
Abstract
Introduction
T
Unique features of HV1
HV1 belongs to a superfamily of voltage-gated ion channels, but it has a number of unique features that distinguishes it from other voltage-gated ion channels (Fig. 1). From a structural standpoint, the HV1 channel lacks the prototypical pore domain of ion channels (114, 122). Voltage-gated ion channels usually consist of six transmembrane (TM) segments with the segments S1-S4 constituting the VSD that detects the changes in membrane potential and S5-S6 forming the ion conductance pore domain responsible for selective ion permeation. HV1 channels only have the first four TM segments (S1-S4), which function both as VSD and as pore, the channel assembling as a dimer (72, 77, 139) with each subunit containing its own permeation pathway (72, 139). From a functional point of view, HV1 channels are uniquely selective for protons with essentially no permeability to other ions, a feat that no other ion channel can achieve (30). This perfect selectivity is conferred by a single acidic residue Asp112 whose neutralization remarkably converts the hHV1 channel into an anion channel (102). This structure enables HV1 channels to efficiently transport protons despite their usually very low concentration (100 nM at pH 7.0) in physiological solutions. HV1 channel gating is also peculiar, with the channel lacking inactivation and sensing both the TM voltage and pH gradient. Activation, therefore, is favored by depolarizing voltages, by acidic intracellular pH, and by alkaline extracellular pH. Three charged Arg residues in the S4 TM segment confer the voltage dependency similar to the VSD of potassium channels, while the structural basis of the pH sensing is not known. HV1 proton currents are also highly sensitive to temperature (33, 74, 75) and have a small (15 fF) unitary conductance (21), which can be accounted for by the low concentration of protons in physiological saline solutions. Finally, from a pharmacological perspective, HV1 channels of most species are potently blocked by Zn2+ at concentrations ranging from 100 nM to 1 mM depending on the extracellular pH and on the presence of other polyvalent cations. In mammals, Zn2+ shifts the current–voltage relationship positively and slows the kinetics of HV1 channel activation, two distinct effects that can be accounted for by the binding of polyvalent cation to two histidine residues (His140 and His193) located at the interface between the channel monomers (77, 101). A number of studies revealing the gating mechanism and structural features of HV1 have been published recently (1, 11, 63, 73, 77, 97, 102, 112, 114, 138), and the known structural elements are shown in Figure1. A detailed discussion of HV1 structural and functional aspects is beyond the scope of this review, and all the pertinent information can be found in a recent exhaustive review (31). Therefore, only structural aspects relevant for the development of HV1 inhibitors will be described in the HV1 Pharmacology section.

Mechanisms and regulation of HV1 activation
The primary mechanism of HV1 channel activation is voltage, the channel being activated by membrane depolarization, but the actual voltage required for activation is not fixed but set by the TM pH gradient (ΔpH, the pH difference between the two compartments separated by the conductive membrane). Thus, both the intracellular and extracellular pH (which is functionally equivalent to the intraluminal pH if the channel is located on an intracellular, vesicular membrane) are primary determinants of HV1 channel activation. When the cytosol becomes more acidic relative to the extracellular or intraluminal side (as a result of either cytosolic acidification or extracellular/vesicular alkalinization), the entire conductance-voltage relationship of the channel shifts by 40 mV to more negative voltages for each unit increase in ΔpH. Conversely, when the extracellular or intraluminal side becomes more acidic than the cytosol (as a result of either cytosolic alkalinization or extracellular/vesicular acidification), the conductance-voltage relationship shifts by 40 mV to more positive voltages for each unitary change in ΔpH (20). In practical terms, this implies that three parameters should be known to determine whether an HV1 will be activated in a given membrane: (i) the membrane voltage (measured on the cytosolic leaflet, relative to the extracellular or luminal side); (ii) the cytosolic pH, whose