
Abstract
Significance:
Myofibroblasts are prototypical fibrotic cells, which are involved in a number of more or less pathological conditions, from foreign body reactions to scarring, from liver, kidney, or lung fibrosis to neoplastic phenomena. The differentiation of precursor cells (not only of fibroblastic nature) is characterized by a complex interplay between soluble factors (growth factors such as transforming growth factor β1, reactive oxygen species [ROS]) and material properties (matrix stiffness).
Critical Issues:
The specific mechanisms of action of ROS remain largely unknown, although evidence suggests that both intracellular and extracellular phenomena play a role.
Ontology of Myofibroblasts
M
In the following sections, we will review the current knowledge about (i) the signature characteristics of these cells, whose identification has often been somehow ambiguous in the past, (ii) their origin, that is, their precursors and the differentiation mechanisms, and (iii) their biomedical importance, that is, the pathologies that myofibroblasts are most commonly associated with.
Identification
Although morphologically similar to fibroblasts (both are spindle-shaped or stellate cells), myofibroblasts are characterized by some structural and biochemical peculiarities (90) that allow distinguishing them from other, more or less close mesenchymal phenotypes. These markers are described hereafter and summarized in Table 1.
ECM, extracellular matrix; TIMPs, tissue inhibitors of matrix metalloproteinases.
Structural markers
1) Alpha-Smooth Muscle Actin (α-SMA)
α-SMA is the actin isoform typically encountered in vascular smooth muscle cells (SMCs); its incorporation in stress fibers (163) allows the generation of higher contractile forces than those achievable with fibers composed by only β- and γ- cytoplasmic actin (93), as e.g. in normal fibroblasts. α-SMA accounts for the distinctive staining pattern obtained by labeling (myofibroblast-rich) granuloma pouches with human anti-smooth muscle sera (76), and there is a general consensus in using α-SMA as the key molecular hallmark of myofibroblasts. It is worth noting that α-SMA is not only the main marker of the myofibroblast phenotype, but also a major determinant of their contractility (6, 91, 92); in addition, other contractile proteins, such as desmin or smooth muscle-myosin, can be expressed by myofibroblast cells (221), but it is the rather occasional presence that makes them quite precise markers of muscle cells rather than of myofibroblasts.
As a caveat, α-SMA is present in several contractile cells of nonmuscular phenotype (99, 205). For example, α-SMA is a classical marker for mature/terminally differentiated pericytes, which many consider cognates to myofibroblasts; however, pericytes also express desmin, although the contemporaneous expression of both markers has been the subject of a long debate and may depend on the degree of pericyte maturation (122, 146, 156). Therefore, the presence of α-SMA and the absence of typical muscle cell markers, such as smooth muscle myosin, are not sufficient to conclusively identify a cell as a true myofibroblast.
2) Vimentin
Vimentin is the main component of intermediate filaments of mesenchymal cells (75) with a variety of functions related to cell adhesion and signaling (103). As a typical mesenchymal marker, it is overexpressed in several forms of myofibroblast differentiation, such as the epithelial–mesenchymal transition (EMT) (242) or the induction of myofibroblast character in the reactive stroma of tumors (222). It is, therefore, often used to confirm the mesenchymal character of α-SMA-positive cells.
3) Extra Domain-A Fibronectin (E D-A FN)
Fibronectin is a dimeric glycoprotein with each monomer composed of structural repeating units (modules) of three different types (100). Modules III lack disulfides in their structure and as a result can unfold upon application of relatively moderate mechanical loads (180), causing a different presentation of specific binding sites such as RGD sequences (118); the exposure of these generally cryptic sites makes these modules mechanically sensitive switches. Whereas the plasma FN is soluble and preserves a strictly dimeric form, the cellular form (cFN) has a strong tendency for fibrillar aggregation, which is related to the presence of alternatively spliced modules III: Extra Domain-A (ED-A) and Extra Domain-B (ED-B). Extra-Domain-A fibronectin (ED-A FN) is important in the regulation of integrin-mediated cell adhesion (129) and activates leukocytes by binding toll-like receptor 4 (TLR-4) (125, 157); these two features are probably the basis of its necessary role in natural wound healing (148) and probably in angiogenesis (8). The ED-B isoform is considered a more reliable angiogenic marker due to its more restricted distribution (36), but its biological roles are yet to be fully clarified (241).
In fibrotic phenomena, ED-A FN is expressed before α-SMA [in the evolution of granulation tissue (196)] and before major collagen deposition [in idiopathic pulmonary fibrosis (120)]; further, by blocking ED-A FN with antibodies (196) or using ED-A FN−/− fibroblasts (149), it is possible to inhibit the transforming growth factor β1 (TGF β1)-mediated myofibroblast transition (see later the section “Differentiation mechanism”), both of which attain the production of α-SMA and collagen I and the overall organization of intracellular stress fibers and extracellular fibronectin (106).
ED-A FN could be therefore considered to be a marker of incompletely differentiated and noncontractile (non α-SMA-containing) cells, which have been defined by Gabbiani as protomyofibroblasts (55).
4) Calponin and caldesmon
Calponin and heavy caldesmon (h-caldesmon) are calmodulin-binding proteins involved in smooth muscle contraction, which have been used as markers of SMCs (74). Due to the significant similarities between the contractile machinery of smooth muscle cells and that of myofibroblasts, the latter are positive for calponin, but they are generally negative for h-caldesmon both in proper myofibroblastic tumors (37) and in proliferative/fibrotic responses (46).
5) Fibronexi
Myofibroblasts can actively contract and transmit the generated force to the surrounding extracellular matrix (ECM) via specialized structures characterized by transmembrane associations of α-SMA-containing stress fibers and ED-A FN; such structures are named supermature focal adhesions in vitro and fibronexi in vivo (60, 202). These peculiar structures allow force transmission and physical continuity between coaligned stress fibers and ECM fibers (92), being therefore part of a positive feedback loop: stress fibers contract the surrounding ECM, and the mechanical tension is sensed by myofibroblasts via the fibronexi, thus maintaining the activated phenotype (92, 202).
6) Adherens junctions containing OB-cadherin (95)
Adherens junctions are points of intercellular contact where the cytoplasmic sides of the junctions are linked to cytoskeletal elements, typically stress fibers in a fashion mediated by catenins and transmembrane cadherins. Adherens junctions are not typically present in fibroblasts in vivo, but develop when they are cultured on hard substrates (95), and may thus be considered to be a rather protomyofibroblast marker. The composition of cadherins participating in adherens junctions can significantly change in the presence of phenotypic transitions involving different mechanisms of locomotion and/or adhesion, for example, when EMT is induced via TGF β1 administration, cells typically move from an E-cadherin-expressing (epithelial and static) phenotype to an N-cadherin-expressing (fibroblastic and migratory) one (24). Similarly, the myofibroblast differentiation is accompanied by a switch from N-cadherin (migratory phenotype) to OB-cadherin (fibrogenic phenotype) (95), where OB-cadherin is specifically useful for increasing the mechanical strength of the junctions (167).
