
Abstract
Extracellular Matrix
Introduction
T
Collagenous proteins
Collagen is a major fibrillar protein present in the ECM that represents approximately 30% of the total protein content in the body; therefore, it constitutes the principal structural protein in mammalian tissues (34). Thus far, 28 collagens encoded by 49 genes have been identified in vertebrates (44, 120). The members of the collagen superfamily, listed next, are classified based on structure and supramolecular organization. For a summary, please see Table 1.
FACITs, fibril-associated collagens with interrupted triple helix.
Fibril-forming or fibrillar collagens include type I, II, III, XI collagen and the more recently discovered type XXIV and XXVII collagen. They are the most abundant and widely distributed collagens in the body. Their role is largely mechanical, as they provide tensile strength to both tissues and organs (118).
Fibril-associated collagens with interrupted triple helix (FACITs) constitute the largest collagen subclass, including type IX, XII, XIV, XVI, XIX, XX, XXI, and XXII collagen. FACITs do not form fibrils themselves, but they bind the surface of pre-existing collagen fibrils contributing to fibril enlargement.
Anchoring fibrils, composed largely of type VII collagen, extend from the basal lamina of epithelial cells and attach to the lamina reticularis by wrapping around the reticular fiber type III collagen bundles and constituting the basement membrane (117).
Network-forming collagens contain multiple disruptions in the triple-helical chains, providing flexibility and enabling them to form linear, axial, and lateral associations within protein networks. Type IV collagen is the most important structural component of the basement membrane (41, 75). Type VI collagen is a heterotrimeric molecule that aggregates into filamentous networks and binds multiple matricellular proteins. Type VIII and X collagen are very homologous heterotrimeric short-chain molecules that form hexagonal networks; however, they exhibit different localization (41).
Transmembrane collagens include homotrimeric type XIII, XVII, XXIII, and XXV collagen, most of which are anchored to the plasma membrane. Transmembrane collagen contains several extracellular domains that can be easily detached from the cell surface. They also act as cell surface receptors and soluble extracellular molecules (118, 149).
Multiplexins include type XV and XVIII collagens. They are basement membrane collagens, endostatin precursors, endostatin-XVIII, and endostatin-XV and are secreted by proteolysis (118).
Finally, type XXVI and XXVIII collagen do not properly fit within any of these subgroups, although both of them contain collagenous domains within their structure (44).
Non-collagenous proteins
Among the proteins belonging to the family of non-collagenous proteins from the ECM are fibronectin, tenascins, laminins, elastin, fibrillins, proteoglycans, and matricellular proteins.
Fibronectin is a high-molecular-weight glycoprotein that binds membrane-spanning receptor proteins named integrins and ECM components such as collagen, fibrin, and heparan sulfate proteoglycans (e.g., syndecans). Fibronectin plays a major role in cell adhesion, growth, migration, and differentiation, and it is important for wound healing and embryonic development (104). Altered fibronectin expression, degradation, and organization have been associated with fibrosis and cancer (104, 146, 152).
Tenascins are large oligomeric glycoproteins (10) that under physiological conditions are not highly expressed in the majority of adult tissues; however, they participate in embryonic development, wound healing, tumorigenesis, and inflammation (91). Tenascins frequently reappear around healing wounds and in the stroma of some tumors (17).
Laminins are glycoproteins that are commonly found in basement membranes, which contain domains that are responsible for the interaction with cell surface integrins (124). Laminins condition cell differentiation, migration, and adhesion as well as the phenotype and survival in almost every tissue (138). Defective laminins lead to muscular dystrophy, lethal skin blistering disease, and nephrotic syndrome (159).
Elastic fibers such as elastin and fibrillins are essential ECM components providing the necessary resilience and elastic recoil to tissues. Both of them interact with collagens and proteoglycans, in addition to the ability of fibrillin to bind transforming growth factor-β (TGF-β) (85).
Proteoglycans are highly hydrophilic molecules that hydrate cells and tissues contributing to ECM assembly. They participate in the regulation of numerous signaling cascades via interactions with growth factors such as fibroblast growth factor (FGF), hepatocyte growth factor (HGF), and vascular endothelial growth factor (VEGF) (10).
Matricellular proteins are a group of ECM components that control cell function and cell-matrix interactions and signaling (9). They include, among others, thrombospondin-1 and -2 secreted proteins, osteonectin, osteopontin (OPN), and the CCN family (cyr-61 and connective tissue growth factor [CTGF]). This key family of proteins determines cell function via interactions with cell surface receptors, growth factors (VEGF, TGF-β, and platelet-derived growth factor [PDGF]), proteases, and structural ECM proteins (2).
ECM homeostasis
The ECM remodeling is a complex but highly coordinated process resulting from the balance between synthesis, secretion, degradation, and reorganization of its components (86). Synthesis and deposition of ECM constituents occur in response to signals conveyed by cell surface receptors, among which integrins appear to play a major role as demonstrated for the pro-fibrogenic effects of OPN (145).
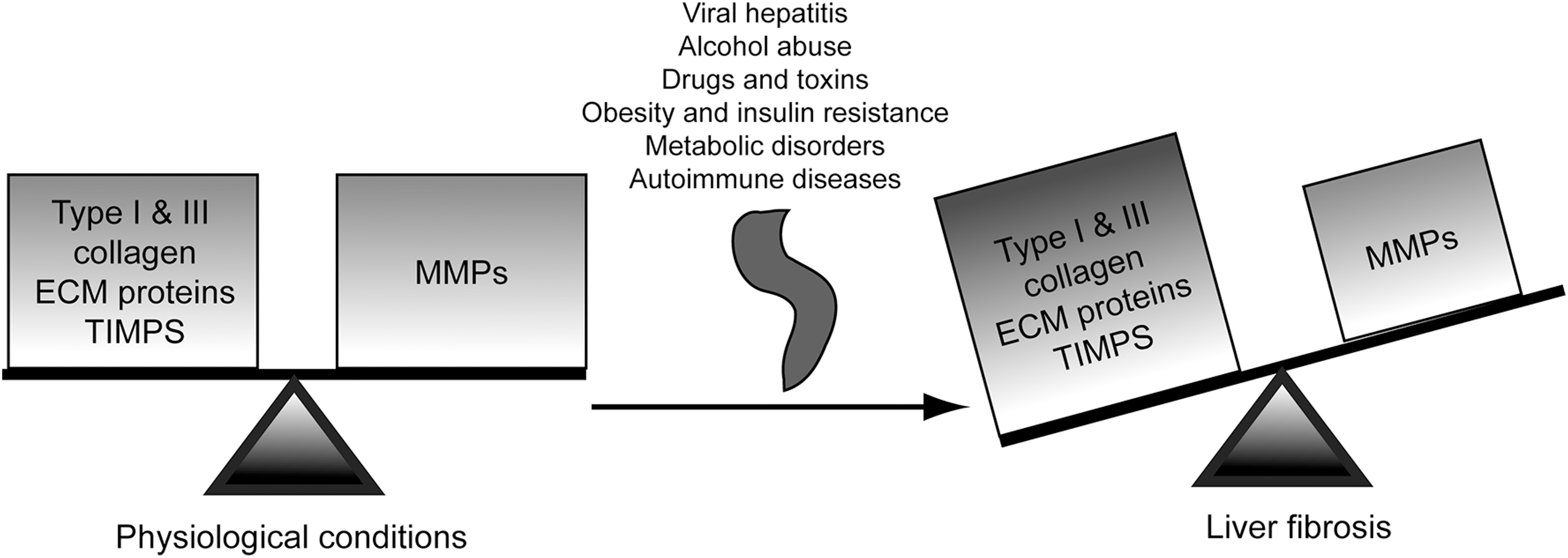
The most relevant ECM-modifying enzymes are the matrix metalloproteinases (MMPs), a superfamily of zinc-dependent endopeptidases (103). MMPs and other proteinases such as plasmin, thrombin, and cathepsins are synthesized as inactive zymogens requiring proteolytic cleavage of a self-inhibitory pro-domain in order to become activated. MMPs' activity is regulated by specific inhibitors such as the tissue inhibitor of metalloproteinases (TIMPs). These two families of enzymes control ECM homeostasis and remodeling that is essential for normal cellular and organ function (11).
However, in the diseased state, impairment and/or disco-ordination in the activity of these enzymes leads to disruption of the physiological ECM dynamics, loss of the normal tissue architecture, and pathological ECM deposition, all of which contribute to the pathogenesis of fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) (21, 35). As an example, MMP-1 decreases in fibrotic livers whereas MMP-2 increases as fibrosis progresses. Likewise, TIMP-1 and TIMP-2 expression significantly increases in fibrosis, thus inhibiting collagen-degrading MMPs and resulting in a net increase in ECM deposition (82) (Fig. 1).
