
Abstract
Introduction
H
The metabolic response to hypoxia is characterized by a shift in ATP production to glycolysis and lactic acid fermentation at the expense of oxidative phosphorylation. This shift is associated with the suppression of apoptosis, as well as a reduction in oxygen-sensing potassium channels (70) and quenching of cytosolic ROS (62). Since anaerobic metabolism is inherently less efficient than glucose oxidation, such cells also show an associated increase in glucose transport and processing to compensate for the loss of ATP (95). All metazoan cells display this so-called “glycolytic shift” when exposed to low levels of oxygen (1) (known as the “Pasteur effect”), and on short time scales, such adaptations serve to improve cell survival and function by striking an optimal balance between cellular energy production and oxidative stress. During chronic or prolonged hypoxia, however, this phenomenon can result in persistent changes in cellular energy metabolism that do not resolve when oxygen supplies are restored. This “Warburg effect” is considered a major component of many chronic pathologies, including cancer (95), pulmonary hypertension (91), and others. Moreover, even when anaerobic metabolism does not persist, the long-term effects of mitochondrial ROS production during hypoxia can be seen in cases of stroke (85), hypoxic-ischemic injury (7), and diabetes mellitus (24, 68). In all such cases, hypoxia has a profound effect on cellular metabolism, and these changes have clinical relevance to a wide range of seemingly disparate diseases.
At the heart of the hypoxic response is hypoxia-inducible factor (HIF), often referred to as the master regulator of the hypoxic response (45). HIF is a heterodimeric transcription factor that is composed of either HIF-1α or HIF-2α and HIF-1β. Under normoxic conditions, HIF-α is targeted by the prolyl hydroxylase (PHD) family of enzymes, which add post-translational modifications to HIF-α for recognition by the von Hippel-Lindau tumor suppressor protein (VHL) (80). After its association with VHL, HIF-α is ubiquitinated and rapidly degraded by the 26S proteasome. This process is oxygen dependent, and in hypoxic conditions, prolyl-hydroxylation of HIF-α is suppressed, allowing for the dimerization of HIF-α and HIF-β (80). A third HIF-α isoform, HIF-3α, lacks the transactivation domain that is common to both HIF-1α and HIF-1β (35). Though its function remains largely unknown, it is thought to serve as a negative regulator of the other HIF-α isoforms (39). Once assembled, HIF selectively targets genes carrying cis-recognition sites, termed hypoxia response elements (HREs), within their promoter regions (96). It is estimated that HIF has upward of 100 distinct transcriptional targets, and is responsible for the vast majority of hypoxia-driven transcriptional changes in the cell (13). However, while the direct effects of HIF are well characterized, the downstream consequences of its stabilization are not fully understood.
Recently, the dynamic regulation of a specific set of endogenous microRNA (miRNA) has been described under low oxygen conditions. Termed “hypoxamirs,” these miRNA are thought to play an essential role in the phenotypic changes that occur after the stabilization of HIF, at both acute and chronic time scales. A handful of these miRNA, such as miR-210, have been shown to carry functional HREs within their promoters (13), and many more have been shown to interact with known HIF-targets and to participate in the regulation of HIF itself (13, 36, 52). At the same time, recent studies have also revealed a role for miRNA in the regulation of mitochondrial metabolism (15, 76). This article will focus on the nexus of these two sets of findings, examining the roles that hypoxamirs play in the hypoxia-induced metabolic response and their implications for the treatment of clinical pathologies driven by hypoxia ischemia.
Hypoxamirs: HIF's Inner Circle of miRNA
miRNA are short, noncoding RNA strands that are 19–23 nucleotides in length. Most have been highly conserved throughout evolution, emphasizing their essential role in cellular function. As a component of the RNA-induced silencing complex (RISC), the primary function of a given miRNA is the down-regulation of its target genes. The miRNA transcript binds to a complementary sequence within the 3′ untranslated region of its mRNA targets, thereby either blocking protein translation or inducing mRNA degradation (13). The site of interaction on the mRNA target is often referred to as the “seed sequence.” It is estimated that upward of 1000 miRNA genes are encoded in the human genome, and between 30% and 60% of all mRNA transcripts are thought to be under some form of miRNA regulation (6).
After transcription of the miRNA gene, a hairpin-looped primary miRNA molecule is formed. This structure is processed in the nucleus, resulting in the production of a smaller miRNA precursor, termed pre-miRNA, which is then exported to the cytoplasm (22, 57). Once there, the RNA endonuclease Dicer removes the pre-miRNA hairpin loop, producing a double-stranded miRNA duplex that can then be incorporated into the RISC. Once the RISC has formed, the active strand of the duplex is fully functional and sheds its antisense (miRNA*) strand. The miRNA* molecule proceeds to degradation or, in some cases, binds its own set of target mRNA molecules (13). These interactions are summarized in Figure 1.
