
Abstract
Introduction
M
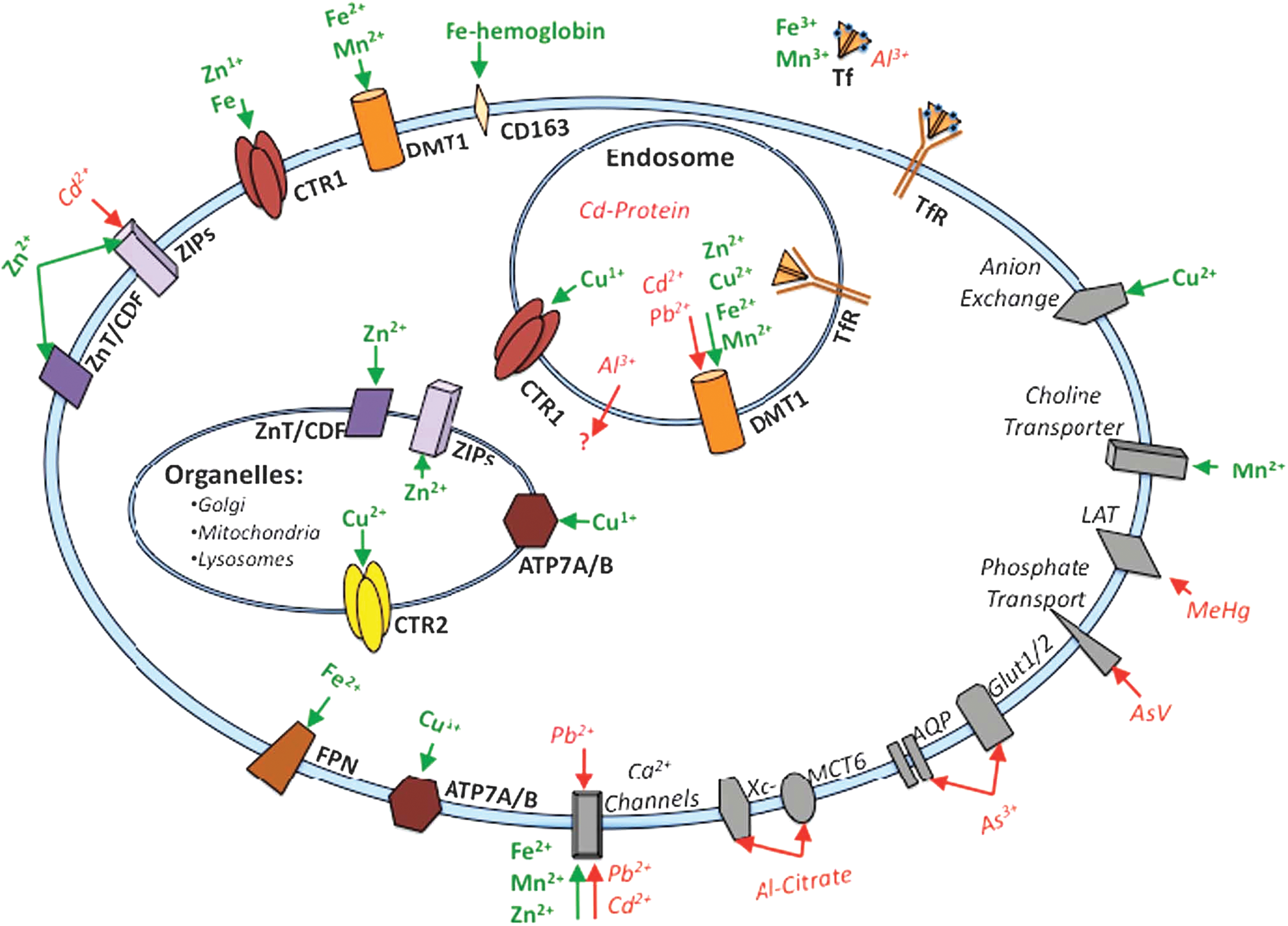
Protein tyrosine phosphatases (PTPs) constitute a large family of signaling enzymes that counteract protein kinases by removing phosphate moieties on tyrosine residues of the target proteins. They play essential roles in intracellular signal transduction, cell growth and differentiation, cell migration, gene transcription, ion-channel activity, immune response, and cell apoptosis (20, 79, 80, 86, 105, 107). In the human genome, there are at least 107 PTP genes, and 81 PTPs of them are active protein phosphatases with the ability to dephosphorylate phosphotyrosine residues (1). PTP activities are dysregulated in many human diseases, including diabetes, obesity, cancer, immune disorders, and osteoporosis (40, 46, 55, 96, 116). Protein tyrosine phosphatase 1B (PTP1B) was the first identified and characterized PTP. It takes part in the negative regulation of signaling pathways mediated by insulin receptors (IRs) and leptin receptors (5, 123). The PTP1B gene null mice exhibit increased insulin sensitivity and obesity resistance with normal phenotypic characteristics, even on a high-fat diet (23), establishing PTP1B as a potential therapeutic target in the treatment of type 2 diabetes and obesity. In addition, leukocyte common related antigen (LAR), PTPα, leukocyte common antigen 45 (CD45), receptor protein tyrosine phosphatase ɛ (RPTPɛ), Src homology phosphatase 1 (SHP-1), T-cell protein tyrosine phosphatase (TCPTP), and Src homology phosphatase 2 (SHP-2) have been conformed to be involved in diabetes mellitus (5, 21, 26). So far, at least 37 PTPs, including SHP-2, phosphatase of regenerating liver-3 (PRL-3), cell division cycle 25 (Cdc25), PTP1B, cell division cycle 14 (Cdc14), and low molecular weight PTP, have been implicated in human cancers (38, 41, 55, 98). Some PTPs (CD45, leukocyte common antigen 148 [CD148], lymphoid tyrosine phosphatase [LYP], SHP-1, TCPTP, and PTP with proline/glutamate/serine/threonine-rich domains [PTP-PEST]) have been linked to lymphocyte activation and autoimmunity (72, 86). Alzheimer's disease is a progressive disease associated with memory loss and impaired cognitive function and characterized by the cortical accumulation of amyloid plaques and neurofibrillary tangles. Amyloid-β accumulation has been demonstrated to be associated with an increase in striatal-enriched PTP levels and activity that in turn disrupts glutamate receptor trafficking to and from the neuronal membrane (115). Because PTPs are implicated in the pathophysiology of various disorders, many of them have been identified as potential