Increased myofibroblasts at the forefront of liver fibrosis
Chronic liver disease is characterized by excessive deposition of ECM proteins in response to persistent liver injury. During fibrogenesis, the ECM undergoes continuous changes in quantity as well as in composition, and the amount of ECM proteins in fibrotic livers increases by six-fold compared with that in healthy ones (5). This increase in ECM deposition elicits tissue remodeling, despite which there is substantial scar formation, distortion of the hepatic architecture, and significant impairment of liver function.
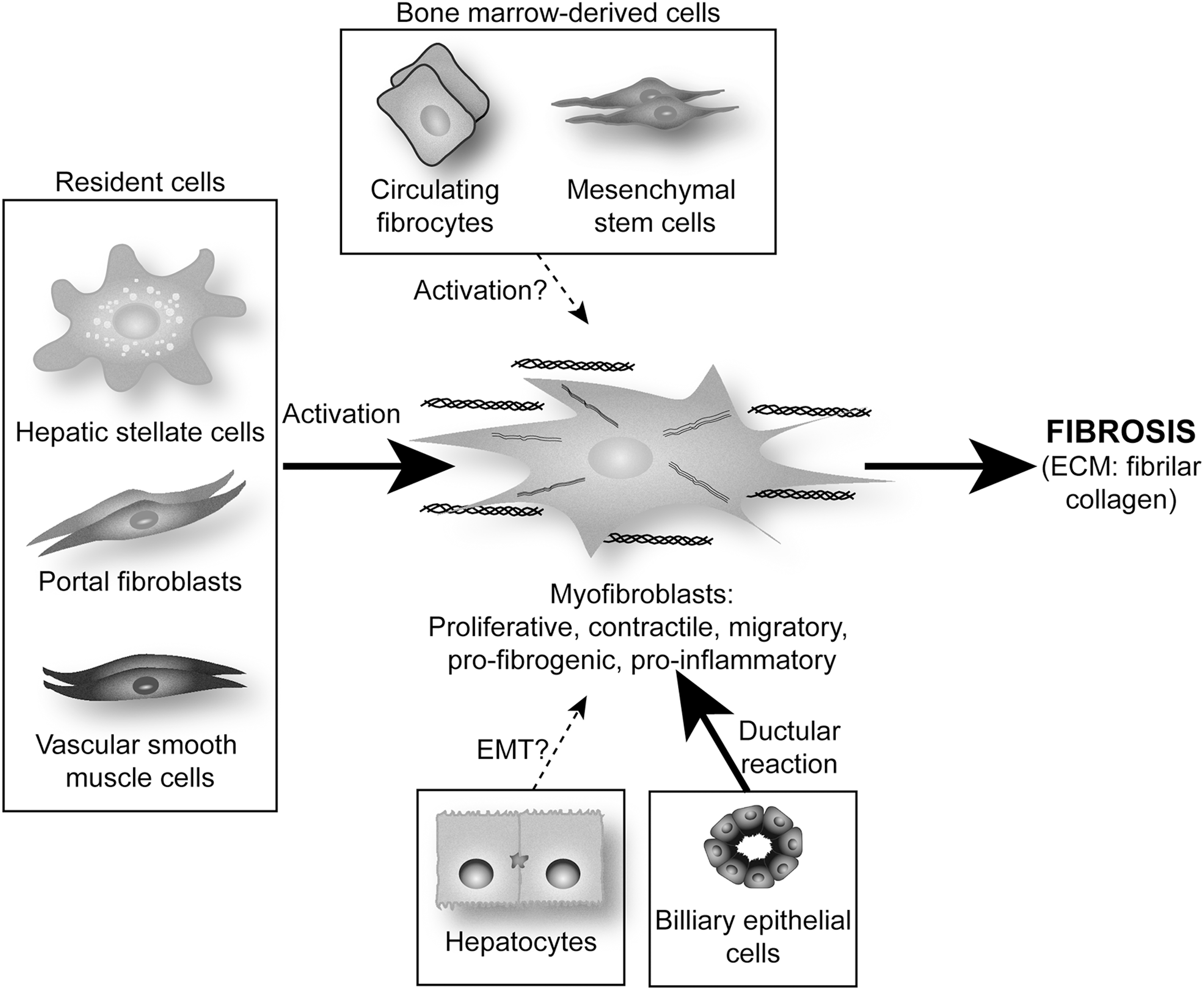
It is now obvious that several cell types participate in the deposition of fibrillar collagen during hepatic fibrogenesis. These pro-fibrogenic cells have diverse origin, but all of them undergo a common process of trans-differentiation and acquisition of a classical myofibroblast (MF)-like phenotype. There are several well-described pro-fibrogenic cells within the liver: resident fibroblasts or MF-like cells mainly represented by hepatic stellate cells (HSC), portal fibroblasts (PF), and vascular smooth muscle cells (101).
In healthy liver, HSC reside in the sinusoidal space of Disse. After chronic injury, they become activated and differentiate into MF-like cells acquiring contractile, pro-inflammatory, and pro-fibrogenic properties (37). Activated HSC migrate and accumulate at the sites of tissue repair, secreting large amounts of ECM, mainly type I collagen and regulating ECM remodeling. Work from our group has dissected how the cross-talk between hepatocytes and HSC (93, 96, 145) and between Kupffer cells (KC) and HSC leads to a fibrogenic response (22, 23, 95, 97 –99).
PF are the other main population of liver cells involved in fibrogenesis (6, 60). The portal connective tissue in normal liver contains only quiescent fibroblasts, with no PF, which only appear when a lesion occurs in the portal zone. Derived from small portal vessels, they express markers that are distinct from HSC [e.g., elastin (60)]. Proliferation of biliary cells is often accompanied by an increase in PF, which form onion-like configurations around biliary structures, acquire an MF phenotype, and are, thus, implied in the early deposition of ECM in portal zones (71).
Immunohistochemical studies have localized most of the different collagen-producing cells in cirrhotic rodent and human livers: (i) portal/septal MF are found either in the inner part of the fibrotic septa or within the expanded portal tracts and originate mainly from PF; (ii) activated HSC are largely located in capillarized sinusoids in the space of Disse between the endothelium and the hepatocytes; and (iii) interface MF, essentially situated at the edge between the fibrotic septa and the surrounding parenchyma, originate from activated HSC (40, 101).
The cell populations involved differ according to the pattern of human liver fibrosis: (i) PF in the septa and HSC at the interface between septa and parenchyma in portal diseases such as chronic viral hepatitis and chronic bile duct obstruction and (ii) HSC and second-layer fibroblasts in centrilobular diseases such as alcoholic liver disease (ALD) and nonalcoholic steatohepatitis (NASH) (101).
In addition to endogenous liver precursors, hepatic MF may also originate from bone-marrow-derived cells such as mesenchymal stem cells (MSC) or circulating fibrocytes (73, 121). Studies by Li et al. and Russo et al. demonstrated that MSC are a source of extra-hepatic MF in the damaged liver (83, 121), although MSC-derived MF represent only a small proportion of the total MF that contribute to ECM deposition during liver fibrosis (73). Further studies are required to establish the quantitative relevance of their contribution to liver fibrosis (60, 73, 83, 121).
In the last few years, although still quite controversial, it has been proposed that hepatocytes and biliary epithelial cells could contribute to liver fibrosis by undergoing epithelial-to-mesenchymal transition (EMT) (18). Several in vitro experiments suggest this possibility. Indeed, TGF-β up-regulates mesenchymal genes such as α-smooth muscle actin (α-SMA) and collagen, and down-regulates epithelial genes such as albumin (60, 151). However, lineage tracing in murine models of hepatic fibrosis such as common bile duct ligation (BDL) or an injection of carbon tetrachloride (CCl4) has shown that hepatocytes and biliary epithelial cells do not undergo EMT after liver injury (25), whereas other reports provide in vivo evidence which suggests that EMT occurs during liver injury (102, 160). Additional studies are needed in order to identify the cell type that undergoes EMT, if any, and their specific contribution to ECM deposition during liver injury (60, 151) (Fig. 2).
Pathological ECM and Liver Disease
Fibrogenesis: the wound-healing response to liver injury
Wound healing or tissue remodeling and repair is a protective mechanism that is activated in response to stress and injury in order to maintain the functional and anatomical integrity of the liver. When injury and the related acute inflammatory reaction cause moderate cell necrosis and ECM damage, tissue repair usually occurs. In response to parenchymal cell loss, hepatocytes restore the physiological liver mass by self-replication regenerating and replacing necrotic or apoptotic cells (59). However, continued injury and deregulation of the normal healing does not repair as effectively; regeneration fails, resulting in liver fibrosis, massive deposition of ECM, scar formation, and, in progressive and persisting injury, hepatic failure (38).
Chronic damage leading to liver fibrosis occurs in response to a variety of insults, such as viral hepatitis, alcohol abuse, obesity, drugs and toxins, insulin resistance, metabolic disorders, venous outflow obstruction, or autoimmune disease. Overall, fibrogenesis is characterized by several key features: (i) immediate damage to the epithelial/endothelial barrier, (ii) continuous hepatocyte injury, (iii) recruitment of inflammatory cells with the subsequent secretion of pro-fibrogenic cytokines and growth factors, (iv) generation of reactive oxygen species (ROS), (v) activation of collagen-producing cells with proliferative, migratory, and contractile features, (vi) matrix signaling to MF, and (vii) marked changes in the composition and quantity of the ECM deposits that distort the normal hepatic architecture by forming fibrotic scars (38).