acidification favors activation at any given voltage; and (iii) the extracellular or intra-luminal pH, whose acidification opposes activation at any given voltage. In most organisms and tissues, the channel opens at voltages higher than +20 mV when the pH on both sides of the transporting membrane is identical (e.g., pHi/pHo 7.0/7.0). This implies that HV1 essentially drives the efflux of protons from cells or the influx of protons inside membrane-enclosed compartments, provided that the voltage and pH are permissive for channel activation. These conditions are, in fact, rarely met in the plasma and organellar membranes of mammalian cells. Under physiological conditions, the cytosolic pH is only slightly more acidic than the extracellular pH, and ΔpH typically averages 0.3 pH units (acidic inside). A channel present at the plasma membrane will, therefore, only be activated at voltages higher than 8 mV (+20mV−(40mV/pH*0.3 pH)=8 mV), a value that is exceeded transiently in excitable cells during action potentials, in phagocytes during the respiratory burst (5, 47, 65, 113), and in eosinophils (6, 41). Since most intracellular organelles are acidic, the conditions are even less permissive for channel activation in endomembranes. For instance, a channel expressed in the membrane of early endosomes will face a luminal pH of 6.0 and a cytosolic pH of 7.0, that is, a ΔpH of −1 pH unit, which will shift the voltage threshold for activation to +60 mV (+20mV−(40mV/pH*(−1 pH))=+60 mV). Whether voltages higher than +60 mV can be reached in the membrane of organelles is not known, as direct recordings of the potential of these structures have not been performed, but at first glance, the pH conditions do not appear permissive for HV1 channel activation. So far, the only evidence for HV1 activity has been gathered either at the plasma membrane or at the membrane of phagosomes, phagocytic vacuoles forming around particles ingested by cells of the innate immune system such as neutrophils and macrophages. In these membranes, channel activation is facilitated by phosphorylation of the channel and by the functional coupling of the channel to nicotinamide adenine dinucleotide phosphate (NADPH)-dependent oxidase (NOX) enzymes, as will be discussed next.
HV1 phosphorylation
The activation of phagocytic cells by phorbol esters strongly alters the gating properties of H+ channels, shifting the current–voltage relationship positively and increasing the speed of channel activation while decreasing the kinetics of channel deactivation on repolarizing voltages. These effects are mediated by phosphorylation of the channel on Thr29, as Thr29Ala or Thr29Asp substitutions completely abolished this potentiation effect (97). Protein kinase C (PKC) (in particular PKC-δ in eosinophils) seems to be the kinase responsible for this modulation (7, 104).
Arachidonic acid
Arachidonic acid (AA) is a polyunsaturated fatty acid, constituent of biological membranes, and a secondary messenger. Early observations indicated that AA directly activates a proton conductance in phagocytes (32, 51, 56, 71, 127, 134). However, AA is also known to activate multiple signaling pathways, including PKC (91), leading to the activation of NOX enzymes. Therefore, the activation of H+ currents on AA treatment, in at least in certain cases, could be explained by an increased activity of the NOX (91).
NOX enzymes
NOX enzymes are a family of seven members (NOX1–NOX5 and DUOX1-2) that produce superoxide (O2−) or hydrogen peroxide (H2O2) using NADPH and molecular oxygen as substrates [for a review, see Bedard and Krause (9)]. These enzymes are widely expressed through multiple species and tissues and have varying functions depending on the localization, expression levels, and developmental stage. Reactive oxygen species (ROS) production by NOX enzymes has been implicated in host defense, redox-dependent signaling and also contributes to the progression of multiple diseases that are characterized by oxidative stress. NOX enzymes share a common structure: NOX1–5 have 6 and DUOX1-2 have seven TM domains. NOX1, NOX2, and NOX3 require cytosolic subunits for catalytic activity; NOX4 is constitutively active; and NOX5 and DUOX1-2 are Ca2+ dependent. NOX2 is probably the best characterized member of the family. It is expressed at high levels in neutrophils, and electron transfer by NOX2 mediates respiratory burst in neutrophils that are necessary for bacterial killing.