7) Tenascin
Tenascin (more specifically tenascin-C) is an ECM glycoprotein that modulates the adhesion of cells to fibronectin; it is typical of developing tissues, while in the adult organism it is found essentially in the ECM of mesenchymal tissue, with a specific association to pathological conditions (infection, inflammation, cancer) (41). In a kidney environment, tenascin expression is upregulated upon TGF β1 stimulation of renal tubule endothelial cells, but not of mesangial cells, and, therefore, it could be considered a marker of myofibroblast-mediated fibrosis due to the endothelial–mesenchymal transition (EndMT) (82); during wound healing, with good localization of myofibroblasts in myocardial repair (218), but only partial colocalization in cutaneous wounds (45, 133), the link with tumor-associated myofibroblasts has been clearly demonstrated in the progression of colon (50) or prostate (222) cancer.
8) Enhanced ECM production
The defining feature of a fibrotic reaction is the excessive deposition of ECM and specifically of its fibrillar components; therefore, it is not surprising that myofibroblasts are primary collagen-producing cells (244). In particular, there is compelling evidence that myofibroblast persistence is linked to the accumulation of collagen type I and III in both in vitro models and biopsies from fibrotic conditions (9, 44, 214, 253). Interestingly, the synthesis of both collagen type I and type III in response to TGF β1 appears not to be a stand-alone pathway, and interactions with other growth factors can modulate the presence or extent of the response (84). Specifically, coincubation of TGF β1 with insulin-like growth factor 2 (IGF 2) appeared to further promote collagen synthesis, whereas coincubation with EGF completely abolished TGF β1-induced collagen upregulation.
Biochemical markers
1) Lysyl hydroxylase 2
The most important elastically active ECM components, collagen and elastin, are typically stabilized via lysyl oxidase (LOX)-induced cross-linking (68, 177); this post-translational process occurs extracellularly and it is based on the enzymatic conversion of primary amines into aldehydes followed by the spontaneous reaction leading to Schiff bases (Fig. 1, top examples); in its simplest version, which is typically encountered in elastin, lysine residues in peripheral positions can be oxidized at their the ɛ-amine group and converted into an allysine structure (more correctly α-aminoadipic-δ-semialdehyde), to finally react with lysines located in specific positions of the helical portions of the macromolecules (Fig. 1, see production of Δ-HLNL).
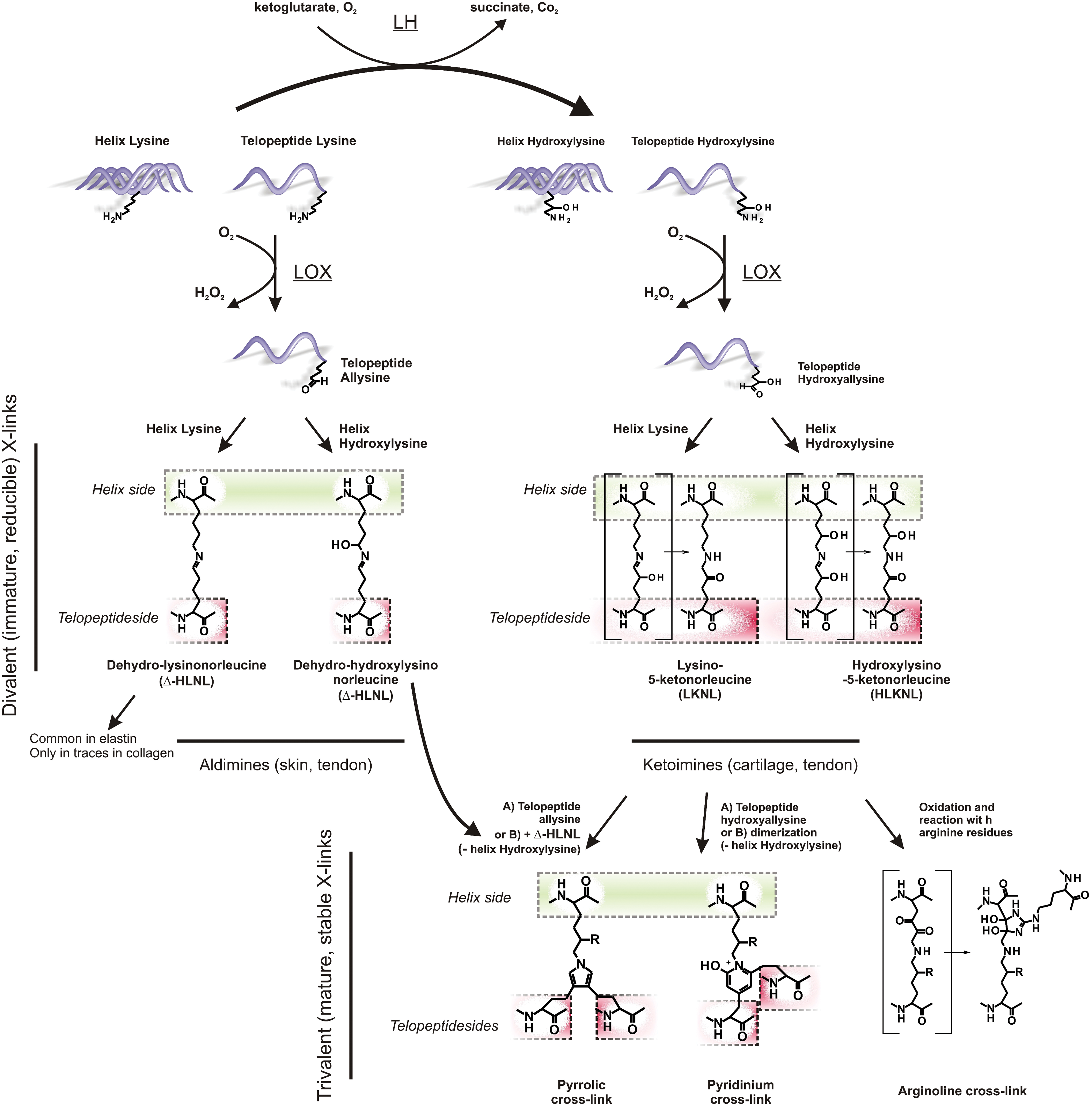
In the stabilization of collagen, however, difunctional cross-links based on lysine/allysine coupling are rather rare (10); on the contrary, the stabilization mechanism is essentially based on lysine hydroxylation by the action of lysyl hydroxylase (LH) in the rough endoplasmic reticulum. The presence of these secondary reactive groups allows for a number of spontaneous rearrangements after amine oxidation by LOX, which eventually produce irreversible, trifunctional cross-links in the mature collagen (66, 67), the most common ones being based on pyridinoline (hydroxyl pyridinium) groups. Importantly, the increase of pyridinoline-based collagen cross-linking is a widely recognized fibrosis hallmark (30, 102, 179); since their biosynthesis is necessarily based on lysine hydroxylation, the latter has been suggested as a marker of fibrosis (229) and therefore also of a putative marker of myofibroblast differentiation. Indeed, the LH splice variant LH2b is heavily overexpressed in different fibrotic conditions such as hypertrophic scars and Dupuytren's disease, as well as in vitro in activated hepatic stellate cells (230); it is noteworthy that the expression of the splice variant LH2a, of the LH1 and LH3 isoforms and also of LOX, was not altered under fibrotic conditions. The inflammation/fibrosis-activated character of LH2b is confirmed by the fact that treatments with a variety of cytokines (TGF β isoforms 1, 2, and 3, as well as interleukin 4, activin A, and tumor necrosis factor α [TNF α]) result in LH2b overexpression (228) and that both LH2b and α-SMA have a concomitant expression burst at day 7 after wounding (223).