The distribution of the fibrous material depends on the cause of liver injury. In human chronic viral hepatitis and chronic cholestatic disorders, the fibrotic tissue is initially located around portal tracts, while in ALD, it is located in pericentral and perisinusoidal areas. As fibrosis advances, progression from collagen bands to bridging fibrosis and to frank cirrhosis occurs; the latter is characterized by formation of regenerative nodules of liver parenchyma that are separated by fibrotic septa (42, 47, 67), liver failure, and portal hypertension, which often require liver transplantation. Thus, emerging antifibrotic therapies aim at inhibiting the increase and activation of pro-fibrogenic cells and at preventing ECM deposition, particularly fibrillar type I and III collagen.
Alcoholic liver disease
ALD is a significant clinical problem and a major cause of morbidity and mortality worldwide. The spectrum of disease ranges from fatty liver to alcoholic steatohepatitis (ASH), progressive fibrosis, and HCC (110). In developed countries, ALD is a major cause of end-stage disease that requires transplantation. Current interventions such as nutritional support with vitamin and zinc supplements, steroids combined with N-acetylcysteine for patients with biopsy-proven ASH, and pentoxifyline for patients with contraindications to steroids have proved beneficial for patients with ALD; however, it remains the most important etiology of half of the cirrhosis-related deaths in the United States.
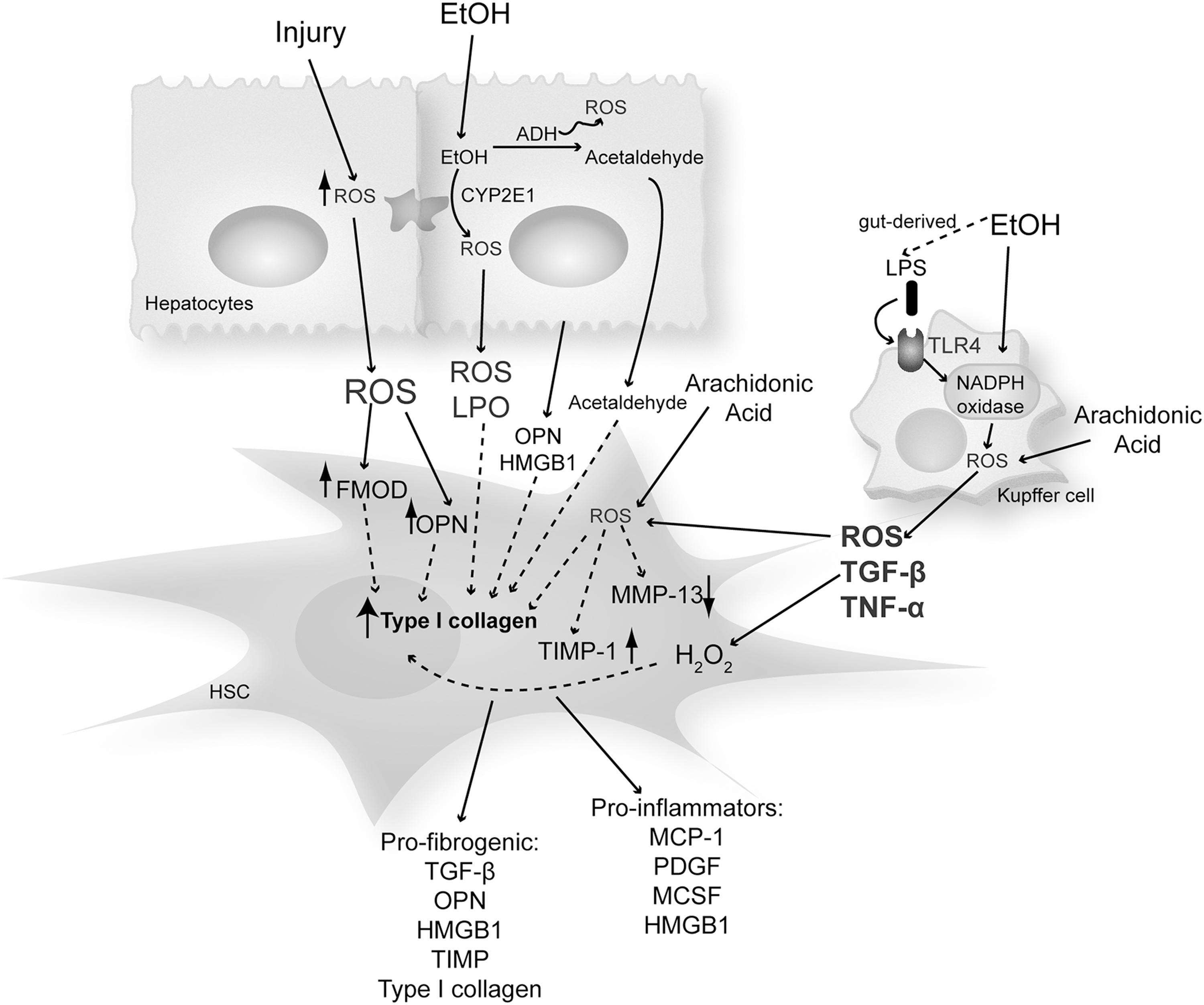
Ethanol exposure causes a state of oxidant stress in hepatocytes, altering the architecture of the endoplasmic reticulum (ER) and the function of the proteasome system. The causative pathological role of both oxidative and nitrosative stress has been extensively demonstrated in ALD as a key factor promoting disease progression.
Many pathways play a key role in how ethanol causes cell injury and induces oxidant stress in the liver. Some of these include redox state changes because of ethanol and acetaldehyde oxidation, generation of acetaldehyde and acetaldehyde adducts, mitochondrial damage, decreased ATP production, membrane damage by the solvent-like actions of ethanol, nutritional interactions, hypoxia, cytokine secretion, KC activation due to an increase in gut-derived lipopolysaccharide (LPS), and an increase in damage-associated molecular patterns such as high mobility group box-1 (HMGB1) that drives both sterile and nonsterile inflammation.
ROS derived from alcohol metabolism in hepatocytes are mainly generated by the mitochondrial respiratory chain, cytochrome P450 isoforms such as cytochrome P450 2E1 (CYP2E1) that metabolizes ethanol per se, xanthine oxidase, and NADPH oxidase (NOX) (16). It is well known that continued alcohol consumption also decreases the antioxidant defense, thus promoting liver injury (15).
Many of these pathways are not exclusive of one another, and it is likely that several of them contribute to the ability of ethanol to induce a state of oxidant stress (see sections Non-alcoholic steatohepatitis and Other etiologies). Moreover, several of these mechanisms and secreted mediators induced by them drive HSC activation, further magnifying liver injury (22, 23, 93, 95, 97, 100, 145) (Fig. 3).
Nonalcoholic steatohepatitis
NASH represents a progressive form of nonalcoholic fatty liver disease. It is characterized by histological liver damage that is similar to ALD in the absence of significant alcohol consumption. Risk factors for NASH include obesity, type II diabetes, hyperlipidemia, total parenteral nutrition, jejuno-ileal bypass surgery, and the use of certain drugs. Although there are no officially approved drugs for the treatment of NASH, metformin has been shown to improve insulin resistance, liver enzymes, and steatosis, whereas its efficiency in liver injury and fibrosis shows mixed results (74).
Oxidative stress markers and lipid peroxidation products are increased, while the antioxidant defense is reduced in the liver and in the plasma from NASH patients (48, 58, 68). In NASH, there is an imbalance in fat synthesis along with an increase in circulating adipokines, elevated ROS, and inflammation leading to fibrosis. Leptin, the adipocyte-derived hormone, promotes hepatic fibrosis by increasing type I(α1) collagen gene (Col1a1) expression in human and rat HSC (125, 135).
Other etiologies
Hepatitis C is a human infectious disease affecting the liver that is caused by an RNA virus from the Flaviviridae family. The hepatitis C virus (HCV) is transmited primarily by the blood. Acute HCV infection is asymptomatic in 50%–90% of cases. Chronic infection is associated with variable degrees of hepatic inflammation and fibrosis progression, regardless of HCV genotype and viral load. Liver disease progression takes place over several decades. Depending on the presence of cofactors, 10%–40% of patients with chronic HCV infection will develop cirrhosis.
Hemochromatosis, a disease characterized by iron overload in the liver, is a hereditary autosomal recessive syndrome with two known mutations in the HFE gene (C282Y and H63D) that disrupt iron metabolism (122). Patients with hemochromatosis develop cirrhosis, as iron deposition catalyzes Fenton reactions that promote the formation of lipid peroxidation-end products. These effects, exacerbated by alcohol consumption, contribute to the fibrogenic response (80).