new targets for novel drug discovery. A number of PTP inhibitors have been developed (6, 9, 45, 68, 81, 93). Some of them have been proven to be efficient in lowering blood glucose levels in vivo or in inhibiting tumor xenograft growth (93). Although PTPs do not contain any metals, their activities are potently inhibited by some metal ions and metal complexes (65). In this review, we will focus on the recent developments in metal-based PTP inhibitors.
PTP Inhibition by Metal Ions
Metal ions are known to strongly inhibit PTPs. Brautigan et al. originally identified Zn2+ as a PTP inhibitor in 1981 (10). They found that Zn2+ completely inhibited the dephosphorylation of phosphotyrosine residues on an unidentified membrane protein at concentrations of 10 μM, while other divalent metal cations had no substantial effect. Zn2+ strongly inhibits many PTPs in vitro (19, 30, 42, 106, 112, 126). The IC50 values of Zn2+ inhibition of some PTPs are in the nanomolar range, ∼17 nM for PTP1B, 200 nM for TCPTP, 98 nM for SHP-1, and 1–2 μM for SHP-2 (35, 36). The study by Maret and colleagues demonstrated that Zn2+ is a reversible inhibitor of the receptor PTPβ, with a remarkably low Ki of 21 pM. The cellular free or loosely bound zinc concentrations in various cultured cells are in the range of tens to hundreds of picomolar, suggesting that cellular Zn2+ may act as a physiological regulator of PTPs and modulates signal transduction (114). Cellular studies have shown that high concentrations of Zn2+ (>100 μM) cause an increase in the tyrosine phosphorylation of proteins such as the epidermal growth factor receptor (EGFR), IR/IGR, c-Src kinase, and extracellular signal-regulated kinase (Erk) (35, 36, 90, 91, 99).
Choi's group investigated the effects of various metal ions on the activity of the vaccinia H1-related protein tyrosine phosphatase (VHR) (50). Among the various metal ions examined, Fe3+, Cu2+, Zn2+, and Cd2+ were found to inactivate VHR, and Cu2+ was the most potent inactivator. The inactivation effect of Cu2+ ion on VHR can be restored by ethylenediamine tetraacetic acid (EDTA). High concentrations of GSH effectively decrease the inactivation effects. The loss of VHR activity is correlated with a decreased 14C-iodoacetate labeling of active-site cysteine. They thus concluded that the highly potent Cu2+ inactivation of VHR is a consequence of the oxidation of the active-site cysteine. Cu2+ also potently inhibits PTP1B, TCPTP, megakaryocyte protein-tyrosine phosphatase (PTP-MEG2), and SHP-1 with IC50 values of ∼0.14, 0.15, 0.20, and 0.18 μM, respectively, but almost does not inactivate SHP-2 (127).