2) Xylosyltransferase-I
The two isoforms of xylosyltransferase (I and II) catalyze serine glycosylation (addition of UDP-xylose), which is the first step in the production of proteoglycans from core proteins (83). These enzymes are used as markers of proteoglycan production since scar/fibrotic tissues contain a higher proteoglycan content than I normal tissues (215). Xylosyltransferase-I has been recently proposed as a marker of myofibroblast content and activity in a given tissue (70); in comparison to α-SMA, xylosyltransferase-I appears to have several advantages: first, at the mRNA level, its TGF β1-stimulated upregulation is larger compared with α-SMA; second, it appears to be nonlinearly sensitive to the myofibroblast density, which may provide further information about their cross-activation; finally, it can be analyzed simply by sampling a fluid phase (it is a soluble marker).
3) Matrix MetalloProteinases (MMPs) and their inhibitors
MMPs are zinc ion-dependent (Zn2+) proteolytic enzymes that form part of a large metzincin superfamily, which includes astacins, A disintegrin, metalloproteases (ADAMs) and ADAMs with thrombospondin motifs. MMPs are usually secreted as inactive proteins from cells [with the exception of MT-MMPs (159)] and are tightly regulated by a group of naturally occurring inhibitors called tissue inhibitors of matrix metalloproteinases (TIMPs) (80, 152). Achieving homeostasis between MMPs and TIMPs is pivotal in ECM turnover and remodeling, whereby failure results in a range of pathological processes, such as tumor invasion, chronic ulceration, atherosclerosis, arthritis, and neurodegenerative diseases (150, 234). TGF β1-induced myofibroblast differentiation has been associated to a reduction in ECM remodeling via modulation of the TIMP/MMP ratio (62). Similar results were obtained by Seeland et al., who reported a reduced collagenase expression and an overexpression of TIMP-1, -2, and -4 in TGF β1-overespressing transgenic mice (194). It has likewise been reported that MMP 1 overexpression following myocardial ischemia in rats could be abolished by TGF β1 treatment (39). A recent article reported the downregulation of MMP 2, also known as gelatinase A, after treatment of fibroblasts with TGF β1 (97). Taken together, these results seem to indicate that TGF β1 exerts its profibrotic activity both by increasing collagen type I and III synthesis and reducing ECM degradation via increased TIMP/MMP ratio (203).
A special case of MMP is represented by MMP 7, which has been reported as cleaving the insulin-like growth factor-binding protein 5 (IGFBP-5) secreted by myofibroblasts and thus possibly acting as a putative myofibroblast-recruiting signal expressed by epithelial cells (88). Incidentally, another member of the insulin-like growth factor-binding protein family, IFGBP 3, is induced via TGF β1 treatment and has been used as a myofibroblast marker (104, 224).
Origin
Myofibroblasts have been traditionally considered mesenchymal cells derived from quiescent residing precursors: sensu stricto fibroblasts, cognate mesenchymal cells such as hepatic stellate cells (181) (HSCs, also known as Ito cells or lypocytes) or pericytes (122), and also cells sharing some mesenchymal features such as mesangial cells (renal cells with smooth muscle phenotype, but also positive to mesenchymal markers, such as vimentin) (109). In the last 20 years, the discovery of a number of different progenitors has considerably changed this view (94). In particular, myofibroblast differentiation is now widely accepted to occur in bone marrow-derived circulating cells generally referred to as fibrocytes (31, 170, 185, 226), in epithelial cells via EMT (123, 155, 247) and also in endothelial cells via EndMT (251). For a comprehensive review of the cellular and molecular aspects of EMT and EndMT and their role in physiological and pathological development, the reader may refer to the recent review by Kovacic et al. (117).
However, at least from the point of view of the involvement of myofibroblasts in inflammatory/fibrotic reactions, the traditional mainly mesenchymal differentiation paradigm seems to hold. For example, in kidney fibrosis, it has been shown that, although the EMT of tubule epithelial cells is a possible contributor to the myofibroblast population, the main progenitors are α-smooth muscle actin-negative (which means possibly immature) pericytes (98); additionally, in transgenic mice with high levels of blood TGF β1, interstitial fibrosis and tubular atrophy were recorded as a consequence of myofibroblast differentiation of activated mesangial cells (116).
In addition, in liver fibrosis, EMT from either hepatocytes (219) or cholangiocytes (193) has been substantially discarded as a significant mechanism of myofibroblast production [while it may act as protective escape phenomenon (166)]; also fibrocytes appear to have a negligible contribution to the overall collagen production in fibrosis (89); similarly, in the case of kidney fibrosis, it appears that the majority of myofibroblasts can be traced to resident, nonepithelial cells, namely, HSCs and portal mesenchymal cells that provide two distinct myofibroblast subpopulations (126).
Differentiation mechanism
It is now rather commonly accepted that myofibroblast differentiation is triggered by soluble molecular mediators and mechanical stimuli, both in vivo and in vitro, and this holds virtually for any type of progenitor cells.