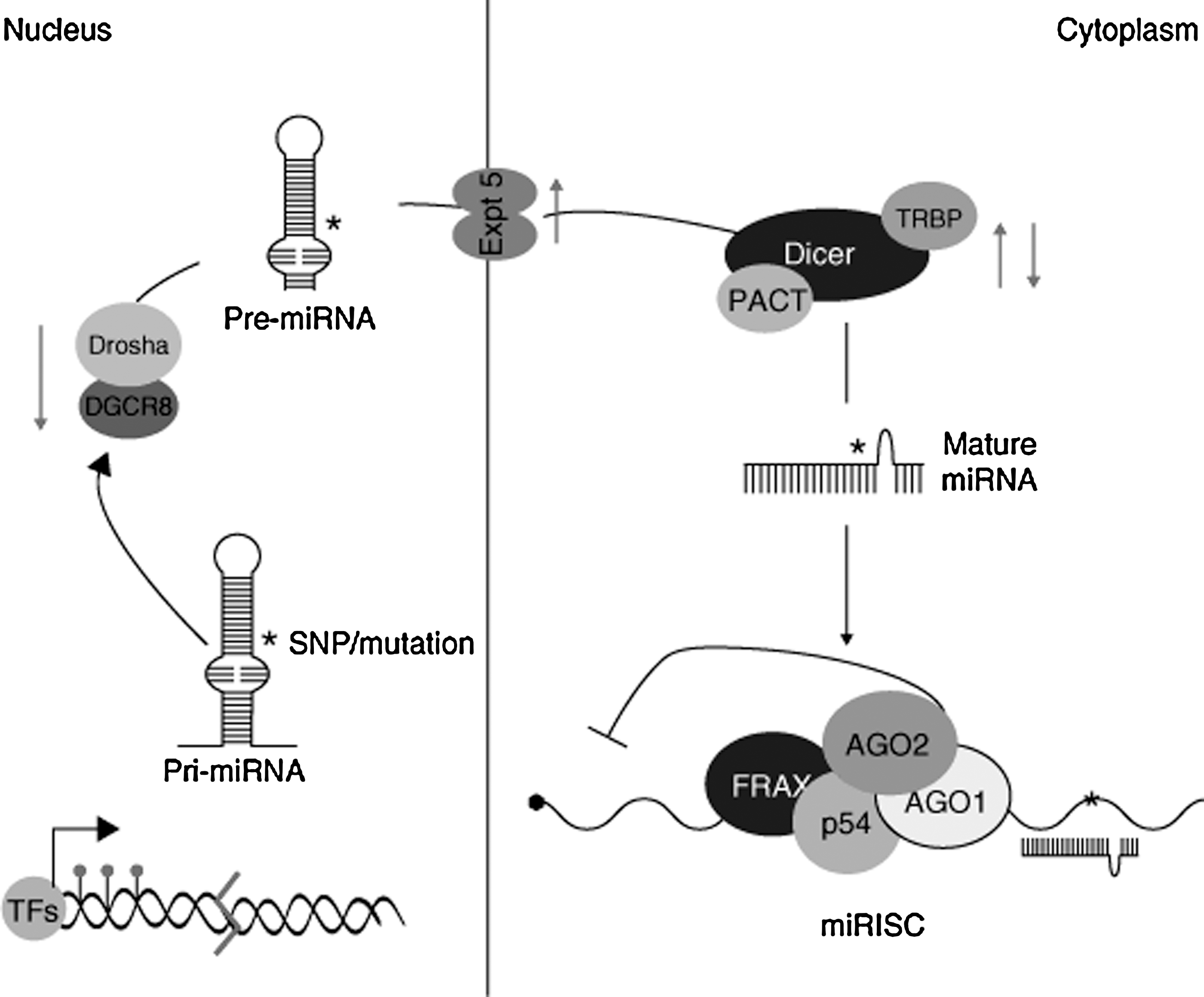
Expression profiling has demonstrated a wide range of miRNA whose expression is altered under hypoxia, in both primary (14, 27) and transformed (11, 33, 36, 52) cell types, although the results can be quite tissue specific. To date, nearly 100 miRNA have been found to show differential expression during hypoxia in some cellular context (13). Though the bulk of miRNA research has focused on cellular miRNA, miRNA levels in the blood have also been shown to correlate with hypoxia and tissue damage in a variety of diseases, including myocardial infarction (42), chronic heart failure (92), and cancer (74, 94). Notably, levels of the hypoxia-induced miRNA, miR-21, and miR-210 are elevated in the serum of patients with ovarian and pancreatic cancer, respectively (74, 94), and levels of miR-21, along with miR-146a and miR-221, become elevated during sustained aerobic exercise in healthy human subjects (4).
It is likely that many of these hypoxia-induced miRNA are indirect targets of HIF, or are otherwise up-regulated by secondary hypoxia-associated conditions such as inflammation and oxidative stress (59). Nonetheless, all of these are potential contributors to the hypoxic program, and several have been shown to participate in metabolic regulation (Table 1). Furthermore, miRNA activity has been uncovered at nearly every level of the mitochondrial response to hypoxia, from HIF stabilization (9, 32, 46, 88), to the induction of anaerobic glycolysis (44, 53, 55, 82, 101), to the suppression of oxidative phosphorylation (13, 28, 56, 71). Thus, hypoxamirs represent key intermediaries between hypoxia, HIF, and the mitochondrial phenotype.
Targeting the Warburg Effect
HIF has long been known to play a central role in Pasteur and Warburg physiology. This section will focus primarily on the role of HIF in the glycolytic shift (as displayed in Fig. 2), in both healthy and pathological tissues, as well as on the set of miRNA that directly target glycolytic enzymes and components of the tricarboxylic acid (TCA) cycle.

Among the prominent mitochondrial enzymes that display differential expression in hypoxia is pyruvate dehydrogenase (PDH). Often referred to as the “mitochondrial gate-keeping enzyme,” PDH is responsible for the fate of glucose after its initial conversion into pyruvate during glycolysis (8). Pyruvate that remains in the mitochondria is converted by PDH into acetyl-CoA, which is used as a substrate for the TCA cycle and the ETC. Pyruvate that is exported into the cytoplasm is instead converted into lactate, a process which marks the second stage of anaerobic respiration. While PDH is not itself a direct target of HIF, HIF modulates its expression via up-regulation of pyruvate dehydrogenase kinase 1 (PDK1) (47), whose phosphorylation of PDH decreases its activity. The importance of this relationship is reflected by the reversion to oxidative phosphorylation in models in which HIF-1α is absent (47). Mouse embryo fibroblasts lacking HIF-1α display suppression of PDK1 expression and persistent accumulation of ROS during hypoxia, thus indicating the lack of normal suppression of glucose oxidation. ROS production is rescued when such cells are transfected with an expression vector encoding PDK1 (47).