Chromium supplementation either improves glycemic control or reduces insulin requirements in patients with type 2 diabetes mellitus (2 –4). An in vitro study demonstrated that 0.1 mM CrCl3 inhibits PTP1B (rat and human) activity to 21%–33% of control activity. The treatment of cultured rat hepatoma cells with 0.1 mM CrCl3 increased the insulin-stimulated tyrosine phosphorylation of the high Mr IR substrate (IRS) proteins by 49% to 7.3-fold over basal phosphorylation levels, without altering basal IR or IRS tyrosine phosphorylation or insulin-stimulated receptor autophosphorylation (31).
Gallium nitrate inhibits membrane PTPs both in vitro and from cells with the IC50 values of ∼6.0 μM (7).
These studies indicate that some metal ions are potent inhibitors of PTPs. Particularly, copper and zinc inhibit most PTPs, with IC50 values in the nanomolar range. Copper and zinc are essential metals. Total cellular copper or zinc concentrations are in the several to a few hundred micromolar range, which is usually considered physiologically significant. The nanomolar IC50 values of the copper and zinc required for inhibiting most PTPs are obviously lower than the concentrations of physiological significance, implying that a minor increase in cellular copper or zinc ions will efficiently inhibit PTPs and increase cellular phosphorylation levels, thereby disturbing metabolic processes. Thus, PTP inhibition is possibly involved in the pathophysiological and toxicological processes of metals. Furthermore, the cellular free zinc may act as a physiological regulator of PTPs to modulate signal transduction.
PTP Inhibition by Metal Complexes
Many metal complexes have been utilized for curing diseases. For example, auranofin is a therapeutic agent used clinically to improve the symptoms of rheumatoid arthritis, and cisplatin is used to treat cancers, especially for testicular and ovarian cancers. Indeed, metal complexes have a wide range of biological effects, such as anticancer, antibacterial, antimicrobial, antifungal, and antiparasitic activities. They have also been tested in the treatment of heart diseases, diabetes, rheumatoid arthritis, and ulcers (25, 27 –29, 47, 60, 73, 113). However, their cellular targets are not well known, impeding the design of more efficient metal-based drugs. With the development of proteomics in the last two decades, the interactions between metal complexes and proteins have attracted more and more interest. Metal complexes have been shown to potently inhibit many enzymes, such as protein kinases, matrix metalloproteinases, telomerases, topoisomerase II, glutathione-S- transferases, histone deacetylases, and the chymotrypsin-like activity of the proteasome and so on (11, 14, 24, 49, 54, 71, 83). These enzymes have become possible targets for a novel metal-based drug design.
The activities of PTPs are potently inhibited by metal complexes. Some metal complexes have been shown to be more efficient than metal ions in affecting cellular tyrosine phosphorylation.
Vanadium complexes
Vanadium mimics the effects of insulin. Organic vanadium complexes exhibit high insulin-mimetic potential with few side effects compared with inorganic vanadium (102, 103, 113). A widely accepted mechanism for this action is the inhibition of PTPs.
An earlier study indicated that pervanadate and peroxovanadium complexes display a higher insulin-mimetic activity and PTP inhibitory activities (8, 58). They inhibit the in situ dephosphorylation of autophosphorylated IRs and EGFRs activating the IR kinase and other kinases in cells (82, 94). The increased PTP inhibition activity is partially related to their ability to induce intracellular oxidation (52). An in vitro study has shown that pervanadate inhibits PTP1B by an irreversible oxidation of the catalytic cysteine-SH group to -SO3H (43).
Although pervanadate and peroxovanadium complexes display higher insulin-mimetic and PTP inhibitory activities, they are more toxic due to their higher valence (92). Vanadate and vanadium complexes are potent PTP inhibitors and efficiently lower blood glucose levels, such as bis(ethylmaltolato)oxovanadium(IV) (BEOV), a bis(maltolato)oxovanadium(IV) (BMOV) derivative, selected for phase 1 and 2 clinical trials (92, 102, 103, 113). The model of PTP inhibition by vanadium compounds is reversible rather than an irreversible oxidation of the catalytic cysteine-SH group (37, 43, 61, 64, 78, 120). The inhibitory abilities of most vanadium complexes (Fig. 2) are shown in Table 1. These data show that vanadium complexes inhibit a great variety of PTPs. BMOV

HCPTPA, human low molecular weight cytoplasmic protein tyrosine phosphatase; HePTP, hematopoietic protein tyrosine phosphatase; HRPTPβ, human receptor protein tyrosine phosphatase beta; PTP, protein tyrosine phosphatase; PTP1B, protein tyrosine phosphatase 1B; SHP-1 Src homology phosphatase 1; SHP-2 Src homology phosphatase 2; TCPTP, T-cell protein tyrosine phosphatase.