Soluble (molecular) factors
TGF β1 is considered to be the most potent myofibroblast differentiation soluble activator, whatever the initial phenotype of the progenitor cells [fibroblasts (57), epithelial cells (250), fibrocytes (1)]. In addition, fibroblast growth factor-2 is capable to trigger EMT and produce myofibroblasts (209) and myofibroblast activation may also be obtained via stimulation of pattern recognition receptors such as TLRs (142), alone or as potentiating agents of TGF β1 (23, 195). In the last 15 years, a general consensus has been reached regarding the mode of action of TGF β1, which is now predominantly considered to be a cytodifferentiating and fibrogenic factor rather than a proliferating and migratory one; specifically, it has been shown that a number of other cytokines can act in a concerted fashion to modulate the action TGF β1: in airways, the fibrosis platelet-derived growth factor (PDGF) appears to have a crucial role to initiate a chemotactic and proliferative response (migration of myofibroblasts), which is then switched to a collagen producing mode by TGF β1 through the downregulation of PDGF receptor α (28). In portal fibroblasts, PDGF inhibits full myofibroblast differentiation (128), and previous reports of PDGF-induced transitions (111) may have been complicated by the use of stiff substrates (see later). It is, however, unclear whether PDGF already determines a transition to a protomyofibroblast phenotype. It has also been demonstrated that the switch between proliferative/migrating and collagen-producing (=stricto sensu myofibroblast) states involves other cytokines in concert with TGF β1: for example, in a connective tissue growth factor (CTGF)-mediated fashion, the epidermal growth factor (EGF) stimulates proliferation and migration (similarly to PDGF), while IGF 2 coinduces collagen production (84). The reports that the EGF/TGF β1 combination is a potent causative factor of EMT do indeed point out that the two cytokines initially transdifferentiate epithelial cells to a migratory mesenchymal phenotype before the transition to a real myofibroblast state.
Finally, very recent evidence has shown that also inflammatory cytokines (TNF α, but not interleukins) can effectively promote myofibroblast differentiation (233)
Mechanical action
Mechanochemical transduction processes are key elements in the natural or regenerative processes in skin (138, 201) as well as several other tissues (79), and it was indeed in skin that mechanical tension was first reported to induce the presence of myofibroblasts (111, 208). A number of cells have demonstrated mechanosensitivity (59, 162), and the mechanical tension-induced transition of connective tissue cells to contractile and ECM-producing phenotypes (110) offers some of the best known cases: for example, fibroblasts exert forces on the ECM with an intensity proportional to its stiffness (137), produce α-SMA in response to mechanical stimulation (254), and rearrange cytoskeletal elements to match the cellular stiffness with that of the substrate (207), which is experienced through several mechanosensitive receptors; among them, α11β1 integrins are known to mediate myofibroblast differentiation (34, 217) and to reduce the chemotaxis induced by PDGF (171). It is noteworthy that other integrins have opposite effects: binding of collagen to α1β1 and to α2β1, respectively, downregulates collagen I synthesis and upregulates matrix metalloproteinases (MMP 1 and MT1-MMP), favoring cell migration (61, 124).
The literature about mechanical effects in the myofibrobast differentiation and fibrotic reactions is vast, and we here want simply to highlight that substrate stiffness is now considered a major inducer of the phenotypic transition: using liver cells as an example, stellate cells preserve a quiescent phenotype when cultivated on a soft substrate (e.g., Matrigel, typically storage shear modulus [G′] <0.4 kPa), but readily differentiate to a contractile one on tissue culture plastics (78) and more generally on any substrate with G′>> 1 kPa (158); the same behavior is shared by portal fibroblasts (128). We address the reader to an excellent recent review by Wells for a comprehensive overview of the field (240).
Combination of soluble and mechanical factors
A number of cellular processes, for example the expression of several transcription factors, are comodulated by mechanical and biochemical stimuli (141). In this frame, it is now widely accepted that mechanically and chemically triggered fibroblast–myofibroblast differentiation pathways are not distinct, but the two classes of stimuli can be regarded as comodulatory aspects of the same phenomenon. For example, the calcium concentration has a major role in stimulating stress fiber contraction in myofibroblasts, but its influence appears to decrease by increasing the stress applied to the matrix (73); similarly, also the effect of TGF β1 on fibroblasts is modulated mechanically, decreasing on softer substrates (7) (Fig. 2).
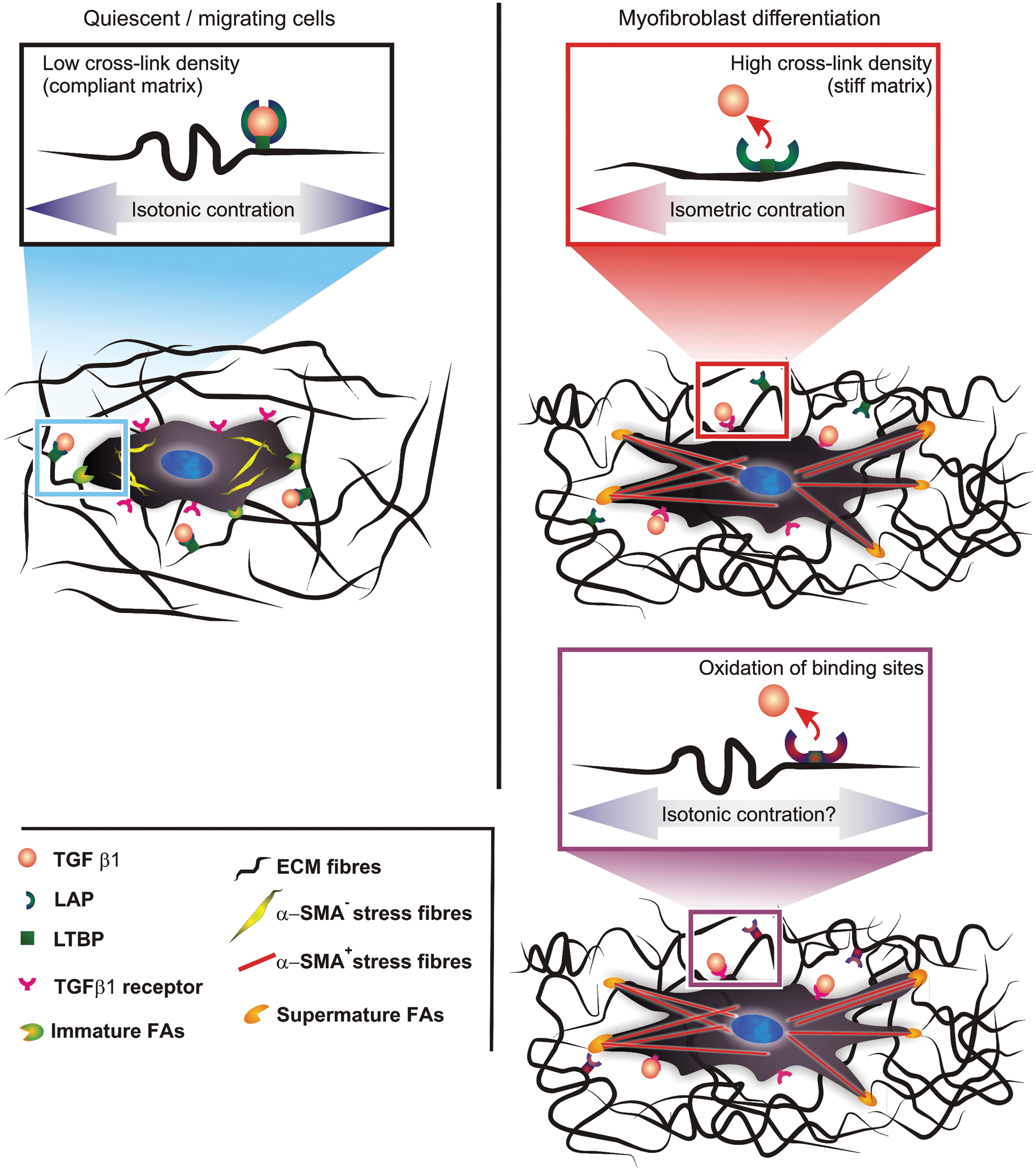
The latter phenomenon has been recently explained by the group of Hinz as related to the liberation of the growth factor from latent pericellular sites; these sites contain an integrin-binding mechanically sensitive component (the latency-associated peptide [LAP]) that would release TGF β1 following integrin-mediated myofibroblast contraction (33, 243). However, also the storage of the growth factor in the ECM increases with the formation of fibronectin fibrillar networks (48), and this may be an additional source of mechanically activated growth factor. The persistence of myofibroblast markers in wounds kept open under mechanical tension (93) may be, at least, partially ascribed to a higher efficiency of TGF β1 liberation.