Lastly, Wilson's disease is an autosomal recessive disorder in copper metabolism that is characterized by excessive copper deposition in the liver, brain, and other tissues. It is caused by mutations in the ATP7B gene that codes for a copper carrier involved in transferring copper from the trans-Golgi system to apo-ceruloplasmin and the transport of copper from the hepatocyte to the bile. Copper deposition causes mitochondrial dysfunction and oxidative stress, hence promoting scarring in the liver (26).
Liver Cell-Specific ECM Regulation
Hepatic stellate cells
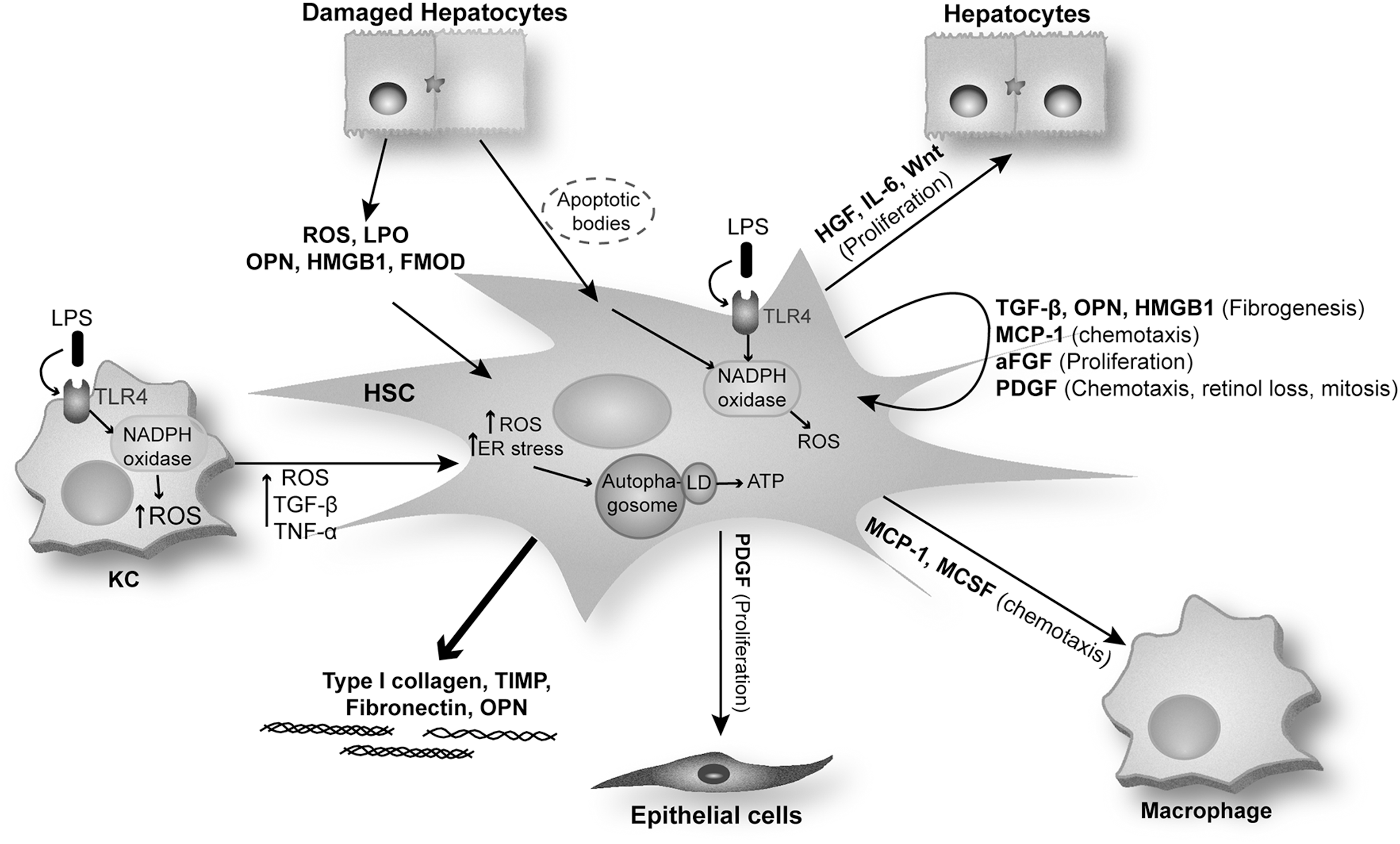
A large majority of the MF present in the damaged liver are HSC derived (37). Quiescent HSC express typical markers of neural cells, including glial fibrillary acidic protein (GFAP), synemin, synaptophysin, nerve growth factor receptor p75, and desmin, in addition to markers that are characteristic of adipocytes, such as peroxisome proliferator-activated receptor gamma (PPARγ) (69, 129). In chronic liver injury, quiescent HSC are exposed to growth factors and pro-fibrogenic cytokines and become activated by acquiring an MF phenotype (37). On activation, HSC proliferate, migrate, and become highly contractile. Moreover, they become pro-inflammatory, as they produce and secrete monocyte chemoattractant protein-1 (MCP-1), PDGF, macrophage colony-stimulating factor (MCSF), and HMGB1, in addition to acquiring pro-fibrogenic properties as they produce pro-fibrogenic factors such as TGF-β, OPN, HMGB1, and TIMP and increase the synthesis and deposition of type I and III collagen and fibronectin (28, 60).
Multiple mechanisms and signaling pathways are involved in HSC activation such as factors released by activated KC (ROS and cytokines) (23, 95), damaged hepatocytes (lipid peroxidation products, ROS, OPN, and HMGB1) (97, 145), and hepatocyte-derived apoptotic bodies (46). Phagocytosis of apoptotic bodies makes HSC more resistant to Fas ligand and tumor necrosis factor-α (TNF-α)-related apoptosis (63), increasing their survival and hence perpetuating the fibrogenic response.
Recently, it was described that autophagy can hydrolyze triglycerides in hepatocytes in a process called lipophagy. Lipophagy has been proposed as significant for HSC activation and, thus, for liver fibrogenesis. Indeed, autophagy stimulates loss of lipid droplets and provides the cellular energy that is necessary for HSC activation (51). Additional studies have shown that autophagy can be induced in activated HSC via oxidative injury and ER stress (52).
Once activated, HSC secrete growth factors (TGF-β and epidermal growth factor), hepatocyte mitogens (HGF, interleukin 6 [IL-6], Wnt), MCSF, stem cell factor, macrophage chemotactic factors (MCP-1 and TNF-α), as well as autocrine factors, including HSC mitogens (PDGF-BB and aFGF), HSC chemotactic factors (MCP-1, PDGF), and HSC pro-fibrogenic factors (TGF-β, OPN, and HMGB1). Activated HSC also respond to mediators that are secreted by other cells, including endothelin-1, OPN, and HMGB1 amplifying the pro-fibrogenic response (37, 45, 145).
TGF-β is a key inducer of ECM secretion by activated HSC. In addition, leptin and angiotensin II have been shown to exert pro-fibrogenic actions in activated HSC (94). Leptin is an adipogenic hormone that activates the JAK-STAT pathway and induces HSC proliferation while inhibiting HSC apoptosis (126). Activated HSC secrete angiotensin II, which induces fibrogenic effects via activation of NOX. Lastly, blockade of the renin-angiotensin axis attenuates fibrosis in experimental models of chronic liver injury (Fig. 4).
Portal fibroblasts
In healthy liver, PF are located adjacent to bile duct epithelia, where they participate in ECM turnover. PF are the first responders in biliary fibrosis after cholestatic liver injury and are later replaced by perisinusoidal HSC (71). PF, unlike HSC, do not express cytoglobin, desmin, or GFAP and they lack lipid droplets. However, they are characterized by expression of highly specific fibroblast markers such as TE7, elastin, IL-6, fibulin-2, ecto-ATPase nucleoside triphosphate diphosphohydrolase-2, p100, α2-macroglobulin, and neural proteins (60). After liver injury, they also undergo activation, increasing the expression of α-SMA, proliferating, and secreting type I collagen. MF differentiation of PF is induced by growth factors (PDGF-BB), pro-inflammatory cytokines (IL-6 and MCP-1), and pro-fibrogenic proteins (TGF-β2, OPN, and HMGB1) that are secreted mainly by damaged biliary epithelial cells (60).
Kupffer cells
KC are resident macrophages that are characterized by high phagocytic, antigen presentation and pinocytotic capacity. KC release a wide array of soluble mediators, including ROS, cytokines, growth factors, cyclooxygenase, and lipoxygenase metabolites, all of which provide physiologically diverse and pivotal paracrine effects on all other liver cells (90, 141). KC are also central to the liver's homeostatic response to injury, as on degenerative changes in hepatocytes they immediately respond to the insult and release mediators to orchestrate inflammatory and reparative responses (90, 141).