HIF is also responsible for the induction of lactate dehydrogenase A (84), a cytoplasmic enzyme that is responsible for the conversion of pyruvate into lactate, as well as for the up-regulation of glucose transporters GLUT1 and GLUT3 (50) and the glycolytic enzyme hexokinase II (HK-II) (1). Positron emission tomography imaging has confirmed that the most malignant tumor cells—often subject to hypoxic microenvironments—show increased glucose uptake and metabolism when compared with their healthy counterparts (8). A similar result has been observed in the pulmonary vasculature of human patients who are diagnosed with pulmonary hypertension, another condition that is strongly associated with hypoxia at the cellular level (97).
While HIF is primarily responsible for the induction of the glycolytic shift, many of its transcriptional targets will continue to maintain this cellular phenotype even in the absence of further stimulation from HIF itself (Fig. 3). MiR-155 and the miR-23a/b cluster are two such examples—both acting directly on enzymes of the TCA and glycolytic pathways (31, 44). Expressed in a variety of tissues, miR-23 is suppressed by the proto-oncoprotein Myc and up-regulated by hypoxia (31, 52). Recently, Gao et al. demonstrated that Myc expression is correlated with high levels of glutaminase (GLS) in human prostate cancer cells (31). By analyzing endogenous target gene transcript and reporter gene construct expression in both gain- and loss-of-function assays, these authors demonstrated that GLS is a direct target of miR-23 and can be rescued by overexpression of either Myc or anti-miR-23 agents (31). Furthermore, GLS catalyzes the hydrolysis of glutamine to glutamate, and in the process, GLS generates α-ketoglutarate, which is used as a source of fuel in the TCA cycle (Fig. 3). In the context of hypoxia, where miR-23a/b is overexpressed, the authors found that this activity results in the suppression of oxidative metabolism by reducing α-ketoglutarate entry into the TCA cycle (40).

A separate hypoxamir, miR-155, has been directly linked to the up-regulation of glycolysis through its suppression of the anti-glycolytic miR-143 (26, 64). While not itself a hypoxamir, miR-143 targets HK-II, the enzyme that is responsible for catalyzing a key transformation early in the glycolysis pathway. This relationship was demonstrated by Fang et al. in small cell lung cancer cells, and may extend to other cell types as well (26). Best known for its induction by inflammatory enzymes such as C-Jun N-terminal kinase (JNK), nuclear factor-κB, and activator protein 1, miR-155 is a widely expressed miRNA that is up-regulated by hypoxia in a variety of cell types (41). Working with human breast cancer cells, Jiang et al. demonstrated that miR-155 suppresses miR-143 expression via degradation of its transcriptional activator, C/EBPβ, thus increasing HK-II expression at the post-transcriptional level (Fig. 3) (44). Supporting the relevance of this relationship, miR-155 has been shown to up-regulate HK-II in this same cell type via indirect activation of STAT3, a factor known to promote the transcription of HK-II (44).
Finally, miR-15a—a hematopoietic miRNA whose deletion is commonly associated with chronic lymphocytic leukemia—has been shown to serve a protective role via down-regulation of the glycolytic enzymes aldolase A and triosephosphate isomerase I (10). miR-15a impairs the rapid glycolysis which is necessary to sustain cells that have suppressed glucose oxidation, and it is concordantly down-regulated under hypoxic conditions (38).
In general, miRNA are reasonable candidates for prolonged regulation of the glycolytic shift, as they possess a remarkably long half life. By examining miRNA decay rates in mouse embryonic fibroblasts after the silencing of Dicer, Gantier et al. demonstrated that miRNA molecules may be approximately 10 times as stable as mRNA, with an average half life of 5 days (30). This finding implicates hypoxamirs as potential contributors to the persistence of Warburg physiology that is characteristic of tumorigenic and hypertrophic cells in cancer and heart disease, respectively, particularly in cases in which HIF stabilization has been nominally resolved.
Targeting Glycolysis Via the p53 Signaling Pathway
The tumor suppressor p53 maintains an antagonistic relationship with HIF during hypoxia—both directly through up-regulation of GLS, and indirectly through its transcriptional target, Tp53-induced glycolysis and apoptosis regulator (TIGAR), a known inhibitor of glycolysis (5). Correspondingly, miRNA that regulate this signaling network are also affected by the induction of hypoxia (Fig. 4). The two most dramatic examples of such miRNA are miR-125b and miR-30d. MiR-125b, a hypoxamir (52) with particular enrichment in the brain and eye, is up-regulated during neurogenesis and thought to play a role in neuronal differentiation (55). Studying both human and zebrafish cells, Le et al. recently demonstrated that miR-125b binds directly to p53, reducing its expression in a dose-dependent manner (55). They were also able to identify a matching miR-125b seed sequence in two transactivational targets—p21 and Bax—as well as seven upstream regulators of p53, demonstrating that the involvement of miR-125b occurs at multiple points in the pathway and is likely to involve synergistic regulatory actions (55). Similarly, Kumar et al. have uncovered an additional hypoxamir, miR-30d, which suppresses p53 and several of its downstream targets, including p21, Bad, Puma, and Gadd45α, in human multiple myeloma cells (53). Since many of these targets are potent regulators of apoptosis, their suppression by miR-30d results in an apoptosis-resistant phenotype (53). Interestingly, miR-30d is expressed in a variety of tissues, most notably pancreatic β cells, and is known to be up-regulated by the presence of glucose (89), a fact that may help in explaining its induction during hypoxia.