Relationship between HV1 and NOX enzymes
During the reaction catalyzed by NOX enzymes, NADPH provides two electrons that are translocated across the plasma membrane to reduce molecular oxygen to O2−, generating two protons in the cytoplasm (Fig. 2). Based on the amount of O2− generated by phagocytes, it has been calculated that NOX activity could lead to a drop in cytosolic pH by more than five units (i.e., to lethal levels around pH 2) (40). A variety of proton extrusion mechanisms prevent this lethal acidification, with HV1 channels probably being particularly implicated in proton extrusion during NOX enzyme activation. The extrusion of protons through an electrogenic channel, rather than through an electroneutral exchanger, has a second advantage that charges are compensated and massive depolarization through isolated electron transport is avoided. The connection with O2− producing NOX was made 5 years after the discovery of H+ channels in snail neurons (137), when Henderson et al. suggested that a voltage-gated proton channel contributes to the pH homeostasis in neutrophils, by counterbalancing acid production and depolarization through the phagocyte NOX (58). In 1993, voltage-gated H+ currents were reported in human neutrophils (32) and in the myeloid cell line HL-60, which expresses an NOX (40), and numerous subsequent studies reported that high levels of NOX expression coincide with high levels of H+ channel activity in phagocytes. A causal genetic link was soon refuted, however, as voltage-gated H+ currents could be recorded in monocytes (103) and granulocytes (37) from NOX2-deficient patients suffering from the chronic granulomatous disease (CGD). However, proton currents in NOX2-containing, but not in CGD, eosinophils were enhanced by phorbol-12-myristate-13-acetate (PMA) (5). A truncated form of NOX2 conferred increased H+ ion fluxes (as detected by fluorescent probes and by patch-clamp) to transfected cells, and mutations of critical histidines within this truncated NOX2 prevented the H+ fluxes (3, 4, 55, 86). These findings revived the interest in the theory that the phagocyte NOX2 might either function as a