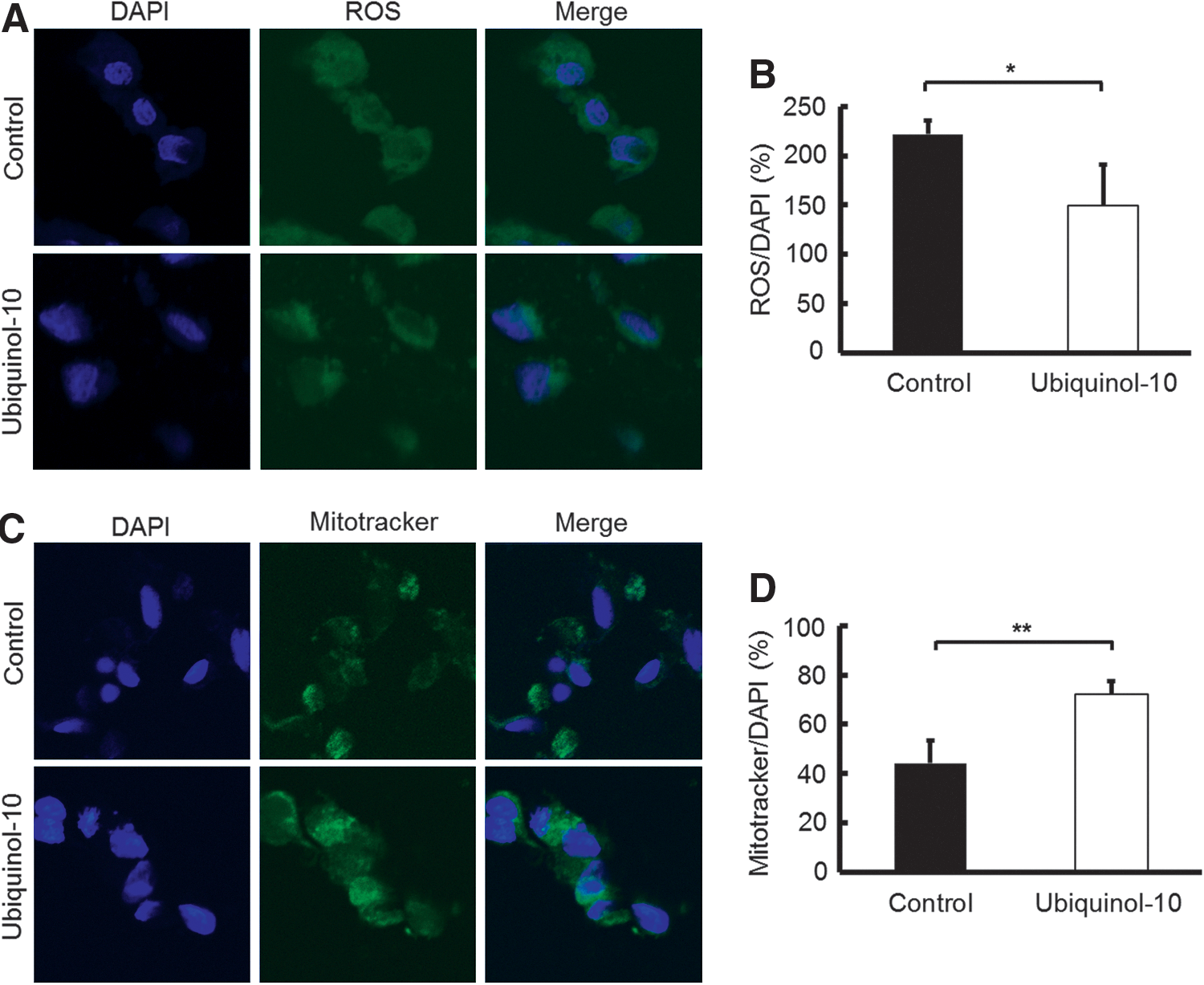
To determine the effect of ubiquinol-10 on mitochondrial metabolic activity, HepG2 cells were treated with ubiquinol-10 and the oxygen consumption rate was determined. Ubiquinol-10 treatment resulted in a significantly increased rate of oxygen consumption (Fig. 6E, F). The NADH and NAD concentrations were also measured in ubiquinol-10-treated HepG2 cells. The NAD/NADH ratio was significantly increased after ubiquinol-10-treatment (Fig. 6G). We next measured the total ROS production using specific fluorescent probes and found that the total ROS levels were significantly decreased by ubiquinol-10-treatment (Fig. 7A, B). The possibility that the effect of ubiquinol-10 might be associated with mitochondria biogenesis was investigated further in HepG2 cells by labeling mitochondria with Mitotracker. HepG2 cells treated with ubiquinol-10 had higher numbers of mitochondria compared with control cells (Fig. 7C, D). Together, these data provide biochemical evidence that ubiquinol-10 increases the amount of mitochondria and regulates mitochondrial metabolic activity.