Biomedical relevance
There exist resident myofibroblast populations in a number of stromata, for example in lymphoid tissue (220), and the permanent presence of myofibroblast-related cells is essential for the correct functioning of several organs, for example pericytes in healthy vasculature (131). However, in nonpathological conditions, myofibroblasts are most often associated to the processes of transient nature: for example, these cells are major regulators of the stages of tissue remodeling and contraction after injury, for example, in cutaneous wounds (132, 221) or in injured vasculature (199). In such environments, myofibroblasts upregulate the production of ECM components, in particular collagen I, which they also heavily cross-link and contract, while at the same time, secreting chemotactic and mitotic signals for epithelial or endothelial cells. It is noteworthy that although the ECM-increased accumulation is typically interpreted as a change in the balance between anabolic (matrix-producing) and catabolic (matrix-degrading) factors, recently, it has also been hypothesized that defective autophagy in myofibroblasts may contribute to the progression of fibrosis (51).
Typically, at the end of an acute inflammatory phase, myofibroblasts disappear through apoptosis (58, 101) or through transition to a quiescent/senescent state (119); after contraction of collagen substrates in vitro, (myo)fibroblasts have been often reported to dedifferentiate to a quiescent, noncontractile state (reduced expression of fibronectin and (pro)collagen I, reduced presence of actin filaments) (144) that has the potential to reverse the symptoms of fibrotic diseases (136). These dedifferentiation phenomena depend strongly on the stiffness of the substrate/matrix the myofibroblasts interact with (206, 238) and are related to the deactivation of the MyoD transcription factor (86). However, the picture of this possible phenotype reversal by biomechanical action is further complicated by reports of a sort of mechanical memory which, for example, would keep lung myofibroblats in an activated state even when they are cultured for weeks on soft substrates (12).
In these cases, the long-term permanence of myofibroblasts is typically accompanied by insurgence of pathological conditions, most often of fibrotic nature, for example pulmonary, hepatic, kidney, cardiac, and corneal fibrosis (16, 54, 96, 120, 198), hypertrophic scars and keloids (113, 154). It is noteworthy that hypertrophic scars and keloids are characterized by a different balance of the other TGF β isoforms (TGF β2—profibrotic; TGF β3—reducing collagen deposition), with a lower TGF β3 expression in keloids and a higher TGF β2 expression in hypertrophic scars (26); this may suggest a different form or a different degree of activation of myofibroblasts in the two conditions.
The association between myofibroblasts and fibrotic phenomena include also foreign body reactions to artificial implants; the literature is particularly rich of examples in relation to silicone materials used, for example, in mammary implants (11), tissue expanders (43), and conduits for nerve regeneration (38). Controlling myofibroblast behavior is also of paramount importance in the use of scaffolds for tissue regeneration: myofibroblast cells are often among the first cells to colonize tissue-engineered materials (151), and often lead to their contraction (21, 227), which is typically the end of their controlled performance.
It is therefore not surprising that myofibroblast deactivation is considered to be an interesting target of antifibrotic therapies (114, 183); several drugs with known antifibrotic effects have demonstrated effects on myofibroblast, for example, tranilast (N-(3′,4′-dimethoxycinnamoyl) anthranilic acid; reduces collagen synthesis) inhibits the myofibroblast-induced contraction of collagen gels (200), N-acetyl-seryl-aspartyl-lysyl-proline [Ac-SDKP, a stem cell-regulating peptide with demonstrated antifibrotic activity (246)] reduced TGF β1 expression and reversed myofibroblast phenotype (245), and the same effects are shown by relaxin (192) (an hormone with known antifibrotic activity) and by peroxisome proliferator-activated receptor gamma agonists (32, 143).
Myofibroblasts are not only linked to fibrotic phenomena. For example, typical signatures of myofibroblasts are common to cancer-associated fibroblasts (also known as peritumoral fibroblasts or reactive stroma) (29). Indeed, it is now accepted that myofibroblast cells can have a significant tumor-supporting role (65) and are generally present at the invasion front of many primary tumors (50). The latter effect may make use of local populations: for example, there are resident myofibroblasts in dental pulp (2), periodontal ligaments (6, 19), and gingiva (6), and myofibroblasts are thought to be major contributors to the evolution of odontogenic tumors (232), therefore an involvement of the local populations appears logical. However, there is a general consensus that in most cases, tumor-supporting myofibroblasts are mainly produced in situ from stromal cells under the influence of tumor-secreted cytokines such as TGF β1 (50, 222). Last but not least, myofibroblasts are now recognized not only as support cells, but also as the central element to both malignant processes, for example, myofibrosarcomas, such as inflammatory myofibroblastic tumor (72), and benign hyperplastic ones, such as benign prostatic hyperplasia (BPH) (20, 224); it is noteworthy that in such hyperplastic processes, the myofibroblast population appears to be partially obtained via EMT (3), differently from liver or kidney fibrosis.