Copper complexes
Copper complexes have been widely explored as therapeutic agents (22, 76). Many new copper complexes have been demonstrated to have great anticancer potentials and relatively low side effects compared with traditional platinum-based drugs (70, 89, 100). Copper complexes decrease the levels of the Alzheimer disease amyloid-β peptide by activating cell signaling pathways, having potentially neuroprotective effects (12, 17). As potential therapeutics, copper complexes are generally thought to generate ROS and damage macromolecules such as DNA (34, 47, 101). Some investigations have indicated that proteasome inhibition is involved in the apoptosis of cancer cells induced by copper complexes (15, 16, 18, 104). Copper complexes have been designed as human immunodeficiency virus type 1 (HIV-1) protease inhibitors (56, 57). Thus, cellular proteins are possible targets of therapeutic copper complexes.
We found that copper complexes (Fig. 3) can potently inhibit PTPs (Table 2) in vitro. Kinetic studies indicated that these complexes inhibit PTPs by a reversible inhibition model. They bind to PTPs at the active site or allosteric sites. This inhibition may not involve ROS, but may be related to the oxidation state of copper, because monovalent copper complexes exert weaker inhibition (110, 111). The structure of copper complexes affects the potency and selectivity of their inhibition of PTPs. The copper complexes

PTP-MEG2, megakaryocyte protein-tyrosine phosphatase.