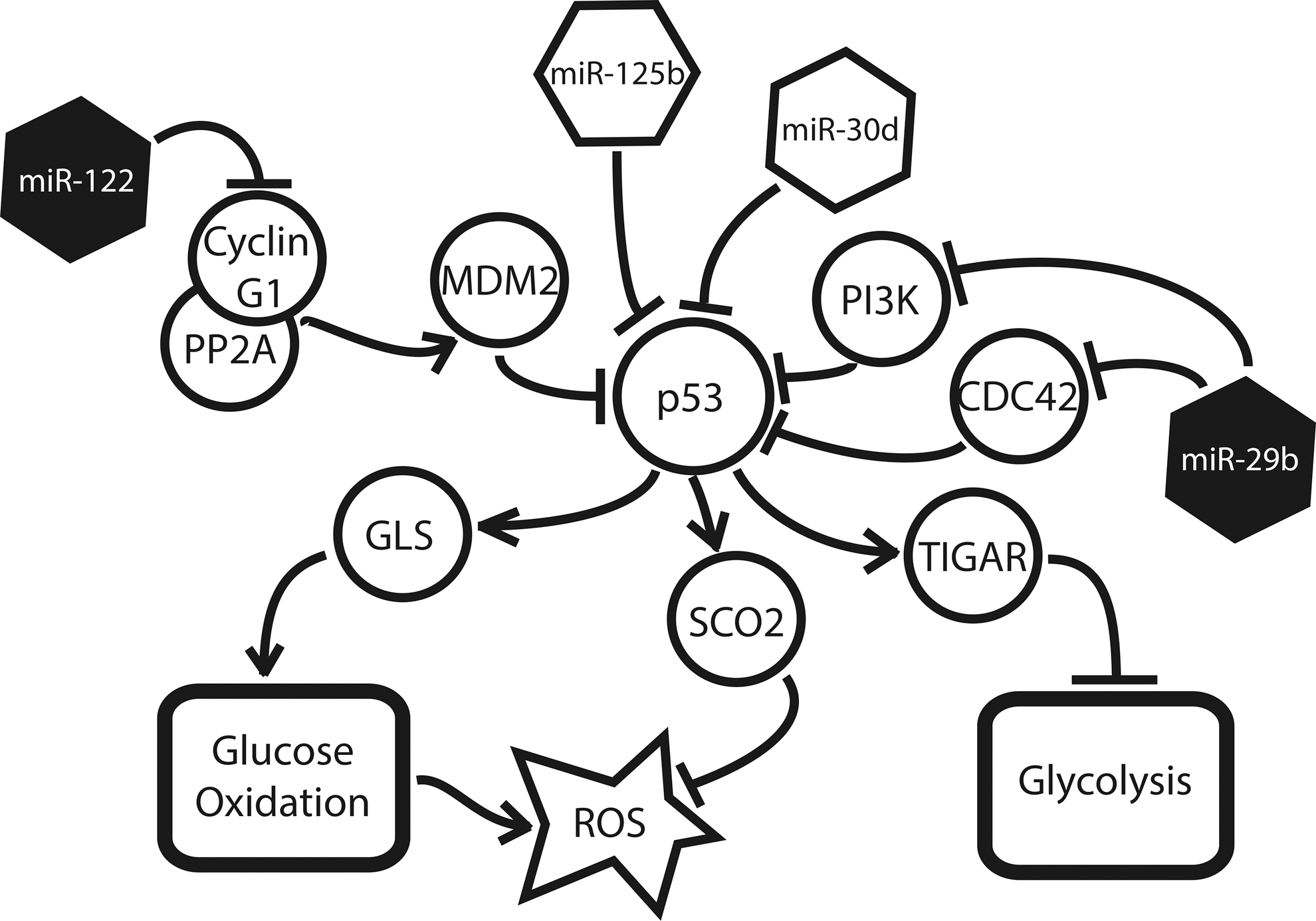
This suppression of p53 has a variety of downstream consequences for the hypoxic cell (Fig. 4). p53 is responsible for the up-regulation of GLS2, which indirectly generates α-ketoglutarate for the TCA cycle as previously described (36). In addition, the p53-induced factor, TIGAR, is a key inhibitor of glycolysis (48). Structurally, it resembles fructose-2,6-bisphosphatase phosphofructokinase-2 (FBPase-2), the bisphosphatase domain of the bifunctional glycolytic enzyme, PFK-2/FBPase-2; accordingly, TIGAR has been shown to suppress FBPase-2, leading to a marked decrease in glycolytic flux (5). As demonstrated by Kimata et al., the inhibition of TIGAR induced glycolysis in hypoxic cardiomyocytes, while its overexpression preserved glucose oxidation, regardless of environmental oxygen tension (48).
Several miRNA known to induce or protect p53 expression have also been shown to be down-regulated in hypoxia (38), further emphasizing the antagonistic relationship of HIF and p53. Both MiR-122, a liver-specific miRNA involved in lipid metabolism and liver homeostasis (25), and miR-29b, a widely expressed miRNA with particular enrichment in pancreatic β cells (73), have been shown to induce p53 under certain cellular conditions. Working with hepatocarcinoma cells, Fornari et al. demonstrated that miR-122 stabilizes p53 expression via suppression of cyclin G1, a protein that is known to enhance the p53 inhibitor MDM2 (29). Similarly, Park et al. found that miR-29b up-regulates p53 levels—both in mouse embryo fibroblasts and in human cervical and colon carcinoma cells—via the suppression of p53 suppressors, CDC42 and PI3 kinase (66).
The relationship between HIF and p53 is further complicated by the role that p53 plays in the quenching of intracellular ROS. TIGAR-mediated suppression of glycolytic flux enables shunting of glucose derivatives into the pentose phosphate pathway, resulting in the production of NADPH, an electron donor that is capable of reducing ROS (specifically H2O2) to H2O (via glutathione peroxidase-dependent glutathione regeneration from glutathione disulfide) and decreasing cytotoxicity (5). p53 is itself responsible for the up-regulation of the factor SCO2, a protein that is responsible for the transfer of copper to the cytochrome c oxidase complex, which, in turn, mediates the reduction of oxygen to water at the final stage of the ETC (60). Excess ROS also induces p53 reciprocally, resulting in a protective feedback mechanism that has been demonstrated in tumorigenic cells (16), as well as cardiomyocytes in this setting of congestive heart failure (48). Thus, the hypoxamir-mediated suppression of p53 expression and activity can result in an increase in intracellular ROS. While this is not itself advantageous to the survival of the hypoxic cell, increased ROS production is known to induce HIF stabilization (91), which, in turn, suppresses ROS by a variety of other means. This functional “bypassing” of the usual mechanisms of oxidative homeostasis is mirrored in the regulation of iron homeostasis by HIF-induced miR-210 (99), as discussed next.