proton channel or be closely associated with it. Cloning of the HVCN1 gene coding for HV1 proton channel in 2006 clarified the situation by showing that the HV1 protein supports H+ fluxes, and that HV1-deficient phagocytes are completely devoid of H+ currents, even in conditions favoring NOX activation (44, 90). Taken together, it is now clear that NOX enzymes are not H+ channels, but they play a modulatory role in H+ channel function. Along these lines, it has been shown that up-regulation of HV1 and accumulation of HV1 channels in phagosomes do not require functional NOX2 (110).
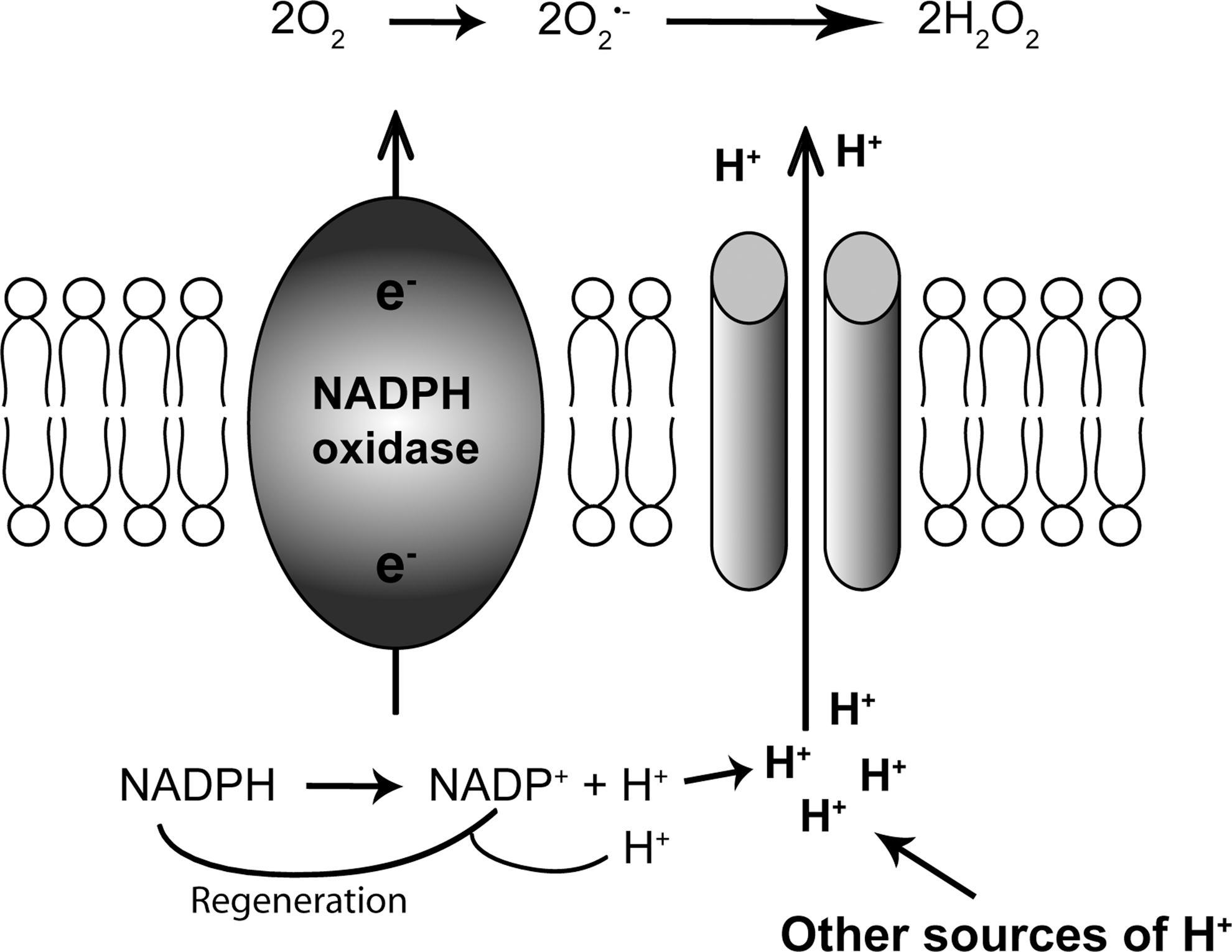
Of note, the functional link between NOX expression and H+ channel activity is well established for NOX2, but from a theoretical point of view it could apply to all NOX isoforms. Indeed, such an association has been described for the Ca2+-activated NOX5 isoform in sperm cells [see section Spermatozoa, (99)]. However, many cell types that exhibit robust H+ channel activity do not show any evidence for NOX expression (e.g., snail neurons, basophil granulocytes). Thus, there are most likely NOX-independent H+ channel functions.
The HVCN1 gene: gene structure and splice variants
Human HVCN1 gene is located in chromosome 12 (12q24.11) and spans 41,127 bp (Fig. 3). It is neighboring with PPP1CC (protein phosphatase 1, catalytic subunit, and gamma isozyme) and with TCTN1 (tectonic family member 1). There is no indication of a thematic gene cluster in the area surrounding HV1.
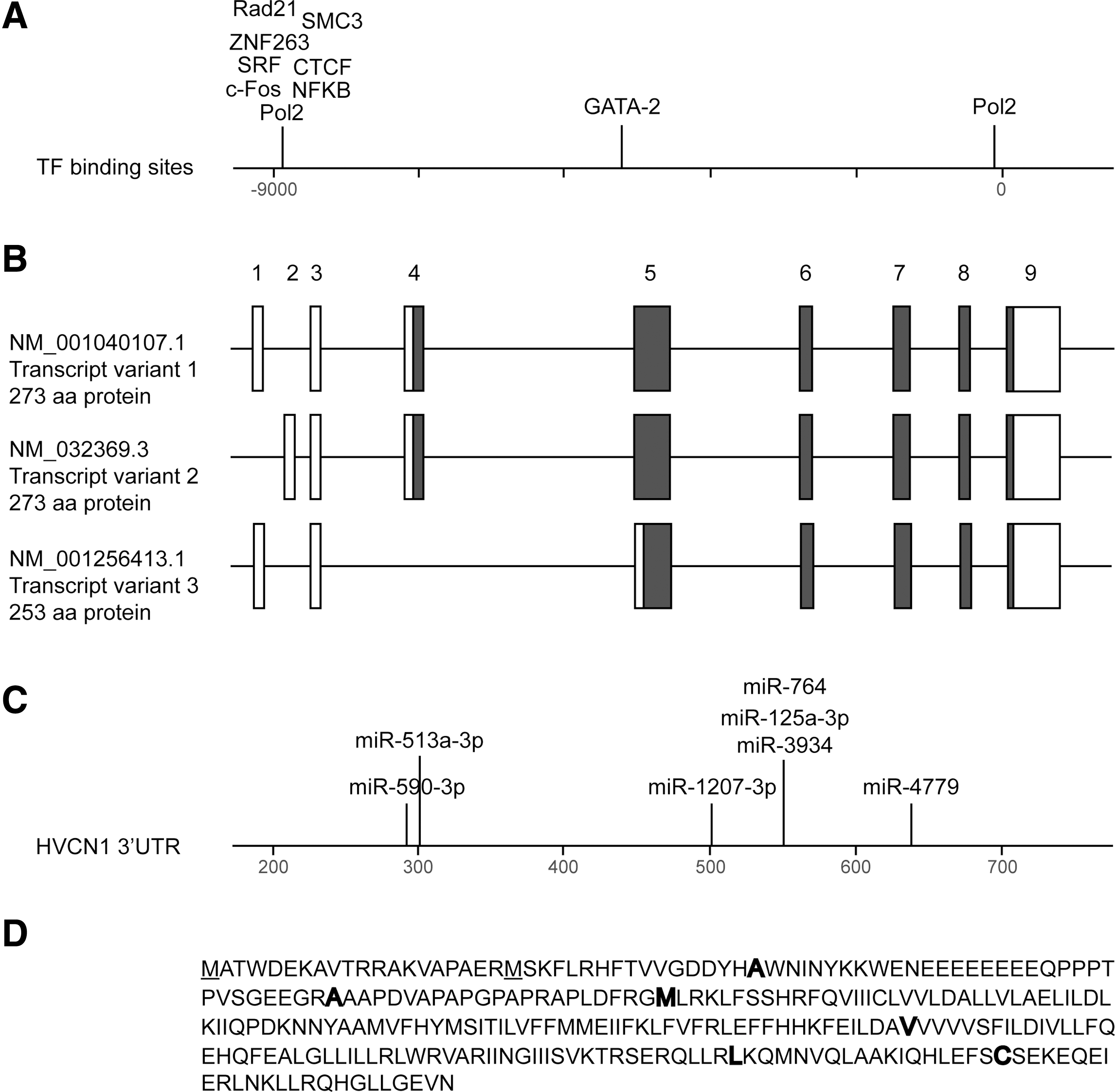
Promoter region
Little is known about the size, localization, and structure of HVCN1 promoter region and transcriptional regulation. There is only one published article describing a transcriptional regulation of HVCN1: The study of gene expression in mouse macrophages stimulated with lipopolysaccharide (LPS) predicts the presence of the Nf-kB site in HVCN1 promoter. Furthermore, ribonucleic acid interference (RNAi)-mediated silencing of nuclear factor kappa B (NF-kB) coactivator B-cell lymphoma 3-encoded protein (Bcl3) leads to a reduction of HVCN1 expression, indicating that the NF-KB/Bcl3 complex might be a part of the transcriptional control of HVCN1 (141). However, chromatin immunoprecipitation–sequencing (ChIP-Seq) data available at the USCS genome browser (