Influx of KC coincides with the appearance of HSC activation markers (64). KC stimulate matrix synthesis, cell proliferation, and release of retinoids by HSC. KC generate ROS, which, in turn, may enhance HSC activation and type I collagen synthesis either by their direct actions on the two type I collagen promoters (Col1a1 and Col1a2) or by inducing pro-fibrogenic cytokines (95). KC also produce nitric oxide, which can counterbalance the effects of ROS by reducing HSC proliferation, contractility, and type I collagen synthesis; however, nitric oxide, when reacting with superoxide radical (O2·−), generates peroxynitrite, which has major profibrogenic effects on HSC (144).
To date, there is limited information on the intercellular communication between KC and HSC, as well as on the molecular mechanisms by which KC-derived mediators modulate the fibrogenic response in HSC. Work from our laboratory (see section: ROS and type 1 collagen) demonstrated a novel dual mechanism that is responsible for the increase in type I collagen in HSC co-cultured with KC (95).
Sinusoidal endothelial cells
Liver sinusoidal endothelial cells (SEC) are nonparenchymal cells that are located in hepatic sinusoids. They control the bidirectional exchange of lipoproteins and other nutrients between the blood and the liver cells via their fenestrations (154). The contribution of SEC to the production of fibrillar collagen is still controversial. However, during the early stages of liver fibrosis, SEC lose their fenestrations, decreasing the exchange of metabolites and increasing the secretion of various basement membrane components (type IV collagen, perlecan, entactin, and laminin), all of which contribute to hepatocyte injury. SEC secrete fibronectin and promote the TGF-β-mediated activation of HSC (151). Moreover, SEC contribute to creating a proinflammatory environment by increasing T cells and decreasing hepatic immunotolerance (19).
Cytokines as Regulators of ECM
Transforming growth factor-β
TGF-β can be secreted by biliary epithelial cells, KC, and activated HSC. All isoforms of TGF-β (TGF-β1, TGF-β2, and TGF-β3) are synthesized and secreted as latent precursors, requiring protein processing to generate their active form (27). In its latent state, TGF-β is usually a part of the network of ECM components along with type IV collagen, fibrillin, and fibronectin. Factors determining the secretion of active TGF-β from its latent complex include MMPs, plasminogen activator, plasmin, ανβ6 integrin, and thrombospondins (45). Once activated, TGF-β signals to MF via the serine-threonine kinase TGF-β receptors, TβRI and TβRII.
In quiescent HSC, TGF-β induces the activity of the inhibitory Smad-7, which acts as a negative feedback loop blocking TGF-β-receptor-mediated Smad signaling and terminating the Smad-controlled signal transduction. It has been shown that a Smad-7 response is absent in activated HSC (133), which, along with the TGF-β autocrine loop, mediates the increase in TGF-β during liver fibrogenesis (Fig. 5).
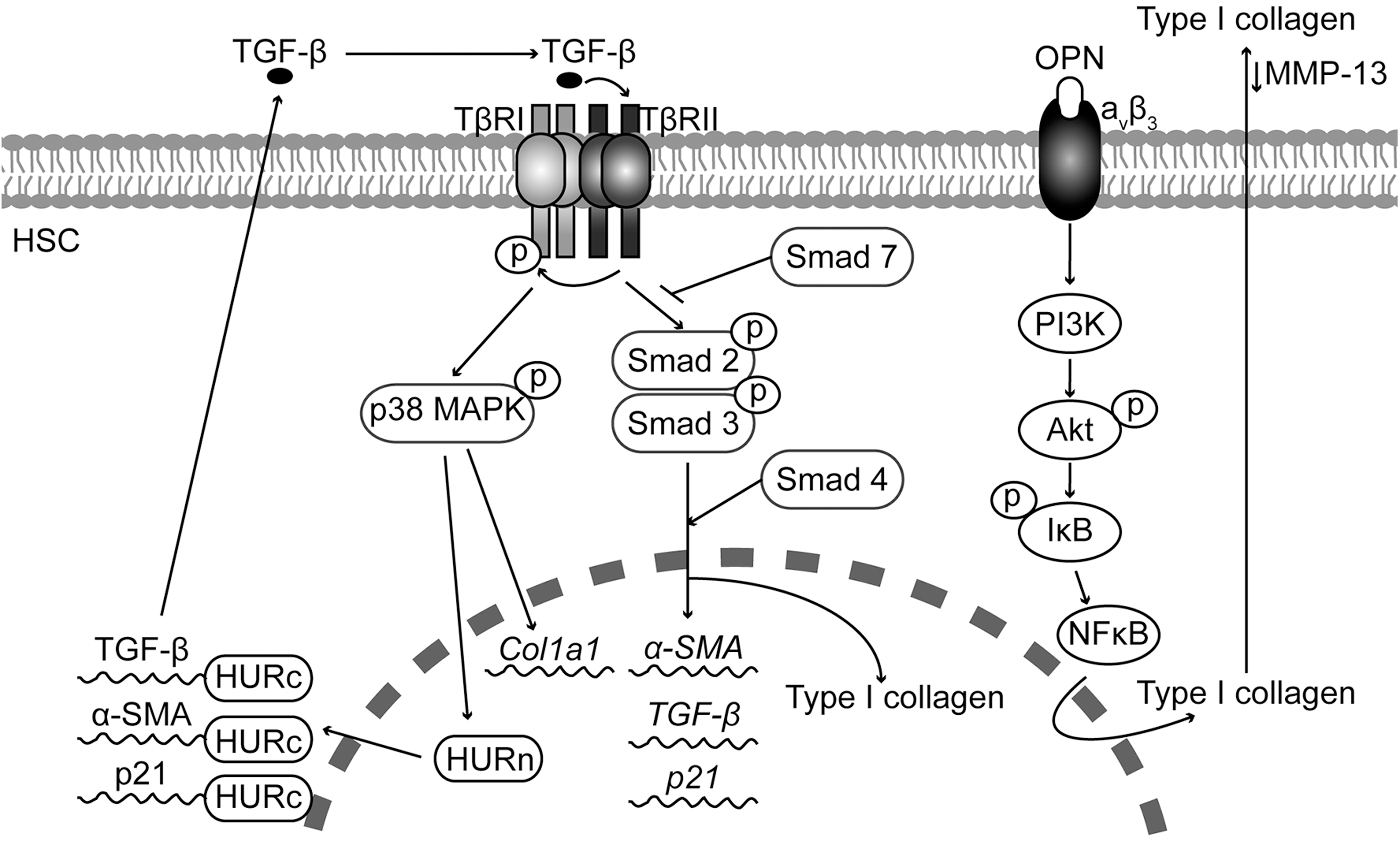
Several studies have identified Smad-3 as the main inducer of collagen expression in HSC in response to TGF-β (55, 127, 128). Likewise, other studies have demonstrated a role for p38 mitogen-activated protein kinase (p38MAPK) in the transcriptional and translational control of the Col1a1 gene in response to TGF-β (148).
The effect of TGF-β appears to be cell-type specific. For example, TGF-β inhibits HSC proliferation. In the BDL model of liver fibrosis, the anti-proliferative action of TGF-β is mediated by an increase in the interaction between the RNA-binding protein human antigen R (HuR) and p21 mRNA, increasing p21 mRNA stability. These effects of HuR are linked to its cytoplasmic localization, which is controlled by TGF-β via p38MAPK (156). In addition, TGF-β also increases the HuR-mediated stability of mytogenic and pro-fibrogenic genes such as α-SMA and TGF-β mRNA (Fig. 5).
Tumor necrosis factor-α
The contribution of TNF-α to hepatocellular injury and inflammation has been widely studied, whereas the role and the mechanisms involved in the TNF-α-mediated HSC activation and collagen production still remain undefined. In vitro studies have shown that TNF-α inhibits Col1a1 mRNA expression in HSC via p38MAPK, CCAAT enhancer binding protein β (C/EBPβ), and C/EBPδ-dependent mechanisms (56). Likewise, TNF-α exerts its anti-fibrogenic effects by regulating ROS production due to a reduction in GSH levels and an increase in nuclear factor kappa B (NFκB) activity (147). Reactive nitrogen species have been shown to switch on early ECM remodeling in HSC via induction of MMP1 and TNF-α (144). Although TNF-α does not participate in HSC activation by increasing α-SMA or TGF-β expression, it plays an important role in proliferation and in regulating TIMP-1 and MMP-9 expression via the TNF-α receptor 1 (TNFR1). This study also shows that the absence of TNFR1 alone or TNFR1 plus TNFR2 leads to a decrease in Col1a1 mRNA expression, suggesting that in some cases TNF-α could be pro-fibrogenic in the liver (136).
Osteopontin
OPN is a phosphorylated acidic glycoprotein that belongs to the family of matricellular proteins which function as adaptors and modulators of cell-matrix interaction. OPN has important roles as an immune modulator and a mediator of wound repair (77). Plasma OPN concentration correlates with the severity of hepatic fibrosis and inflammation in HCV-infected as well as in alcoholic individuals. OPN promotes fibrosis progression in patients with NASH and has prognostic value in hepatitis B virus (HBV)-related HCC (111, 132, 157, 162). Likewise, our laboratory has recently demonstrated that HCV-cirrhotic patients have increased co-induction of collagen and cleaved OPN compared with healthy individuals (145).