Targeting the ETC and Iron Homeostasis Via miR-210
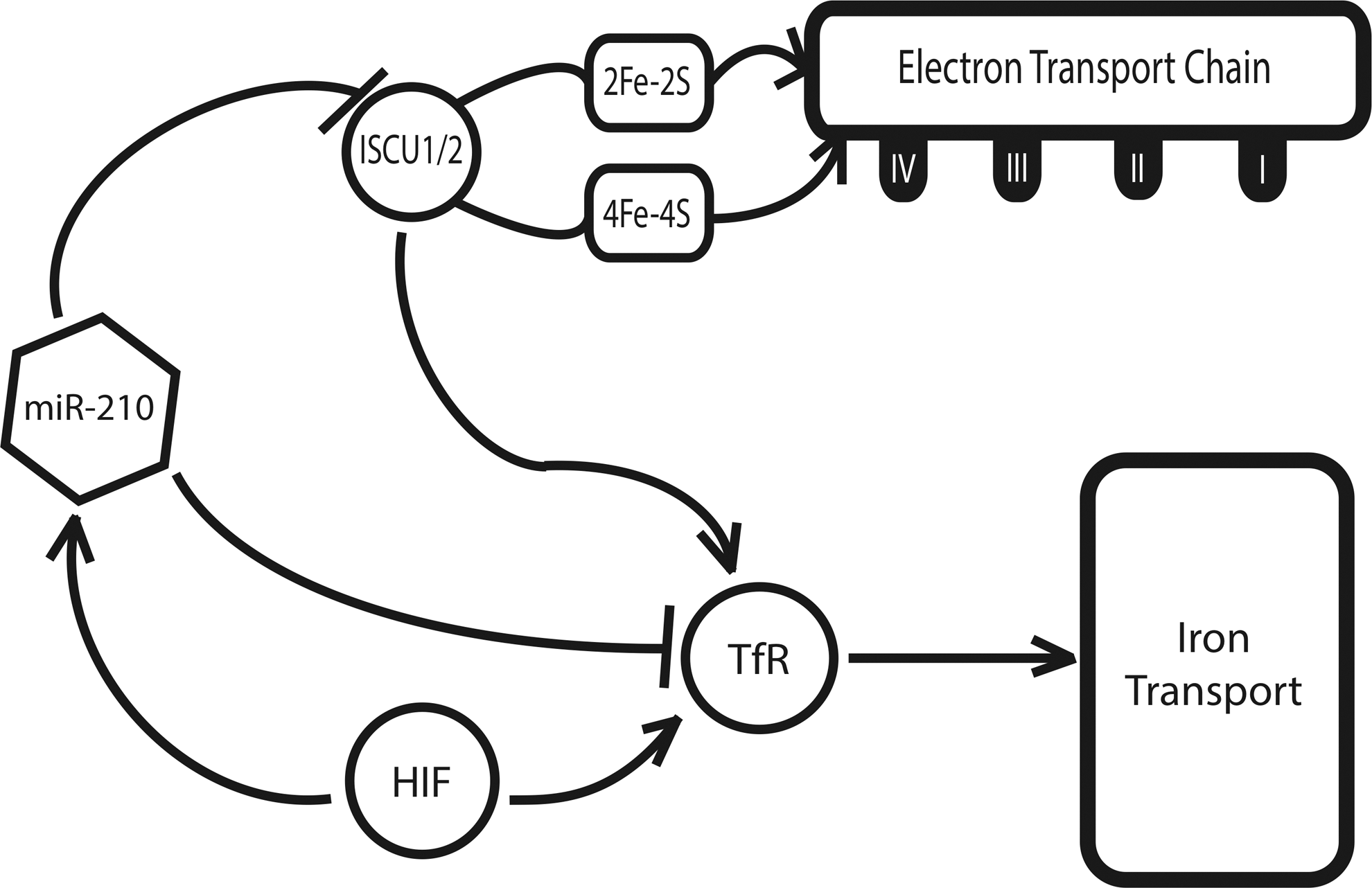
While the majority of the hypoxamirs described thus far are known to be differentially expressed during hypoxia, most are not direct targets of HIF-dependent transcriptional activation (13). A notable exception to this point is the hypoxamir miR-210, a ubiquitous factor that is reliably and robustly induced by HIF across a wide range of primary and transformed cell types (11, 33, 36, 52). miR-210 is also known to be induced in a variety of clinical pathologies that are associated with hypoxia, including preeclampsia (69, 102), ischemia (72), and pulmonary hypertension (14). At a metabolic level, our group found that miR-210 directly targets the iron-sulfur cluster assembly proteins ISCU1 and ISCU2 (14), members of a family of proteins that play a key role in the assembly of [4Fe-4S] and [2Fe-2S] iron-sulfur clusters (58).
Serving essential roles in electron transport and cellular redox state (54), iron-sulfur clusters are an important component of the mitochondrial respiratory complexes in the ETC, and they are incorporated into TCA cycle enzymes, such as aconitase and succinate dehydrogenase (78). Corresponding with a down-regulation of ISCU1 and ISCU2, there is a disruption of intact iron-sulfur clusters, as assessed by electron paramagnetic resonance spectroscopy. In turn, through repressing ISCU1/2 during hypoxia, miR-210 decreases the activity of prototypical iron-sulfur enzymes controlling mitochondrial metabolism, including Complex I and aconitase; decreases mitochondrial respiration; and consequently, improves cell survival in the acute setting (14). These results are consistent across pulmonary arterial endothelial cells (14), tumorigenic cells of breast cancer (28), and trophoblasts in human placental tissue (56), reflecting the potential relevance of this finding to pathologies as disparate as pulmonary hypertension, preeclampsia, and cancer. In addition, miR-210 has been found to down-regulate other related mitochondrial targets, including the NDUFA4 subunit of ETC Complex I and the subunit D of the succinate dehydrogenase complex (ETC Complex II) (17, 71), thereby potentially producing a synergistic effect on inhibiting electron transport and mitochondrial respiration.