Copper complexes have been found to inhibit PTPs and increase phosphorylation in cells (84, 122). Treatment with bis(thiosemicarbazonato) copper complexes substantially enhances phosphorylation of EGFR in U87MG-EGFR (human astroglial U87MG cell line transfected with the epidermal growth factor receptor) and HeLa cells (cells derived from Henrietta Lacks). However, Cu2+ alone does not have this function. The PTP activity inhibited by bis(thiosemicarbazonato)copper complexes at lower concentrations (5 μM) can be partly restored by EDTA or dithiothreitol, but not at higher concentrations (25 μM). The authors consider that the inactivity of PTPs is possibly due to oxidative damage of the enzymes. The proposed mechanism (Fig. 4) is that once the complex enters the cell and encounters the intracellular reducing environment, the Cu2+ ion is reduced and dissociated from the complex, releasing Cu+ that inhibits PTP1B phosphatase activity and leads to sustained phosphorylation and activation of EGFR, which elicits the release of growth factor and cytokine (84).
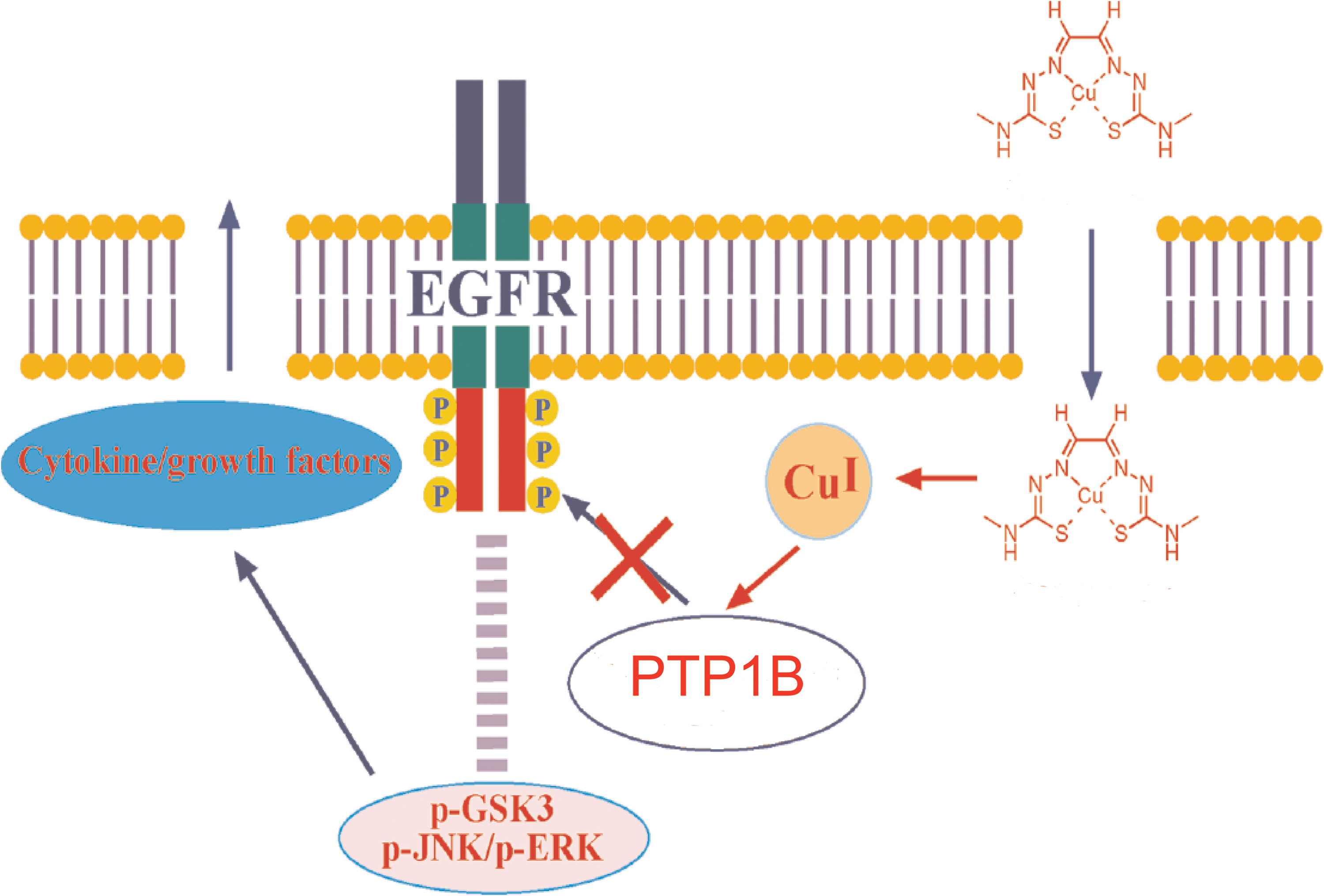
Because complex

Gold complexes
Gold complexes have been used for many years in the treatment of autoimmune and inflammatory diseases, including rheumatoid and psoriatic arthritis, pemphigus vulgaris, and bronchial asthma (95). They also display anticancer, anti-HIV, and anti-AIDS, as well as antiparasitic properties. Their cellular targets are involved in DNA, thioredoxin reductase, proteasome, and topoisomerase I (73, 87, 95, 117, 118). In 1997, Wang et al. first investigated the PTP inhibitory activity of disodium aurothiomalate
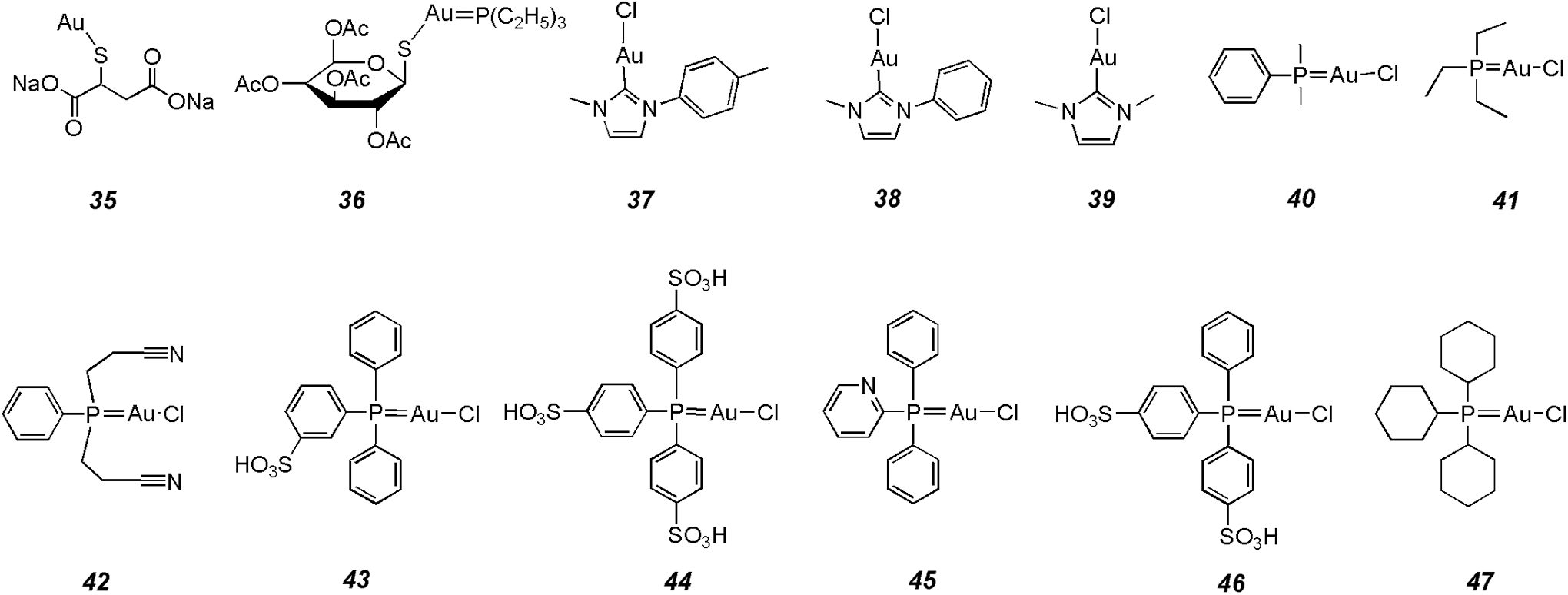
CD45, leukocyte common antigen 45; LYP, lymphoid tyrosine phosphatase; PTP-PEST, PTP with proline/glutamate/serine/threonine-rich domains.