Significance of Reactive Oxygen Species for Contractile/Fibrotic Cells
As initial caveats, it is worth noting that, (i) as many relevant articles on the topic rely on the use of human or animal biopsies, the biochemical analysis of reactive oxygen species (ROS) may underestimate the presence of extremely important, but unstable species such as superoxide, whose influence can often be estimated only indirectly through the use of scavengers; (ii) a large amount of data are obtained from in vitro models were cells are cultured on very stiff substrates, such as tissue culture polystyrene; despite this important difference, it is often possible to replicate the typical in vivo behavior of myofibroblasts: for example, the group of Berger has shown that myofibroblasts also in vitro produce glycoprotein hormone alpha (184) and are sensitive to vardenafil both in vitro and in chorioallantoic membranes (252). However, from a quantitative point of view, in vitro results should be taken cautiously, since mechanical effect may lead to an overactivation of myofibroblasts in comparison to what occurs when in the contact of softer and more physiological matrices.
Phenomenology
We here examine the influence of ROS (hydrogen peroxide, superoxide, OH radicals, etc.) on the behavior of contractile cells.
Effects on muscle cells
ROS/RNS (free radicals) are well-known by-products of the contractile activity of skeletal muscles (49, 140) and their accumulation is reportedly linked to the development of muscle fatigue. However, there is now burgeoning evidence that ROS also play more complex roles, which inter-relate with other signaling molecules; for example, PDGF stimulation has been shown to increase the intracellular concentration of hydrogen peroxide in vascular SMCs (213), but also to elicit bladder SMC contraction through the upregulation of the gap junction protein connexin 43 (127). A comprehensive dissection of this field falls beyond the aim of the present review, and has been exhaustively described for what attains to the ROS-dependent signaling of skeletal muscle cells (13), above all after physical activity (172) and during contraction (176), and of SMC signaling (42), with specific regard to the ROS effects on migration (189), proliferation (210), differentiation (211), and contraction (231, 239). Very interesting opportunities appear to have opened with the discovery of the X-ROS signaling in skeletal and heart muscles, where ROS production is triggered by the application of mechanical tension on cells (microtubules working as mechanotransducing units) and, in turn, controls Ca signaling and thus also the rhythm of contraction in striated cells (173).
Specific effects on myofibroblasts
A very significant body of evidence points to the involvement of oxidative stress in the fibrotic degeneration of different organs (169), in particular, lungs (191) and liver (161), to the possibilities of antioxidant therapies (112) and to the relative facility of replicating the results also in in vitro models (35).
As a common and first important point (later developed in paragraph about NADPH oxidases [NOX]), ROS can induce the myofibroblast transition in cardiac fibroblasts (47), prostatic fibroblasts (188), lung fibroblasts (87), and stellate cells (190); they typically do so in combination with TGF β1 and modulating its action. It is worth noting that ROS not only influence (myo)fibroblast behavior, but are also produced by them as extracellular (paracrine but also autocrine) signaling agents (237); therefore, exogenous ROS may not be necessary for inducing such a transition.
There is also much evidence that ROS play a pivotal role in myofibroblast differentiation via EMT, with an identical relation with TGF β1. For example, in tubular epithelial cells, TGF β1 could cause a burst release of cellular ROS (178), while on the other hand, H2O2 induces EMT (85) and a wide range of antioxidants/ROS-scavengers inhibit the TGF β1-induced EMT (85, 178). In mammary epithelial cells, MMP 3-induced EMT is mediated by ROS (superoxide) and is reversed with the antioxidant N-acetyl cysteine (NAC) (175).
In relation to the EMT, it has been recently suggested to be a ROS-induced survival mechanism, a “Redox-based escape mechanism from death” (160, 166). This hypothesis would both provide an interesting explanation for the EMT, and also for apparently contradictory data that oxidative stress may increase ECM remodeling instead of ECM deposition (204): using neonatal and adult rat cardiac fibroblasts in vitro, Siwik et al. demonstrated that exposure to xanthine oxidase (XO, an enzyme capable of generating molar equivalents of H2O2 and superoxide in the oxidation of xanthine to uric acid) or directly to H2O2 dramatically increased the MMP activity, decreased collagen synthesis, and increased fibronectin synthesis, indicating a transformation from a matrix-depositing phenotype to an actively remodeling, fibronectin expressing one.
Biochemical and biophysical mechanisms
In the following paragraphs, we discuss the individual mechanisms proposed to be on the basis of the effects of ROS on fibrotic reactions. Figure 3 presents a graphical summary of the effects of hydrogen peroxide, which are discussed in the next two sections.