Discussion
A widely accepted hypothesis of how aging leads to age-related diseases is that oxidative damage accumulates in tissues (17). In mammals, increased resistance to oxidative stress that can be provided by antioxidants is consistently associated with delayed aging, resistance to age-related diseases, and enhanced longevity (60). Ubiquinol-10 is regarded as one of the most important antioxidants. Ubiquinol-10 biosynthesis decreases with age and its deficit in tissues is believed to be associated with degenerative changes appearing over the course of aging (5, 22). Our experimental evidence indicates that aging, indeed, results in an increase in oxidative damage to DNA, proteins, and lipids and a decrease in the ratio of GSH:GSSG in the liver (Fig. 1). Therefore, we speculate that ubiquinol-10-mediated modulation of the antioxidant defense system may play a central role in reducing oxidative stress in tissues, and, in turn, retard the effects of aging.
Sirtuins are proposed to be regulators of aging and age-related diseases (7, 56). SIRT1, the most studied member of the family, plays an important role in several processes ranging from cell cycle regulation to energy homeostasis. In this study, we found that age-related decreases in SIRT1 levels were suppressed by dietary ubiquinol-10 supplementation (Fig. 2), which also decelerated the progression of age-related diseases and senescence (Fig. 3 and Supplementary Fig. S1), but no life span extension was observed (Supplementary Fig. S1F). This latter finding may be because Sirt1 has so many important functions in mammalian physiology and metabolism, and global up-regulation of SIRT1 can exert opposing effects, or, alternatively, global up-regulation may, indeed, slow overall aging, but has no effect on life spans in the mouse strains tested.
SIRT3, the most studied mitochondrial sirtuin, deacetylates a number of mitochondrial proteins and might also play a role in regulating ATP production (42, 55). A large body of evidence suggests that CR reduces the age-associated accumulation of oxidative damage of proteins, lipids, and DNA by enhancing the activity of mitochondrial antioxidant defense systems (29, 71). Some analyses showed that CR increases SIRT3 levels, which resulted in lower ROS production and decreased amounts of oxidative damage to mitochondria (62). SIRT1 expression has also been reported to be positively regulated by CR (14) and exercise (72). Interestingly, SIRT3 expression was shown to be regulated by the activity of the ERRα-dependent coactivator PGC-1α that binds to the SIRT3 promoter (16), while SIRT1 was convincingly shown to deacetylate PGC-1α to increase its potential to activate transcription (38). With ubiquinol-10 supplementation, SIRT1 protein levels increased in liver tissue, resulting in increased mitochondrial biogenesis in response to activated PGC-1α-dependent effects on mitochondrial biogenesis and respiration after its activation by SIRT3. Thus, SIRT1 activates SIRT3 by promoting ERRα-dependent PGC-1α activation (Fig. 8). Here, we found that age-related decreases in SIRT3 levels were suppressed by ubiquinol-10 supplementation (Fig. 2).