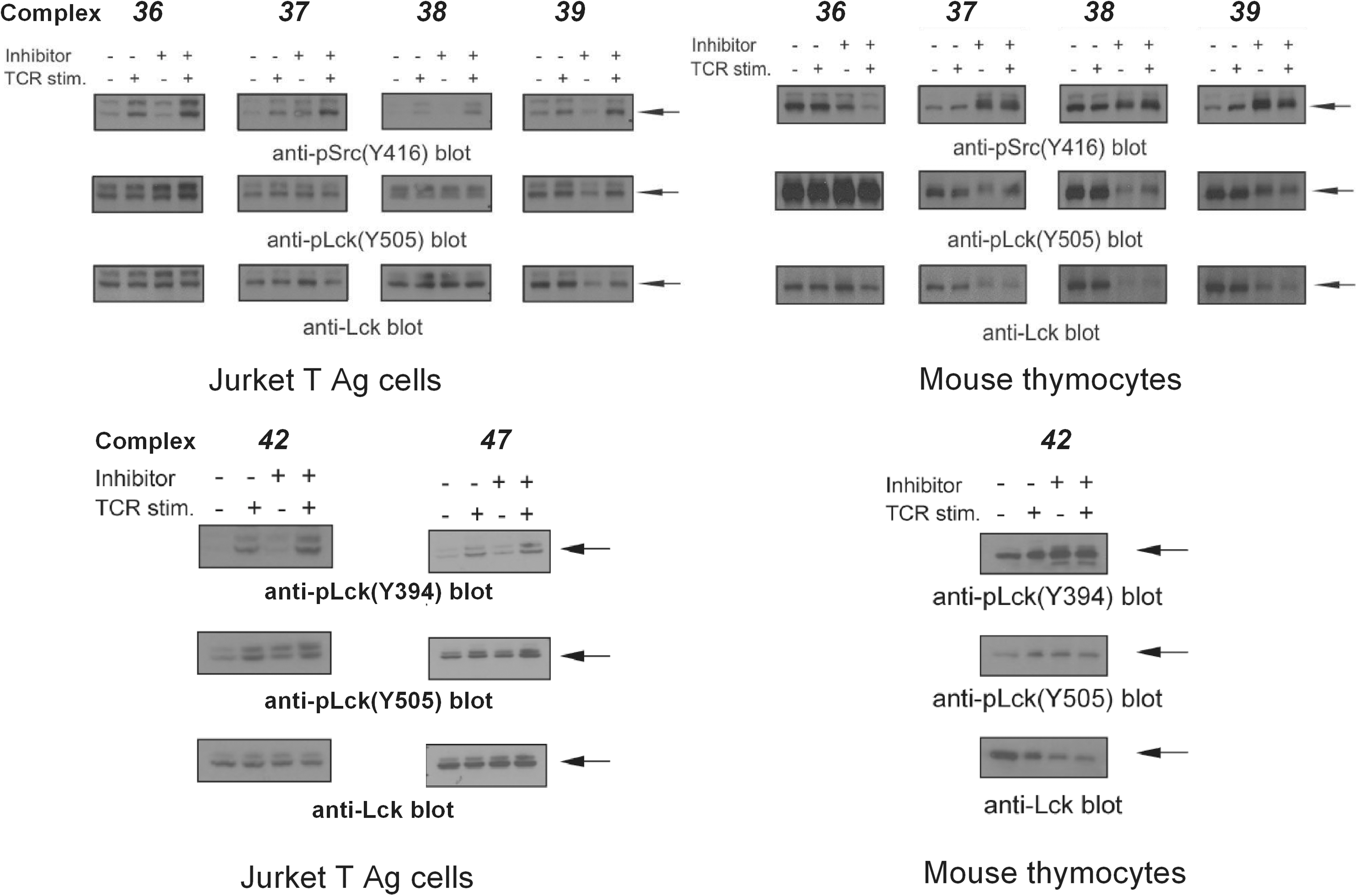
Barrios' group investigated the inhibitory activities of many gold(I) complexes on PTPs (48, 53). Like vanadium and copper complexes, the structures of gold(I) complexes obviously affect their inhibitory potency and selectivity on PTPs. However, their potency seems weaker than vanadium and copper complexes, usually in the micromolar range in vitro. Investigations into the model of inhibition indicate that the complexes reversibly and competitively inhibit PTPs. A comparison of the inhibition activities of gold(I) complexes (Fig. 7) on several PTPs is shown in Table 3. Four gold(I) N-heterocyclic carbine complexes
Cellular investigations of PTP inhibition by gold(I) complexes in a Jurkat T antigen human T-cell line and primary mouse thymocytes revealed that the complexes