The role of miR-210 in the regulation of iron-sulfur clusters in hypoxia is complicated by the need for strict control of iron levels within the cell (Fig. 5). Excess iron promotes the generation of free radicals and is ultimately toxic (17), while iron insufficiency induces hypoferric anemia (87). Specifically, genetic deficiencies of iron-sulfur biogenesis can lead to toxic increases in mitochondrial iron, such as that seen in Friedreich's ataxia (75). In part, such overload is mediated by an increase in the expression of the transferrin receptor 1 (TfR). Recently, Yoshioka et al. have shown that TfR is an additional direct target of miR-210 in hypoxia (99), enabling greater control by this miRNA over the amount of iron that fluxes through the cellular membrane at a given time. Thus, miR-210 is able to induce a suppression of ETC function via ISCU without an associated toxic increase in intracellular iron.
Targeting Mitochondrial Apoptosis Via the AKT/HK-II Signaling Pathway
Persistent resistance to apoptosis, another hallmark of Warburg physiology, is a direct result of the HIF-mediated glycolytic shift (Fig. 6). Kv1.5—a member of the redox-sensitive voltage-gated family of K+ ion channels (Kv)—is known to be activated by H2O2, the byproduct of superoxide reduction by SOD (12). When glucose oxidation is suppressed, ROS production is low and relatively little H2O2 is present in the mitochondria and the cytosol. Referred to by Michelakis and colleagues as the “mitochondria-ROS-Kv channel axis,” this system is thought to serve as an important oxygen sensor for the cell, with low levels of H2O2 promoting closure of Kv1.5 channels and retention of intracellular potassium (63). Potassium, which can serve as an anti-apoptotic mediator via inhibition of caspase activity, is, thus, retained in the cell, promoting a pro-survival phenotype during periods of hypoxic stress (77).
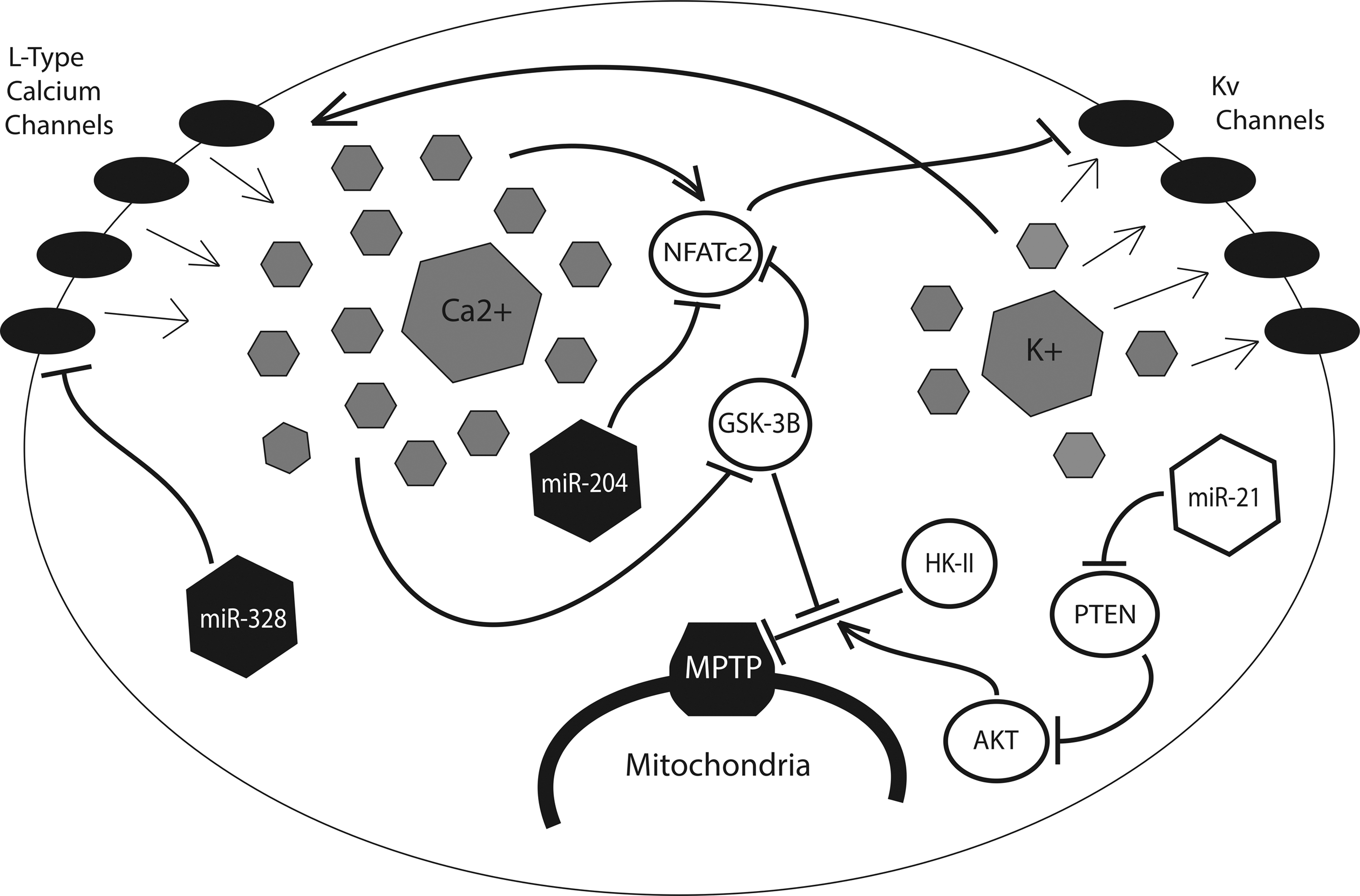
The resulting cellular depolarization triggers the opening of voltage-gated L-type calcium channels, enabling an influx of Ca2+ into the cell (62). High intracellular calcium, in turn, promotes the transcription of nuclear factor of activated T cells (NFAT), which is responsible for the further down-regulation of Kv1.5 potassium channels (8). A direct consequence of this electrical remodeling is the hyperpolarization of the mitochondrial membrane, and the associated dysregulation of the mitochondrial permeability transition pore (MPTP), the large nonselective channel that allows for the passage of proapoptotic mediators from the mitochondria into the cytoplasm (63). HIF-mediated up-regulation of HK-II exacerbates this effect by binding to and inhibiting the voltage-dependent anion channel (VDAC), a key component of the MPTP (63). The proto-oncoprotein Akt contributes as well, by inducing the transport of HK-II across the mitochondrial membrane (8).