Several in vivo studies, including our own, have shown that OPN expression increases in CCl4-, BDL-, and thioacetamide (TAA)-induced liver injury and fibrosis (81, 145). Furthermore, Opn−/− mice were less susceptible to CCl4- and TAA-induced liver injury and fibrosis, whereas transgenic mice overexpressing OPN in hepatocytes (Opn HEP Tg) developed spontaneous liver fibrosis and were more vulnerable to CCl4-induced liver injury and fibrosis (145). Hepatocytes, oval cells, billiary epithelial cells, and HSC appear to be the main source of OPN in the liver (145). In vitro studies demonstrate that OPN expression is up-regulated in culture-activated HSC and treatment with OPN induces HSC activation, promotes HSC invasion and migration as well as type I collagen production (81, 145). The pro-fibrogenic effect of OPN is mediated by integrin aνβ3 binding and signaling via the PI3K-pAkt-NFκB pathway (145). Thus, OPN acts as an autocrine and paracrine mediator of the fibrogenic response to liver injury (Fig. 5).
Reactive Oxygen Species
ROS play a crucial role in the pathophysiology of every liver disease. Oxidative stress is a key noxious factor in ALD and NASH. The major sources of ROS in the liver are hepatocytes, KC, and neutrophils. Hepatocytes are implicated in the detoxification of drugs and toxins by the cytochrome P450 enzymes, which require oxygen activation, resulting in ROS generation. KC and neutrophils typically generate ROS via NOX activation in response to pathogens and inflammatory situations. ROS generation is enhanced by alcohol consumption.
ROS and type I collagen
Over the last few years, our laboratory has studied how ROS contribute to type I collagen induction in HSC. Studies using HSC lines transduced to overexpress CYP2E1 as an endogenous generator of ROS indicate that under oxidative stress conditions, Col1a2 mRNA expression is regulated transcriptionally and by mRNA stabilization (98, 99). Likewise, treatment with a polyunsaturated fatty acid such as arachidonic acid along with ethanol to recapitulate events occurring during ALD linked CYP2E1-dependent oxidative stress to induction of cyclooxygenase-2 and the actions of arachidonic acid and ethanol on the activation of the type I collagen gene in HSC (100).
In a follow-up study, to evaluate possible fibrogenic effects of CYP2E1-dependent generation of ROS, a model was developed using co-cultures of HepG2 cells that either express or do not express CYP2E1 with HSC (97). The results from this study suggest that increased translation of the Col1a1 and Col1a2 genes by CYP2E1-derived ROS is responsible for the increase in type I collagen protein which is produced by the CYP2E1-expressing HepG2 cells co-culture (97).
To assess whether fish oil, as a source of polyunsaturated fatty acids from the n-3 series, could synergize with ethanol to promote type I collagen up-regulation in vivo, Col1a2 promoter-βGal transgenic mice were fed a diet that was enriched in fish oil in the presence of ethanol or dextrose and co-cultures of hepatocytes and HSC were established. These studies suggested that fish oil (mainly n-3 polyunsaturated fatty acids) synergizes with ethanol and induces type I collagen, transactivating the Col1a2 promoter through a lipid peroxidation-PKC-PI3K-pAkt-NFκB-driven mechanism in the absence of overt steatosis and inflammation (96).
The impact of KC on the HSC fibrogenic response was examined in an in vitro co-culture model of primary KC and HSCs. Co-culture with KC induced a more activated phenotype and greater proliferation compared with HSC cultured alone. Co-cultured HSC showed elevated phosphorylation of p38, which when inhibited by catalase, anti-IL-6 and siRNA-IL-6 blocked TIMP-1 up-regulation and type I collagen accumulation. Thus, these results unveil a novel dual mechanism that is mediated by hydrogen peroxide (H2O2) and IL-6 by which KC modulate the fibrogenic response in HSC (95). A follow-up study unveiled synergism between arachidonic acid and ethanol to the mechanism by which KC mediate ECM remodeling and suggests that even if chronic ethanol consumption sensitizes HSC to up-regulate anti-fibrogenic signals, their effects are blunted by a second hit such as arachidonic acid (22).
Recent studies suggest that dietary factors such as protein intake can also modulate ROS-mediated collagen synthesis. HSC cultured in histidine-free medium decrease ROS production via the general control nonderepressible 2 (GCN2) and eukaryotic initiation factor 2α (eIF-2α) activation pathway (3) and lower type I collagen synthesis. Furthermore, Gcn2−/− mice showed more collagen deposition and liver injury after acute and chronic CCl4 administration (4). In contrast, leucine regulates type I collagen synthesis in HSC via increased production of ROS from NOX and mitochondria; leading to activation of the PI3K/Akt pathway, which up-regulates the transcription of type I collagen (106, 107). This demonstrates that protein intake could play a role during the progression of liver fibrosis.
Matrix-associated molecules and oxidant stress
Lately, several matrix-associated molecules have been implicated in the ROS-driven HSC fibrogenic response. As described earlier, work from our laboratory identified that OPN, an oxidant stress-sensitive cytokine, plays a major role in liver fibrosis (145).
Recently, we established that fibromodulin (FMOD), an oxidative stress-sensitive proteoglycan, regulates the fibrogenic response to liver injury in mice (93). Our studies demonstrate that liver samples from patients with cirrhosis had higher levels of Fmod mRNA and protein than controls. Moreover, BDL, CCl4, and TAA treatment increased levels of FMOD protein in wild-type mice. Infection of HSC with an adenovirus that expressed FMOD increased the expression of type I collagen, indicating activation of HSC. Recombinant FMOD promoted proliferation, migration, and invasion of HSC, contributing to their fibrogenic activity. Lastly, co-culture of hepatocytes or SEC with HSC increased ROS in the culture medium, along with type I collagen and FMOD, which was prevented by catalase (93).
Metalloproteinases and oxidant stress
In an in vitro O2· − generating system, Galli et al. evaluated the role of MMP-2 in HSC proliferation and invasiveness (39). Using xanthine and xanthine oxidase as a generator of O2· − , they showed an increase in MMP-2 and in proteins from its activation complex such as MT1-MMP and TIMP2, which provide a negative feedback mechanism, induce HSC proliferation, and increase their invasive potential. The addition of GM6001 (a broad-spectrum MMP inhibitor), OA-Hy (an MMP-2 inhibitor), or vitamin E and superoxide dismutase polyethylene glycol (antioxidants) blocked these effects (39).
Fibrosis Resolution
Apoptosis
Apoptosis of activated HSC is the best-characterized mechanism of fibrosis resolution. Watson et al. identified NFκB as a key regulator for HSC survival and proliferation by maintaining the expression of the anti-apoptotic molecule Bcl-2 (150). Inhibition of the NFκB pathway increases HSC apoptosis by JNK up-regulation both in humans and in rodents (150).
C/EBPβ has been linked to HSC survival and apoptosis in liver fibrosis. C/EBPβ activates caspase 8-dependent apoptosis, which is inactivated by ribosomal S-6 Kinase (RSK) phosphorylation. Buck and Chojkier demonstrated that the nonphosphorylatable form in C/ebpβ Tg mice protects them from CCl4-induced liver fibrosis by inducing apoptosis of HSC. Treating wild-type mice with an RSK-inhibitory peptide induced HSC apoptosis, prevented liver fibrosis progression, and promoted regression (12).
Farnesoid X receptor (FXR) is a ligand-regulated transcription factor that acts as a sensor for bile acids. Fiorucci et al. demonstrated that an FXR-small heterodimer partner regulatory cascade modulates TIMP-1 and MMPs' expression in HSC and promotes resolution of liver fibrosis (33). This mechanism blocks the anti-apoptotic signaling of TIMP-1 and modulates the sensitivity to other apoptotic signals in HSC. In vivo treatment with an FXR ligand protected against liver fibrosis and helped fibrosis resolution (33).
Cannabinoids and their receptors have been linked to the resolution of liver fibrosis due to their apoptotic effects in MF. The cannabinoid receptors CB1 and CB2 have opposing roles in liver fibrosis. Stimulation of CB2 receptor in HSC promotes growth arrest via the COX2 pathway and apoptosis involving oxidative stress in a cannabinoid concentration-dependent manner (66). CB1 receptor plays a different role, as Cb1−/− exhibit less liver fibrosis than CCl4-injected wild-type mice. HSC isolated from Cb1−/− mice undergo apoptosis in serum-deprived medium, whereas normal HSC maintain their viability. Moreover, pharmacological blockade of the CB1 receptor decreases HSC proliferation and reduces the number of MF in vivo (137). Furthermore, cannabinoids induce HSC apoptosis that is mediated by ER-stress activating ASK1/JNK (84).