We demonstrated that ubiquinol-10 supplementation suppressed age-related decreases and acetylation of SOD2 (Figs. 2B and 4B), which is a major mitochondrial antioxidant enzyme. Decreased SOD2 activity causes elevated ROS production, mitochondrial membrane potential loss, and early cell apoptosis (27). SIRT3-dependent SOD2 deacetylation increased the specific activity of SOD2 and its ROS-scavenging capacity. We also confirmed that ubiquinol-10 supplementation suppressed age-related acetylation of IDH2 (Fig. 4C). Data from other groups showed that IDH2 activation is induced in response to increased ROS levels and enhances the activity of the glutathione antioxidant defense system in mitochondria, and that SIRT3 could enhance deacetylation and activation of IDH2 (62). Hence, SIRT3 may modulate oxidative damage in the liver by enhancing the activity of mitochondrial antioxidant defense systems. The capacity of PGC-1α to promote mitochondrial biogenesis is related to its actions on the transcription of NRF1 and NRF2, both of which are up-regulated by PGC-1α. NRF1 and NRF2 are key trans-acting elements in mitochondrial biogenesis that regulate the transcription of several nuclear-encoded genes, including TFAM, which is itself a vital coordinator of the transcription and replication of the mitochondrial genome. Here, we demonstrated that ubiquinol-10 affects the anti-oxidative system by activating expression of genes related to mitochondrial function, including SIRT1, SIRT3, PGC-1α, SOD2, IDH2, NRF2, and TFAM in the liver and that these effects likely arise from an enhancement of mitochondrial activity (Figs. 2, 4, and 8).
The primary function of mitochondria is ATP production, which is conducted by mitochondrial RC complexes. Mitochondrial complex I is a transmembrane protein complex that oxidizes matrix NADH to NAD+, and reduces membrane-bound ubiquinone-10 to ubiquinol-10. We found that complex I and IV activity decreased during aging, and that ubiquinol-10 supplementation suppressed this decrease (Fig. 4D, E). The activity of mitochondrial electron transfer is suggested to depend on the concentration of ubiquinone-10 in the inner mitochondrial membrane (25, 47). Therefore, the suppression of age-related decreases in complex I activity after ubiquinol-10 supplementation may be due to an increase either in the inner mitochondrial membrane CoQ10 concentration or in the complex I protein expression levels. Since the complex I activity did not depend on the presence of ubiquinone (MS141; MitoSciences, Eugene, OR), ubiquinol-10 supplementation likely affects complex I expression levels. In addition, age-related decreases in complex I (NADH dehydrogenase [ubiquinone] 1 β subcomplex subunit 8, NDUFB8) and complex IV (mitochondrially encoded cytochrome c oxidase I, MTCO1) protein levels were suppressed in mitochondria fractions from SAMP1 mice fed a ubiquinol-10-supplemented diet (Supplementary Fig. S3). Ubiquinol-10 supplementation also inhibited the reduction in mtDNA levels that was seen during aging for mice fed a control diet (Fig. 4F). Taken together, these results show that ubiquinol-10 may slow the progression of age-related diseases, including hearing loss by enhancing mitochondrial activity and mitochondrial antioxidant defense system activity and increasing ATP production (Figs. 3 and 4). Previous studies showed that the mean ABR hearing threshold at low frequencies was significantly lower in ubiquinone-10-supplemented control mice (61, 62). Similar to SAMP1 mice, ubiquinone-10 suppressed AHL in C57BL/6J mice by increasing antioxidant activities via SIRT3 activation.