Several miRNA have been shown to play a role in this process. In addition to the miR-155/miR-143/HK-II signaling pathway previously described (44), miR-21, a widely expressed hypoxamir known to be up-regulated in a variety of cancers (3, 67), as well as in tissue ischemia (61), pulmonary hypertension (93), and cardiac fibrosis (79), has been shown to up-regulate Akt through suppression of its negative regulator phosphate and tensin homolog (81). The resulting inhibition of VDAC reduces ion flux between the mitochondria and the cytosol, resulting in mitochondrial hyperpolarization and an increased threshold for the opening of the MPTP. Intriguingly, Zhang et al. have demonstrated that miR-21 also modulates cellular ROS levels by targeting SOD2 and SOD3, the proteins which are responsible for reduction of more reactive free radical anion superoxide to the less reactive hydrogen peroxide (101). By promoting the accumulation of ROS, miR-21 up-regulation induces a cytosolic environment that mimics that which would occur after a period of hypoxia, while the additional role of miR-21 in MPTP regulation ensures that the cell is prepared to withstand such cytotoxic effects without succumbing to apoptosis.
Two additional miRNA, miR-204 and miR-328, have been shown to be protective against the induction of Warburg physiology. Both miR-328 and miR-204 are down-regulated in the hypoxic cellular environment (2, 90). Furthermore, miR-328, a widely expressed miRNA with particular enrichment in hematopoeitic and vascular cells, is known to be ectopically suppressed in hypoxia-induced pulmonary hypertension (90). As a result, miR-328 can directly inhibit the expression of L-type calcium channels, thereby preventing some of the electrical remodeling that is associated with the hypoxic mitochondria (90). Similarly, miR-204, a miRNA primarily expressed in the nervous system, targets NFAT for degradation, preventing its inhibition of redox-sensitive Kv1.5 channels (2).
Feedback Control of HIF Stabilization
In addition to their role as downstream effectors of the hypoxic response, many hypoxamirs regulate HIF itself, creating a series of regulatory feedback loops that serve to modulate HIF up-regulation during and after periods of oxidative stress (Fig. 7). Kelly et al. have recently uncovered an additional function of miR-210, demonstrating that it down-regulates the factor, glycerol-3-phosphate dehydrogenase 1-like (GPD1L), a novel protein which has been shown to modulate HIF stabilization (46). GPD1L suppresses cytosolic HIF by increasing the activity of the PHDs responsible for prolyl hydroxylation of HIF-1α. As previously mentioned, prolyl hydroxylation by PHDs tags HIF-1α for recognition by VHL, which, in turn, leads to its ubiquitination and proteasomal degradation. By targeting GPD1L, miR-210 allows for cytosolic HIF to remain high, resulting in a positive feedback mechanism between HIF and HIF-induced miR-210.
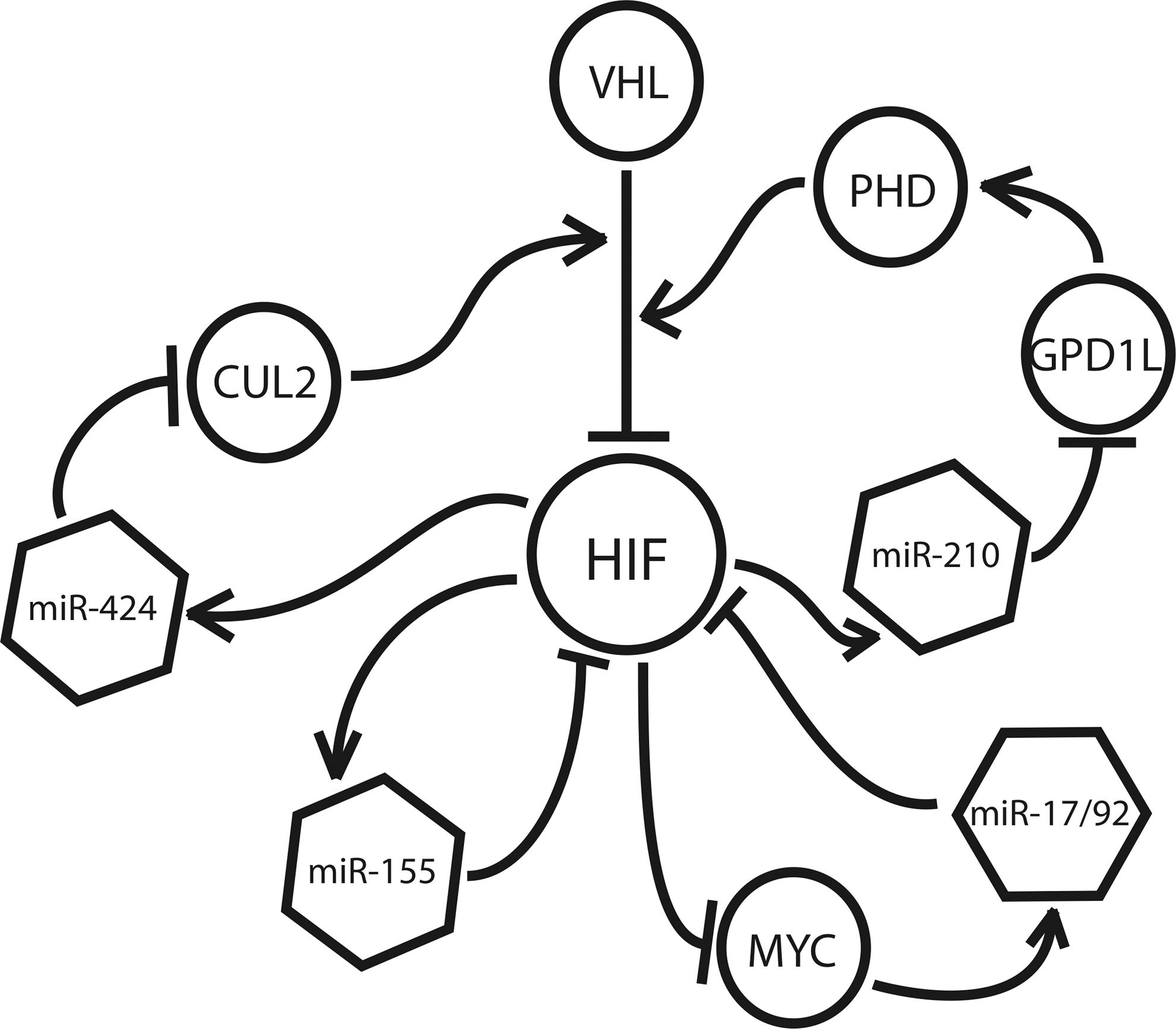
Following a similar principle, hypoxia-induced miR-424 has been shown to target the protein cullin 2 (CUL2), a member of the scaffolding complex that links prolyl hydroxylated HIF-α to the E3 ubiquitin ligase complex, triggering its recognition by the 26S proteasome (37). miR-424 is expressed throughout the vasculature, and it is implicated in the induction of angiogenesis (32). By targeting CUL2, miR-424 destabilizes the E2-ligase assembly, resulting in the up-regulation of HIF (32). Conversely, a negative feedback mechanism is seen in the case of miR-155, which promotes the resolution of HIF during transient hypoxia by targeting HIF-α directly, bypassing the ubiquitin-proteasome pathway altogether (9). Recently, Bruning et al. showed that, when hypoxia exposure is brief, intestinal epithelial cells demonstrate a transient up-regulation of HIF-α, followed by its rapid degradation at the hands of a number of factors, including miR-155 (9).