Splice variants and exon usage
Three distinct transcript variants are found for human HVCN1 according to the National Center for Biotechnology Information (NCBI) Gene database (Fig. 3B): variant 1 (NM_001040107.1) represents the longest transcript and encodes a protein of 273 aa (isoform 1). Variant 2 (NM_032369.3) differs in the 5′ untranslated region (UTR) compared with variant 1. Variants 1 and 2 encode the same protein. Transcript variant 3 (NM_001256413.1) differs in the 5′ UTR, lacks a portion of the 5′ coding region, and initiates translation at a downstream start codon compared with variant 1. The resulting protein (isoform 2) is shorter (253 aa) and has a distinct N-terminus compared with isoform 1. Based on currently available literature, the 273 aa variant appears to be the most widely expressed, while the shorter isoform has been found only in B-lymphocytes so far and its function is not clear (16). Exon usage for the different splice variants is as follows: Exon 1 and 3 code for the 5′ UTR of variants 1 and 3, while exon 2 and 3 code for the 5′ UTR of variants 2. Exon 4 contains the translation initiation ATG for splice variants 1 and 2, while exon 5 contains the alternative translation initiation site used by splice variant 3. Exons 6–9 code for translated sequences of all three isoforms. The exon 9 contains the stop codon and codes for the 3′ UTR.
Microribonucleic acid
The following conserved sites for microribonucleic acid (miRNAs) binding are predicted on 3′ UTR of HVCN1 by TargetScan (
Single-nucleotide polymorphisms
A database search (
SNPs for human HVCN1 found in SNP database on NCBI. The following filters were applied: coding nonsynonymous; stop gained; validated by frequency or genotype data: minor alleles observed in at least two chromosomes. NetPhos 2.0 Server was used for prediction of phosphorylation sites.
HVCN1, hydrogen voltage-gated channel 1; SNP, single-nucleotide polymorphism; NCBI, National Center for Biotechnology Information.