Ruthenium complexes
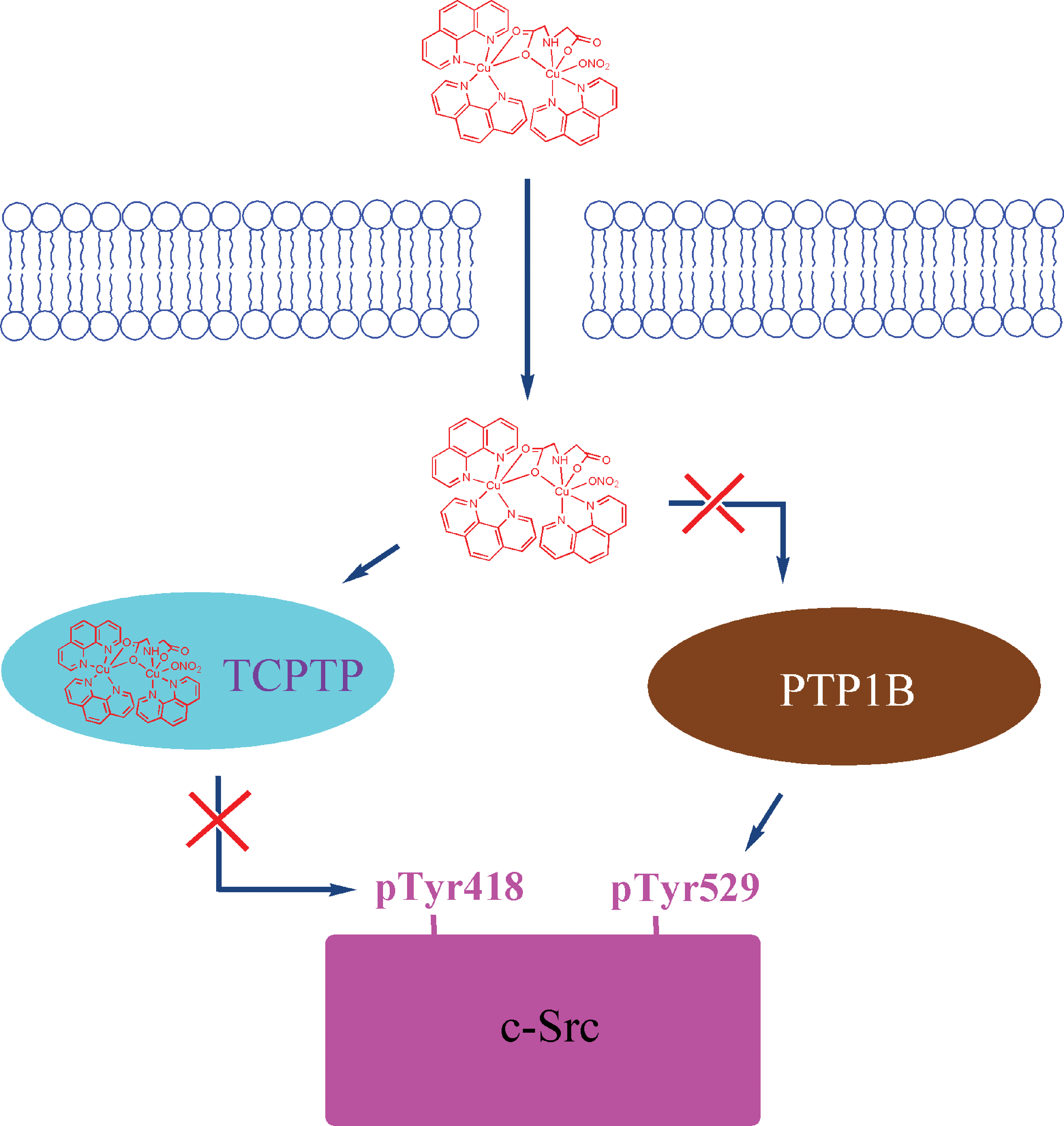
In the last four decades, many ruthenium complexes have been synthesized and tested for antitumor properties (59, 60). Similar to cisplatin, ruthenium complexes are expected to reduce tumor growth by a mechanism of interaction with cellular DNA (60). Recent studies suggest that ruthenium complexes interact with enzymes such as protein kinases, topo II, and glutathione S-transferases (33, 49, 71). A cocrystal structure of a ruthenium complex and GST P1-1 demonstrated that the complex binds to the enzyme at the dimer interface, close to the thiol of Cys101. Some ruthenium complexes were designed as selective inhibitors of PTPs.