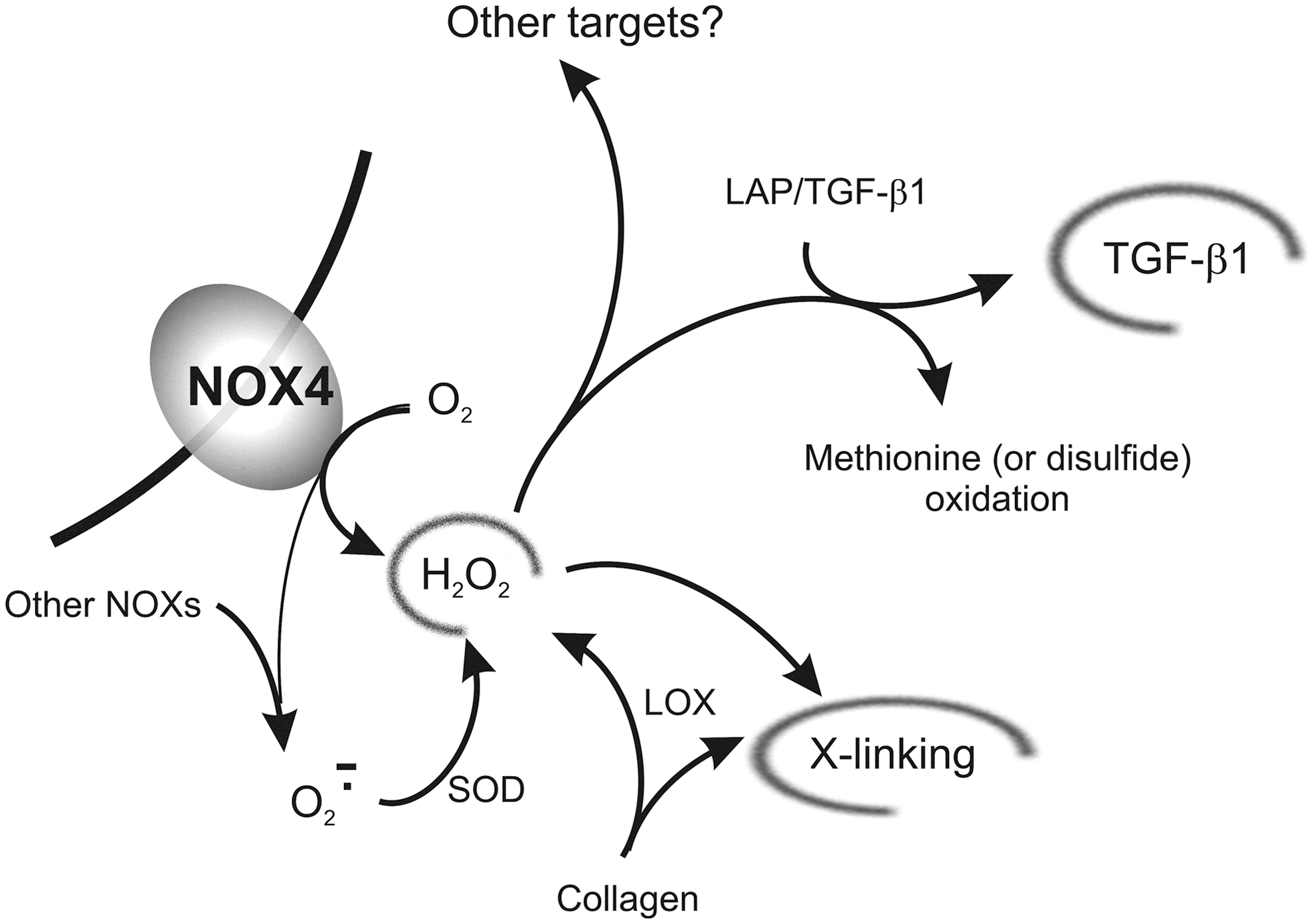
NADPH oxidases
NOX is a seven-member family of enzymes whose primary action is ROS production, typically starting with oxygen activation in the form of superoxide; NOX2 and NOX4 are possibly the best studied homologs in this family, due to their heavy involvement in a number of inflammatory or neoplastic proliferative, migratory, and fibrotic processes (164, 165, 235). NOX2 is often considered to be the most important member of the NOX family; it is heavily expressed in leukocytes, but found also in a variety of other cells (18), including activated fibroblasts/myofibroblasts (216). The peculiarity of NOX4 is that it is the only member of the family associated to the constitutive production of hydrogen peroxide, rather than superoxide (139).
Fibroblasts do feature a NOX machinery to produce superoxide and the ensuing ROS cascade, but there are significant differences from what occurs in leukocytes (primarily based on NOX2): for example, it has been found that fibroblasts from NOX2-deficient (chronic granulomatous disease) patients are capable of ROS production (64), therefore, these stroma cells seem to feature an additional major source of ROS. TGF β1 treatment has been shown to increase the general NOX activity and to specifically cause the overexpression of both NOX2 and NOX4, but only the knockdown of NOX4 caused a reduction of α-SMA and ED-A FN expression, as well as a reduced response to TGF β1 (27); this concept has further developed in the literature and there is now a general consensus of the existence of a very close association between NOX4 and fibrotic pathologies based on the modulation of TGF β1 signaling (15), which has been confirmed in a large number of recent studies (4, 47, 87, 134, 145, 188, 190, 249). It is worth noting that although NOX4 primarily produces extracellular ROS, its activity is heavily influenced by intracellular (mitochondrial) ROS; their inhibition attenuates the sensitivity to TGF β1 and presents antifibrotic effects (105).
Having said that, much is unknown in mechanistic terms. For example, the precise biochemical target of H2O2 is yet to be discovered; the reader is referred to a good discussion on this subject in a recent review by the group of Berger (187). Additionally, although H2O2 produced by NOX4 is generally assumed to be the main signaling molecule, other ROS also have a role in fibrotic reactions, for example, superoxide (174), either directly or in the form of further decomposition products. In fact, superoxide dismutase (SOD), possibly the best scavenger of superoxide, but also a producer of H2O2, was shown not only to exhibit general antifibrotic effects (52, 63), but also to specifically target myofibroblasts, reducing TGF β1 expression and reverting their phenotype (53, 236). Conversely, SOD inhibition has the opposite, fibrotic effect and stimulates collagen production while reducing the TIMP/MMP ratio (130).
ROS interactions with ECM
In the ‘90s, the group of Gabbiani showed that LOX expression and therefore also H2O2 production in the ECM may predate the myofibroblast transition in hepatic fibrosis induced by bile duct ligation (cholestatic fibrosis) (56); this would suggest that a possible source of myofibroblast-inducing ROS is indeed the enzymatic cross-linking of collagen (Fig. 2).
The possibility also exists that H2O2, in addition to its biochemical signaling, may have a biomechanical influence. For example, the LAP has a functional methionine-based redox center, whose oxidation by ROS can trigger a conformational change leading to the release of TGF β1 (107). The oxidation of methionine or of disulfide bonds, which is key to the stability of the LAP/TGF β1 complex (40), may be on the basis of the ROS-triggered release of TGF β1 from LAP complexes, also seen in acellular systems, for example, using asbestos (168) or ionizing radiation or metal ion-catalyzed ascorbate reaction (14).
Oxidation reactions, such as the production of α,β diketones in a ketoimine-based collagen cross-links (Fig. 2, bottom right), may also play an important role in the stiffening of the ECM (69), and thus, also in the mechanosensitive release of TGF β1 from its complex with LAP.
Figure 3 presents a graphical summary of the effects of hydrogen peroxide, as discussed in the last two sections.
Other forms of ROS-related signaling
An intracellular chloride channel (CLIC4) was found to be heavily upregulated in breast fibroblasts after TGF β1 treatment (182); the influence of TGF β1 was modulated by ROS levels (115). Further studies on ovarian cancer models demonstrated that antioxidant treatment could block the TGF β1-induced CLIC4 overexpression and fibroblast–myofibroblast differentiation (248).
Nitric oxide
It has been postulated that an imbalance between nitric oxide (NO) and ROS is a critical factor in the development of fibrotic conditions, for example, it has been shown that TGF β1 could at the same time stimulate ROS synthesis via NOX and reduce NO levels via transcriptional repression of inducible nitric oxide synthase (iNOS) (81, 147).