While ubiquinol-10 suppressed age-related decreases in the expression of SIRT1, SIRT3, and PGC-1α, its direct effect remains unclear. cAMP plays a key role as an intracellular second messenger for transduction events that occur in response to a number of extracellular signals (30). We observed that age-related decreases in cAMP levels were suppressed by ubiquinol-10 supplementation (Fig. 5A). The CREB is probably one of the best understood phosphorylation-dependent transcription factors. Increases in either calcium or cAMP levels can induce the activation of CREB, which leads to the transcription of target genes that carry cAMP-responsive elements in their promoters (30). A recent study indicated that increases in cAMP production were responsible for AMPK activation (41). Activated AMPK triggers both phosphorylation and deacetylation of PGC-1α, with the latter being catalyzed by SIRT1. SIRT1 activation follows AMPK-dependent increases in NAD+. Phosphorylation of Thr 172 in the activation loop of AMPK is required for AMPK activation, and several groups have demonstrated that the LKB1 directly mediates this event (58). LKB1 is well-documented as the critical upstream kinase required for AMPK activation. The effect of cAMP is proposed to involve protein kinase A (PKA) phosphorylation and activation. In addition, genetic and biochemical findings indicate that LKB1 exerts its effects by phosphorylation related to PKA. Aging might result in lower levels of AMPK phosphorylation due to age-related decreases in cAMP. In our experiment, ubiquinol-10 supplementation lessened the age-related decrease in the ratio of phosphorylated AMPK/AMPK and phosphorylated LKB1/LKB1 (Fig. 5). However, we cannot exclude the possibility that ubiquinol-10 may also induce AMPK phosphorylation through calmodulin-dependent protein kinase kinase and intracellular calcium. Taken together, ubiquinol-10 may induce some pathways that activate mitochondrial function by increasing cAMP levels and activating CREB, LKB1, and AMPK (Fig. 8). Nevertheless, as discussed earlier, the mechanism of ubiquinol-10 action other than mitochondria activation should be considered to explain in greater detail its beneficial effect on aging. For example, the ubiquinol-dependent antioxidant system of the plasma membrane was suggested to be important for preventing the accumulation of oxidative damage and regulating the externally initiated ceramide signaling pathway (37). In fact, antioxidant activity of CoQ10 in liver plasma membrane was confirmed in aged rats fed a polyunsaturated fatty acids plus CoQ10 diet (4). Furthermore, a recent study showed that ubiquinol in endothelial cell Golgi compartments may be an important cofactor to maintain endothelial nitric oxide synthase in a coupled conformation that is required to produce physiological nitric oxide (NO), an important mediator of cardiovascular homeostasis, and, eventually, quench leaking-uncoupled electrons (34).
In summary, we propose that ubiquinol-10 may induce pathways that activate SIRT1 and PGC-1α by increasing cAMP levels and activating CREB and AMPK. SIRT1 can activate SIRT3 via ERRα-dependent activity of PGC-1α. SIRT3 modulates oxidative damage in the liver by enhancing the activity of mitochondrial antioxidant defense systems, resulting in significantly delayed senescence and age-related diseases in SAMP1 mice fed a diet supplemented with ubiquinol-10. We postulate that this may be a mechanism of aging retardation promoted by ubiquinol-10, which suggests that ubiquinol-10 may be useful for treating age-related diseases. In future studies, we will use Sirt1−/− and Sirt3−/− mice to decipher the direct effect of ubiquinol-10 supplementation other than mitochondria activation. In SAMP1 mice and various cell lines, we can determine the effect of ubiquinol-10 on other tissues, including adipose and muscle tissues.
Materials and Methods
Animals and cell culture
Methods involving animals and cell culture are described in detail in the Supplementary Data.
Measurement of oxidative damage
Oxidative damage to lipids (malondialdehyde) and proteins (protein carbonyls) were measured by western blot analysis. DNA oxidation levels (AP sites) were determined using a DNA damage quantification kit (Dojindo, Rockville, MD) according to the manufacturer's instructions. GSH and GSSG concentrations were determined with the GSSG/GSH Quantification Kit (Dojindo) according to the manufacturer's instructions.
Real-time reverse transcription-polymerase chain reaction analysis
Total RNA was extracted from the livers of 2-, 7-, 13-, and 19-month-old SAMP1 mice using TRIzol Reagent (Invitrogen, New York, NY), followed by treatment with DNA-Free (Applied Biosystems, Foster City, CA) to remove contaminating DNA and then subjected to reverse transcription using an Omniscript RT kit (Applied Biosystems, New York, NY) with random primers. Quantitative real-time RT-PCR analysis was carried out using an ABI PRISM 7500 Sequence Detection System with SYBR Green (Takara Bio, Tokyo, Japan). We assessed the stability of some genes and found that glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin are the most stable genes in our experiment and, as such, were suitable for use as reference genes. The forward and reverse primer sequences are listed in Table 1.
mtDNA quantitation
To quantify the amount of mtDNA per nuclear genome, we used the following primers. To quantify nDNA, we used a primer set that detects the platelet/endothelial cell adhesion molecule 1 (Pecam 1) gene on chromosome 6. Quantification of the relative copy number of mtDNA to nDNA was carried out using analysis of the difference in threshold amplification between mtDNA and nDNA (ΔΔC(t) method) using real-time PCR. The forward and reverse primer sequences are listed in Table 1.