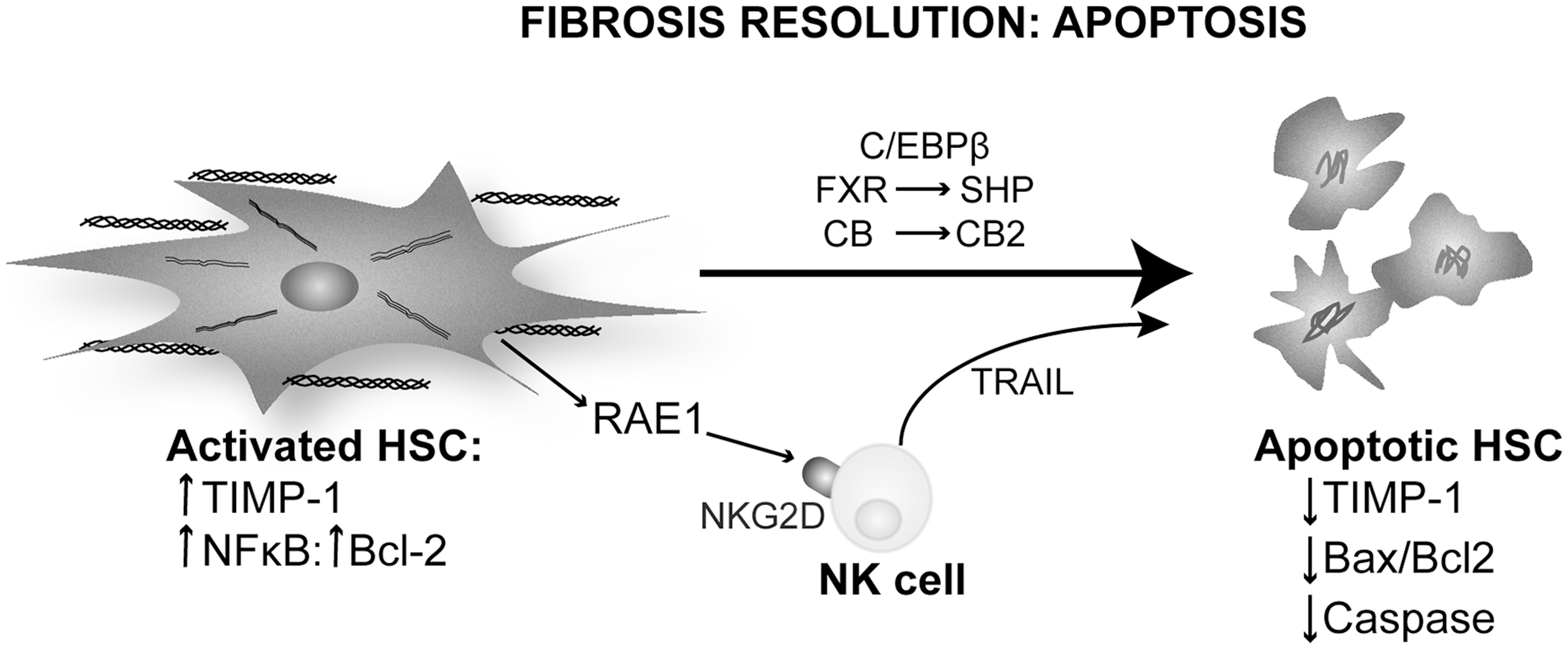
Natural killer (NK) cells are a part of the innate immune response and have been long implicated in apoptosis. Radaeva et al. showed that intrahepatic NK contribute to the resolution of liver fibrosis by targeting activated HSC (114). Activated but not quiescent HSC express RAE1, a ligand for the NK cells receptor NKG2D. An interaction of RAE1 with NKG2D triggers a cytotoxic response in NK cells that express TRAIL (death receptor activator) and activates HSC apoptosis (114). Glässner et al. confirmed this finding in human cells. However, there is a loss of NK cell activity with progression of fibrosis in human liver disease (43) (Fig. 6).
Senescence
Cellular senescence is an irreversible form of cell-cycle arrest that is triggered by stress-related events such as DNA damage, chromatin disruption, oxidative stress, and telomere dysfunction. Cells that enter senescence remain viable, and their metabolism is still active as they express senescence-associated secretory phenotype (SASP) but they do not respond to mitogens. Cellular senescence was first described in fibroblasts that experience replicative exhaustion in culture. Senescent fibroblasts are characterized by increased expression of inflammatory cytokines and MMPs as well as by down-regulation of selected ECM proteins (50). They are eventually cleared by NK cells, making senescence a candidate event to study the mechanisms driving fibrosis resolution.
Human HSC undergo senescence in culture after 9–15 passages due to shortening of telomeres. HSC undergoing senescence show a similar phenotype to skin fibroblasts with production of pro-inflammatory cytokines and chemokines, increased MMPs' expression, and reduction in ECM proteins.
Krizhanovsky et al. described this mechanism in vivo in CCl4-induced liver injury (78). This work identified senescence markers such as β-Gal, p53, p21, p16, and Hmga1 that co-localize with HSC markers such as α-SMA and desmin in fibrotic areas from mouse liver. p53−/− and p16−/− mice lack a senescence program and develop more fibrosis than wild-type mice due to slower resolution on cessation of CCl4 administration. These mice also show as many α-SMA-positive HSC as do transgenic mice expressing a p53 silencing construct under the control of Gfap to target HSC. This suggests that p53 and p16 are key players in regulating HSC proliferation in liver fibrosis and in clearing HSC during the resolution phase (78).
IL-22 induces HSC senescence in vivo and in vitro. Indeed, IL-22 promotes survival in HSC by inducing senescence rather than proliferation as it occurs in epithelial cells. Challenge with IL-22 modulates the expression of SASP by up-regulating pro-inflammatory genes along with MMP-9 and down-regulating TIMPs' expression both in vivo and in vitro. The mechanism involves up-regulation of p53 and p21 (senescence markers) by IL-22 and appears to involve STAT3 and SOCS3, as deletion of these factors abolishes the p53 and p21 response to IL-22 and blocks HSC senescence (76).
A recent study shows that the matricellular protein CCN1, highly expressed in human cirrhotic livers, plays a role in MF senescence after liver injury (70). Ccn1−/− mice develop more liver fibrosis and senescence markers such as β-Gal and p16 when compared with wild-type or Ccn Tg mice. In addition, CCN1 engages integrin α6β1 and activates ROS production via the RAC1-NOX1 pathway. The authors suggest that an increase in ROS could lead to DNA damage and, thus, activate the p53 and p16 pathway to induce senescence (70) (Fig. 7).

Reversal to quiescence
Although still a controversial issue, experimental evidence suggesting differentiation of MF to a more quiescent phenotype has been proposed in CCl4 or BDL-treated rats 1 year after cessation of the insult (72). This was demonstrated by decreased type I collagen and TIMP-1 expression along with increased active MMPs (36, 105).
Macrophages are important for the reversion of activated HSC to quiescent cells, as macrophages clear several mediators that are essential for HSC activation. A study showed that depletion of macrophages during reversal of fibrosis delays matrix degradation (5). Scar-associated MMP-13-expressing macrophages have been targeted as the responsible subpopulation that is involved in the reversal to quiescence (30, 57). Models of chronic liver injury have also revealed that CCR-2 is indispensable for macrophage recruitment during liver fibrosis and that Ccr2−/− mice elicit sustained TIMP-1 and reduced MMP-13 expression, thus enabling a persistent fibrogenic response (92). During reversal to quiescence, there is a rapid decline in TIMPs' expression, tipping the balance between MMPs and TIMPs and resulting in increased scar degradation (59).
Reversibility of liver fibrosis in humans: assessment and therapy
Regression of liver fibrosis is typically defined as a decrease in total matrix content (57). The majority of patients show a variable degree of reversibility of fibrosis after antiviral treatment (triple therapy with peginterferon alfa, ribavirin, and telaprevir or boceprevir), but there is no complete reversal to quiescence (36). So far, the assessment of regression largely relies on histopathological scoring of liver tissue obtained by needle biopsy, an invasive procedure that only provides a limited and static measurement of fibrosis (14). Over the past years, novel tests have been developed to assess the degree of stiffness noninvansively, for instance, transient elastography (FibroScan, now FDA approved for noninvasive liver diagnosis) and serum biomarkers of fibrosis such as the FibroTest® algorithm (14). Therefore, the availability of novel diagnostic techniques for monitoring the progression of liver disease will facilitate the evaluation of reversal from fibrosis and also of the efficiency of antifibrotic therapies.