Chatterjee et al. found that [RuIII(EDTA)(OH2/OH)]1−/2−

Ong et al. synthesized a series of organoruthenium complexes containing difluoromethyl- phosphonate functional groups and evaluated their inhibition activity on PTPs (74). Only compounds containing the phosphonic acid moiety were efficacious. Among them, organoruthenium complexes, [(η6-cymene)Ru(benzimidazole-phosphonic acid)Cl]Cl
Stibium complexes
Sodium stibogluconate is a drug used in the treatment of leishmaniasis. It selectively inhibits SHP-1 with 10-fold stronger potency than against SHP-2 and PTP1B, but does not inhibit dual specificity protein phosphatase 1/mitogen-activated kinase phosphatase 1 (DUSP1/MKP-1) phosphatase. The concentration completely inhibiting the SHP-1 activity is about 10 μg/ml. In Baf3 cells, the complex augments IL-3-induced Janus family kinase 2/Stat5 tyrosine phosphorylation and proliferation, consistent with its inhibition of SHP-1 in vitro (77).
Iron complexes
Recently, Olivier's group investigated the ability of a mononuclear dicitrate iron complex [Fe(cit)2H4-x](1−x)− to inhibit the PTP activity in macrophages (32). Their results demonstrated that the complex alone was able to inhibit the total PTP activity in macrophages, whereas the inorganic salts FeSO4 or FeCl3 did not inhibit the PTP activity in macrophages. The inhibition of the PTP activity in macrophages by this complex was not related to the generation of ROS via Habber Weiss and Fenton chemistry. The complex inhibited recombinant SHP-1, TCPTP, PTP1B, and PTP-PEST. Treatment with the complex led to protein hyperphosphorylation and enhanced mitogen-activated protein kinase (MAPK) signaling in response to LPS (Escherichia coli, serotype 0111:B4) stimulation.
Gallium complexes
Gallium complexes, digallium trisulfate, gallium tris(acetylacetonate), and gallium tris(D-gluconato) are potent PTP inhibitors. The IC50 values for their inhibition of membrane PTPs are ∼8.4, 21.7, and 60.1 μM, respectively (7).
Innovation
Protein tyrosine phosphatases (PTPs) are involved in the pathogenesis of a number of human diseases and are recognized as potential new targets for drug discovery. The inhibition of PTPs by metal complexes demonstrates that metal complexes, especially the V, Cu, and Au complexes, are potent PTP inhibitors. Some copper and gold complexes are good selective inhibitors of one or two specific PTPs and selectively improve the phosphorylation of the corresponding substrates in cells. The data suggest that the exploitation of metal-based therapeutic agents targeting specific PTPs is promising.
Conclusions and Future Directions
The in vitro studies of PTP inhibition demonstrate that some metal ions and metal complexes are potent PTP inhibitors. Metal atoms in metal complexes play an important role in the binding of PTP, while ligand structures influence their inhibitory potency and selectivity. Although metal complexes are usually expected to exchange their metal ion in the cell, some metal complexes are stable enough to penetrate the cell membrane and enter cells, selectively binding to their targeting PTPs and influencing the related cell signaling cascades. These findings suggest that PTP inhibition is possibly implicated in the pathophysiological and toxicological processes of metals, and that PTPs may be cellular targets for metal-based therapeutic agents. However, how metal complexes bind to PTPs and what roles the metals and their ligands play in this binding are still not known. Therefore, using structural biology methods, such as X-ray crystallography, to study the structural basis of the interactions between metal complexes and PTPs will be important to facilitate a comprehensive understanding of the structure–activity relationship and help us to design novel metal-based therapeutic agents targeting specific PTPs.
Footnotes
Acknowledgments
This work was supported financially by the National Natural Science Foundation of China (21171109 and 21271121), SRFDP (20111401110002 and 20121401110005), the Natural Science Foundation of Shanxi Province of China (2010011011-2 and 2011011009-1), and the Research Project supported by Shanxi Scholarship Council of China (2012-004 and 2013-026).