Finally, the miR-17/92 cluster, a ubiquitously expressed set of miRNA known to be induced by the proto-oncoprotein Myc, has been shown to engage in an antagonistic relationship with HIF (88). Working with human lung cancer cells, Taguchi et al. found that members of the miR-17/92 cluster target HIF-1α directly, and decrease its expression in a dose-dependent manner (88). Conversely, HIF has been shown to suppress miR-17/92 expression via inhibition of Myc (19, 21), a function that is likely responsible for the marked down-regulation of this miRNA cluster under hypoxic conditions (15). Taken together, these mechanisms illustrate that the role of miRNA in hypoxia is not restricted to that of downstream effectors, as many hypoxamirs are actively involved in modulating HIF expression and activity throughout the hypoxic program.
Innovation
Hypoxia and consequent metabolic dysfunction underlie a vast array of cellular pathologies. Hypoxia-relevant microRNA (hypoxamirs) offer insights into the metabolic phenotypes of myriad disorders in which hypoxia is known to play a causative role. Their pleiotropic nature makes them intriguing therapeutic targets, and with the aid of new tools such as microRNA mimics and antagomiRs, the role of hypoxamirs in hypoxia-induced metabolic disease may be interrogated and exploited for therapeutic effect. In addition, next-generation computational algorithms may be used to predict new gene targets of validated hypoxamirs and will continue to provide a context for known microRNA-target relationships in a metabolic setting.
Conclusions and Future Directions
Ongoing discoveries involving the often surprising biology of miRNA represent a new dimension of our understanding of metabolic regulation. As a result, these findings may offer miRNA as potential therapeutic targets in the treatment of human diseases. In particular, the discovery of hypoxamirs in metabolic regulation provides new insights into the persistent metabolic dysfunction which is seen in a wide variety of pathologies that share hypoxia as a common causative feature. With the advent of a variety of antisense miRNA inhibitors to suppress miRNA as well as miRNA replacement therapy to restore deficient miRNA function, these factors may represent new opportunities for the treatment of cancer, pulmonary hypertension, hypoxic-ischemic injury, and many others.
In addition, the pleiotropic nature of many hypoxamirs may help pave the way for a better understanding of the function of HIF and its metabolic actions in contexts other than hypoxia. Several hypoxamirs are already known to have robust functions in related cellular contexts, such as inflammation, infection, and tumorigenesis. In this way, hypoxamirs may provide a link between hypoxia and metabolism, and their sibling pathobiologies. Similarly, given the wide “network effect” of each miRNA that can target several genes in a single pathway or related network of genes (100), we predict that many hypoxamirs may harbor multiple, but as yet undiscovered, metabolic targets which affect more robustly the hypoxic cellular response.
Finally, the conserved nature of the miRNA seed sequence has made possible the development of several computational algorithms, such as TargetScan and DIANA (51, 65), that match mRNA transcripts to targeting miRNA (34, 49). With the use of such algorithms, it is possible to generate a network of predicted target interactions for a given miRNA. These types of in silico analyses can help in lending context to those targets that have been validated in vivo and in vitro. Perhaps more importantly, they may themselves point to targets that are not considered canonical components of the pathways under investigation, thus expanding our understanding of the cellular and systemic contexts in which they occur.
Footnotes
Acknowledgments
This work was supported by NIH grants HL61795, HL48743, HL070819, HL108630 (to J.L.), and HL096834, the Lerner, Harris, and Watkins Funds, Gilead Research Scholars Fund, and the Pulmonary Hypertension Association (to S.Y.C.). The authors thank Stephanie Tribuna for assistance with the preparation of this article.