The same group later demonstrated that an increase in NO levels caused by endogenous iNOS upregulation could counter ROS formation in both human Peyronie's disease (PD) biopsies and a rat model mimicking such pathological conditions, with overall antifibrotic effects (71). Similar effects and fibroblast apoptosis were also observed in PD with administration of L-arginine (the NO precursor) or phosphodiesterase inhibitors—pentoxifylline and sildenafil (which restore cyclic guanosine monophosphate (cGMP) and/or cyclic adenosine monophosphate (cAMP) and thus the downstream NO signaling) (225). The mechanism of action of NO may involve the direct scavenging of ROS, resulting in the production of peroxynitrite, which would be responsible for the observed accumulation of nitrotyrosine in PD tissue (17, 153). There are, however, two pieces of evidence that appear to point in the direction of different mechanisms: (i) peroxynitrite is a profibrotic factor itself, since in fetal lung fibroblasts it increases TGF β1 release and collagen gel contraction, both in 3D and 2D cultures (212); (ii) the ROS-scavenging activity of NO would not apply to H2O2, which is the predominant ROS generated by NOX4. Therefore, it has been hypothesized that H2O2 and NO may indirectly interact in an antagonistic fashion, as detailed in the recent review of Sampson et al. (187).
Summary and Prospective Therapeutic Applications
To sum up, there is increasing evidence that ROS play a key role in the differentiation of several cell types to the contractile myofibroblast phenotype and in the persistence of the differentiated phenotype. Most studies to date point to a pivotal role of ROS, in particular H2O2 (and possibly superoxide), in the dynamic interplay between contractile (myofibroblast) and non-contractile (fibroblast) species (Fig. 4); appropriate pharmacological therapies could therefore be used to tip this balance in favor of a noncontractile/nonfibrotic phenotype by modulating the effect of ROS. The two main avenues explored to date have targeted the direct scavenging of ROS via the use of reducing agents [e.g. through selenium (252), NAC (25), or enzymatically via SOD (236)] or the inhibition of NOX [e.g. using diphenyleneiodonium (25)]. In an alternative perspective, NOX can be seen as a part of the delicate balance, where NO/cGMP signaling is in indirect competition with ROS-based one, and the potentiation of the first [e.g. using phosphodiesterase inhibitors (252) or soluble guanidin cyclase stimulators (22)] may be the key for a weakening of the second.
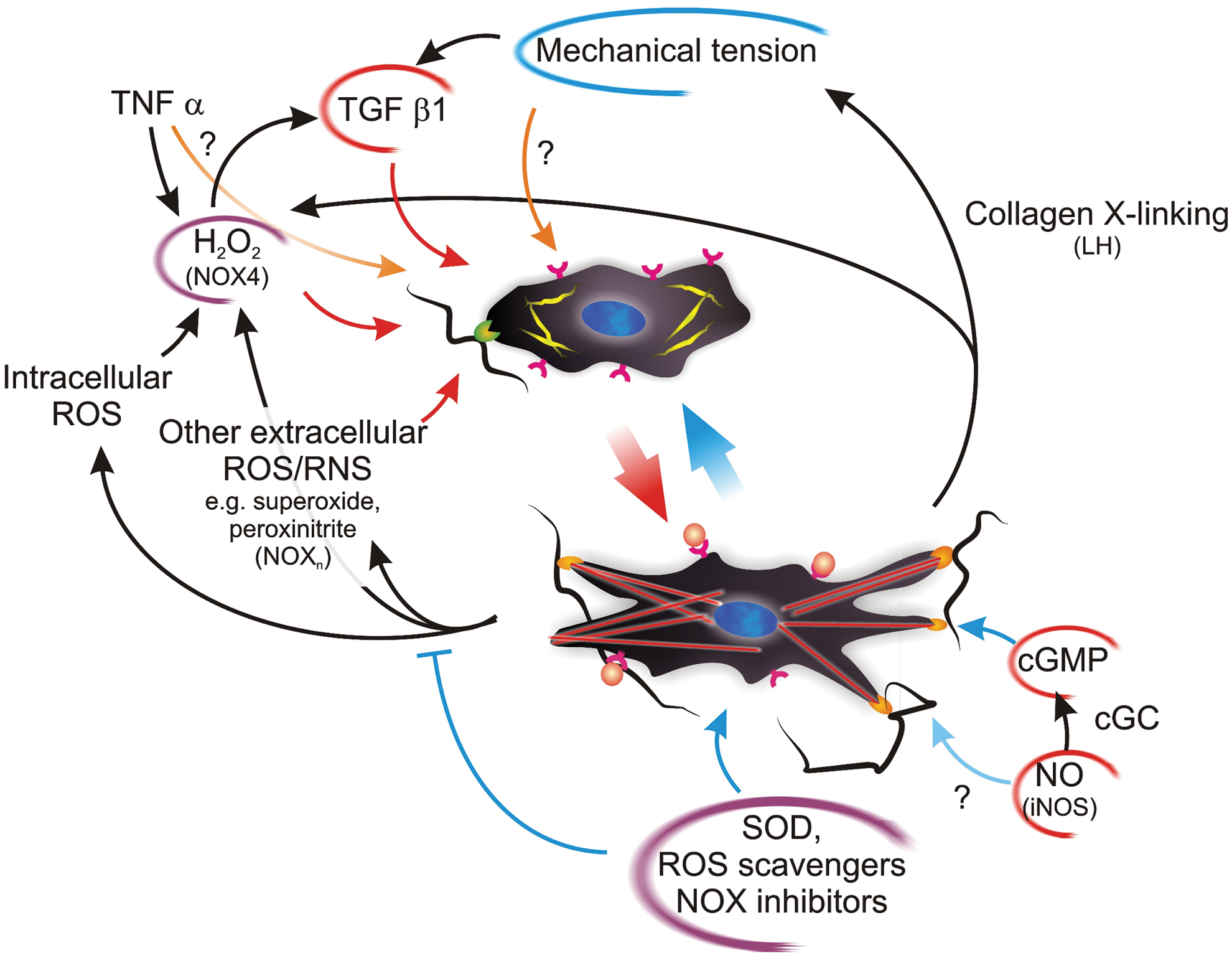
However, the in vivo extension of in vitro or ex vivo pharmacological data is an issue yet to be seriously tackled: the high cross-link density (a barrier to diffusion) and the poor vascularization of fibrotic (scar) tissues may significantly affect and hamper the availability of both systemically and topically administered active principles. Therefore, much has to be done not only in a more detailed biochemical/biophysical understanding of the factors potentially reversing the myofibroblast differentiation, but also in developing an effective approach for their targeted delivery.