Mitochondrial complex activity assay
To determine the activity of mitochondrial complex I, a microplate assay kit for complex I activity (MitoSciences) was used with 50 μg mitochondrial proteins. Complex I was immunocaptured on microplates, and the activity was determined from the oxidation of NADH to NAD+. Complex I activity was measured by the increase in absorbance at 450 nm and expressed as the change in absorbance per minute per microgram of protein. The cytochrome c oxidase (COX) enzyme (Complex IV) was immunocaptured within the microplate wells and determined colorimetrically by absorbance changes at 550 nm.
Cyclic AMP measurement
Cyclic AMP levels were determined using a cyclic AMP chemiluminescent kit (Cell Signaling Technology, Danvers, MA) according to the manufacturer's instructions.
Measurement of ATP levels
ATP was extracted from livers using the AMERIC-ATP kit (AMERIC, Tokushima, Japan). Extracts (10 μl) were added to 90 μl luciferase reagent and measured immediately. Based on the maximum light intensity of the resulting bioluminescence measured in a luminometer (Luminescencer PSN AB2200; ATTO, Tokyo, Japan), a calibration curve with varying ATP dilutions was also prepared. Data were normalized as ATP μmol/g protein.
Measurement of ROS
The oxidative stress levels in HepG2 cells were measured by a commercial kit according to the manufacturer's instructions (Total ROS/Superoxide Detection kit; Enzo Life Sciences, New York, NY). HepG2 cells were sedimented by centrifugation at 400 g for 5 min and then washed. The washed cells were incubated with 1 μM ROS/Superoxide Detection Mix for 60 min at 37°C. Green and blue fluorescence intensities were measured using a microplate fluorescence reader (BioTek, Winooski, VT) at excitation/emission wavelengths of 488/520 and 350/470 nm, respectively.
Mitochondria staining
MitoTracker® Green FM (Cell Signaling Technology) was added into the culture media at a final concentration of 200 nM. HepG2 cells were incubated for 30 min, and then visualized by fluorescence microscopy (Axiovert 200 M; Carl Zeiss, Jena, Germany).
Respirometric assay
For respirometric analysis, oxygen consumption was measured using a commercial kit according to the manufacturer's instructions (MitoXpress®-Xtra; Luxcel, Cork, Ireland). The MitoXpress oxygen probe was added to the culture media at a final concentration of 3 μM. Samples were then covered with 100 μl of pre-warmed (37°C) heavy mineral oil and read kinetically for 1 h at 30°C on a fluorescence plate reader (SpectraMax Gemini; Molecular Devices, Sunnyvale, CA) at excitation/emission wavelengths of 380/650 nm, with a measurement cycle of 1 min.
NAD/NADH analysis
NAD and NADH were analyzed independently in extracts of whole cells (1×106) prepared as previously described (57). NAD and NADH concentrations were determined using a Fluorimetric NAD/NADH Ratio Assay Kit (AAT Bioquest, Inc., Sunnyvale, CA).
Statistical analysis
We used the StatView software package (Abacus Concepts, Berkeley, CA) for data analysis. Real-time PCR, western blot, and ELISA results are presented as the mean±SD, body weight, degree of senescence, and food intake results are presented as the mean±SE. Nonparametric Mann–Whitney tests were used for in vivo results, as a normal distribution could not be assumed and Kaplan–Meier curves were used for animal survival. Body weights were compared by repeated measures ANOVAs. A p-value less than 0.05 was considered significant.
Footnotes
Acknowledgments
The authors thank Masayasu Kitagawa, Taizo Kawabe, Masanori Kato, and Shuka Matsumoto (Frontier Biochemical & Medical Research Laboratories, Kaneka Corporation) for determining the mitochondria concentrations of CoQ9 or CoQ10 and preparing the mouse diet. They also thank Kiyoshi Matsumoto (Research Center for Human and Environmental Science, Shinshu University) for technical assistance and care of mice. This research was supported in part by Grant-In-Aid 2012 from the Japanese Coenzyme Q Association.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.