The current therapy for reversing liver fibrosis focuses on reducing inflammation and the subsequent liver injury, in addition to targeting HCV replication (29). A number of dietary hepatoprotectants have been used to attenuate or neutralize the upstream inflammatory responses and, thus, HSC activation. Among these, silymarin (found in milk thistle), curcumin (turmeric), resveratrol (a component from red wine), and coffee have been tested (29). Promising results in experimental and clinical research have been given by vitamin E (123), a novel HGF mimetic agent (Refanalin), and caspase inhibitors (29, 155). Of particular interest is the development of PPARγ ligands (also known as glitazones) with the goal to reverse the biochemical and morphological features of HSC activation and, therefore, maintain HSC quiescence (79). Other molecules that are currently being tested in randomized controlled trials include inhibitors of 3-hydroxy-3-methylglutaryl-CoA reductase such as statins or their combination with sartans, angiotensin II subtype 1 receptor antagonists, which block HSC proliferation and collagen deposition in patients and in experimental models of liver fibrosis (139). Finally, the development of drugs that are capable of stimulating the degradation of accumulated scar tissue, particularly in patients with advanced disease, is an attractive approach. Indeed, in animal models, collagenolytic MMPs or TIMP-1 scavengers have been delivered systemically via adenoviral vectors and have been proved to stimulate ECM degradation (137).
Epigenetics, an Evolving Field
Epigenetic regulation of HSCs
Epigenetics is a process that alters gene activity without changing the DNA sequence, leading to heritable modifications. These modifications are natural and essential for normal development, differentiation, and tissue-specific gene expression. However, epigenetic abnormalities could occur due to environmental factors such as toxins, drugs, or diseases such as cancer, autoimmune disease, age-related illness, neurological disorders, and ALD (8, 31, 32, 65, 87).
Recent studies have highlighted the regulatory effect of epigenetic modifications on gene expression in HSC and on the risk of developing liver fibrosis (131, 161). Quiescent HSC acquire either an adipogenic or a myogenic phenotype depending on the balance between clusters of genes during their differentiation (142). Under normal conditions, adipogenic genes are dominant and quiescent HSC differentiate into adipogenic cells. When liver injury occurs, the prevalence of myogenic genes such as type I collagen, TIMP-1, or α-SMA promotes the differentiation of adipogenic HSC to myogenic HSC (5, 37). Moreover, aberrant expression of a series of histones and chemokines in activated HSC can aggravate inflammation and generate ROS and that increase HSC differentiation into MF, consequently enhancing liver fibrosis. The degradation of ECM is also affected by epigenetic modulation of matrix-associated enzymes such as MMPs (108).
The study of the methylation status of the promoters from fibrosis-related genes has recently become a hot topic over the past few years. For instance, the methylation status of SP1, a gene related to immunity, is down-regulated after CCl4-induced liver injury (49). Mann and collaborators observed that the offspring of mice exposed to CCl4 developed tolerance to liver injury (88). They hypothesized that genes related to inflammation and differentiation are hypermethylated, whereas most anti-fibrogenetic genes are hypomethylated (Fig. 8).

Epigenetic regulation of MMPs in HSC alters ECM degradation (113). Impaired histone deacetylation during HSC differentiation leads to increased histone deacytelase-4 and suppression of several MMP genes in quiescent HSC, accordingly silencing MMP activity and contributing to scarring (113). Taking into account the direct interplay between TIMP-3, MMPs, and ECM, methylation of TIMP-1 and TIMP-3 could be considered in the future as a reliable predictor of liver fibrosis (108).
Liver disease and epigenetics
A primary goal for preventing liver fibrosis is to discover accurate methods for early epigenetic diagnosis besides developing noninvasive methods to promote reversibility. In order to do so, we should dissect whether epigenetic alterations occur before proteomic deviations or vice versa. This is of clinical relevance, as epigenetic modifications can regulate HSC genes (131). Preliminary data suggest an association between hypomethylation of the PPARγ gene promoter and mild fibrosis in NAFLD patients (161). Recent studies show that inhibitors of DNA methyltransferase reduce the rate of DNA hypermethylation (143) and that a methyl-deficient diet regulates gene methylation and the development of liver fibrosis (140). Lastly, a recent study demonstrated that the necdin-Wnt pathway could be regulated to mediate anti-adipogenic HSC trans-differentiation via epigenetic repression of PPARγ, which may have therapeutic application. Silencing necdin either with shRNA or with rosmarinic acid and baicalin reverses activated HSC to quiescent cells in a PPARγ-dependent manner by suppressing canonical Wnt signaling (158).
Epigenetic modifications triggered by alcohol appear to play a relevant role in the development and progression of ALD. This has been described in parenchymal and nonparenchymal liver cells and contributes to fat accumulation and inflammation (87). In summary, the regulation of epigenetic alterations in key genes may be of therapeutic potential in the future to reverse liver fibrosis and hopefully, HCC. However, further understanding of the mechanisms of transgenerational inheritance in humans needs to be met.
New Tools to Study ECM
Microarrays
Microarrays have advanced the study of liver fibrosis and cirrhosis in humans and in animal models by identifying differentially expressed genes. A study comparing the expression of 22,000 genes from liver biopsies of HCV-infected patients at different disease stages identified 219 differentially expressed genes (1). Other studies have used DNA or cDNA microarray techniques to investigate the alteration of gene expression in liver fibrosis in in vitro or in vivo models (62, 112, 134). Similar analyses were performed by Mas et al. to identify changes in gene expression in patients with HCV-induced and with alcohol-induced liver cirrhosis (89). This study showed that genes involved in virus and immune responses as well as those regulating apoptosis were increased in HCV-induced cirrhosis whereas genes from the cytochrome P450 superfamily were found to be decreased in alcoholic cirrhosis (89). Lastly, Iizuka et al. demonstrated changes in gene expression according to HCV serotypes (54). Thus, the etiology of liver disease conditions gene signatures. However, the microarray technology has not been widely adopted as a routine clinical test not only due to the cost but also due to the lack of correlation between mRNA and protein expression (119).
Proteomics
Proteomics is a large-scale analysis of protein composition, structure, function, activity, and interaction with other molecules in a given biological system. However, post-translational modifications add a significant layer of complexity to interpret the results from this approach. The ECM components undergo vigorous post-translational modifications conditioning protein function (13). For example, type IV collagen, the major basement membrane component in the liver, undergoes glycosylation, phosphorylation, and ubiquitination, enabling collagen to interact with other ECM proteins and cells. By using ECM cataloguing, Rashid et al. showed that the components of the fibrotic tissue are unique to their localization. They used SDS-PAGE separation and mass spectrometry-based proteomic analysis to dissect the protein–protein interaction network and compare cell type-specific ECM niches between human foreskin fibroblasts and HSC cell lines (115). For example, fibulin-2 is an ECM protein in the protein network in the HSC ECM but is one of the least interacted in the human foreskin fibroblast protein network. This suggests that the composition and organization of the ECM protein network is tissue specific.
A limitation of this study is the lack of direct comparison between quiescent and activated HSC, as this could give a better picture in the changes in HSC in health and disease. Matrigel, a commercial basement membrane matrix, resembles the ECM environment and maintains HSC in quiescence (130). Although Matrigel could be used to reverse HSC activation, it would also add other proteins to the cell-derived ECM and alter the results from the mass spectrometry analysis. Consequently, alternative approaches are needed to study the transition of HSC in disease.
In a pilot study by Poon et al. (109), a new serum proteomic fingerprint associated with liver fibrosis was identified. Using surface-enhanced laser desorption/ionization protein-chip arrays technology, they constructed an artificial network fibrosis index that changes with the stage of liver fibrosis (109).
Although an active effort is in place for the search of noninvasive, cost-effective, and disease- and organ-specific liver fibrosis biomarkers, no reliable parameter is currently available for clinical diagnosis. A limiting factor may be the low reproducibility of the proteomics technology. This pitfall is due to a lack of standardized sample preparation protocol, for example, in sample purification and fractionation before analysis.
Innovation
This review drives from the composition and sources of the extracellular matrix (ECM) in healthy and injured liver to the potential mechanism driving fibrosis resolution such as myofibroblasts' apoptosis, senescence, and reversal to quiescence, thus compiling new advances in the diagnosis and treatment of liver fibrosis. It compiles a general and specific point of view of the role of ECM deposition in an oxidant environment in liver disease.
Future Directions
Liver biopsy is still the gold standard in the assessment of liver disease, but it is an invasive procedure and may cause clinical complications. Variations in diagnosis may also arise from sampling error and inter-observer observation (7, 20, 116). Careful integration of all the existing data from imaging techniques, genomics, transcriptomics, and proteomics analyses will provide a comprehensive view of changes in gene and protein expression, structure, function, and interactions in the disease state. The potential of systems biology for identifying specific and unique patterns of liver disease based on etiology may facilitate the diagnosis, prognosis, and, ultimately, treatment. As a result, increased accuracy in these strategies is required to correlate potential biomarkers with disease.
Footnotes
Acknowledgments
US Public Health Service Grants 5R01 DK069286, 2R56 DK069286, and 3 R56 DK069286-06S1 from the National Institute of Diabetes and Digestive and Kidney Diseases (N. N.); US Public Health Service Grants U01 AA021887-01, 5 P20 AA017067, 5 P20 AA017067-01S1, and 5 P20 AA017067-03S1 from the National Institute on Alcohol Abuse and Alcoholism (N. N.).