Methods to measure HV1 proton channel activity
There are essentially two ways to measure the function of voltage-gated proton channels: direct electrophysiological recordings of voltage-gated proton currents or indirect recordings of proton channel activity, obtained by measuring the changes in either the intracellular pH, the extracellular pH, or the pH of intracellular organelles. The two approaches have been extensively used, and each of them has their own advantages and disadvantages. Electrophysiological recordings offer a high degree of control and provide high-quality data but are technically demanding, while simpler pH recordings are amenable to high-throughput screens but lack specificity and rely on indirect means to control the voltage and pH-dependent channel opening. Patch-clamp recordings provide optimal control of the membrane voltage and yield a wealth of information on the channel biophysical properties and (when mutations are studied) on its ionic permeability profile. The approach also enables to impose a defined TM pH gradient by applying extracellular and intracellular solutions of known pH, using either the whole-cell or the inside-out configuration of the patch-clamp technique. Perfect control of the cytosolic pH is impossible to achieve in the whole-cell mode due to the large proton fluxes flowing across open HV1 channels. However, due to its perfect proton selectivity, the HV1 channel itself is a very accurate pH sensor and the reversal potential of the currents can be used to determine accurately the TM pH gradient (30). In contrast, pH recordings are performed in intact cells whose plasma membrane voltage and cytosolic pH varies dynamically. Regardless of whether channel activity is inferred from changes in extracellular pH (measured with electrodes) or cytosolic pH (measured with fluorescent dyes such as 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein [BCECF] or SNARF-1 or with genetically encoded pH indicators), indirect methods should be employed to impose the depolarizing voltage and/or an outwardly directed pH gradient required to activate HV1 channels. Voltage control is usually achieved with ionophores such as gramicidin (a nonselective cation ionopore that will clamp the membrane voltage to 0 mV) or valinomycin (a potassium-selective ionophore which will clamp the membrane voltage to the K+ equilibrium potential). Control of the cytosolic pH is usually done by transiently exposing cells to ammonium (NH4 +), whose conjugated base NH3 is membrane permeable and, thus, reaches the cytosol where it is protonated to NH4 +, thus acting as an effective proton sink. On external ammonium washout, the cytosolic NH4 + that has accumulated is converted back to NH3, which diffuses out of cells, with the released intracellular protons causing a rapid acidification of the cytosol to pH<6.0 depending on the concentration of NH4 + and cell size (118). The outwardly directed pH gradient that is thus generated promotes the activation of HV1 channels, provided the membrane potential is clamped at depolarizing voltages with ionophores. By following the recovery of the cytosolic pH, one can then obtain an indirect readout of the activity of plasma membrane voltage-gated proton channels, as long as the other plasma membrane acid extrusion systems are inhibited, either pharmacologically (e.g., with amiloride derivatives to inhibit sodium-proton exchangers) or by using specific ionic conditions (e.g., bicarbonate-free solutions to inhibit bicarbonate transporters). To ensure that the pH recovery is mediated by HV1 channels, the kinetics of pH recovery is usually measured in the presence and absence of a known HV1 channel inhibitor such as Zn2+. As can be appreciated, this approach relies heavily on pharmacological tools and on manipulations that are rather toxic for cells and does not provide an accurate voltage and pH control. However, despite their limitations, pH recordings have provided much insight into the physiological role of HV1 channels and are now gaining renewed interest for high-throughput screens for HV1 inhibitors. Inhibitor validation, however, will require electrophysiological recordings of either native proton currents or HV1 channels exogenously expressed in mammalian transgene recipient cell lines (e.g., HEK293, chinese hamster ovary [CHO], etc.) or in other types of expression systems, such as Xenopus laevis oocytes.
Physiological Role of HV1 Channels
Voltage-gated H+ currents are detected in a wide range of species and tissues (31). HV1 best-described function so far is the regulation of NOX2 activity in phagocytes. The regulation of activity of other NOX enzymes and NOX-independent functions starts emerging (Table 2), but is less well documented. Our understanding of the function of HV1 was facilitated by the generation of HV1-deficient mice. Two groups independently created HV1 knockout by using gene-trap technology (107, 115). HV1-deficient mice do not have major spontaneous phenotypes or malformations. They do not show a readily detectable alteration of behavior, and their life cycle and reproductive capacity are normal. In vitro, ROS production and bacterial killing by HV1-deficient phagocytes is reduced, but in vivo models of infections were not aggravated in HV1-deficient mice. It should be noted, however, that in aged HV1-deficient mice, an increased tendency toward autoimmunity has been observed. In addition, in vivo infections in aged HV1-deficient mice have so far not been investigated.
Most likely not an Hv1-mediated proton current, as it was (i) not inhibited by Zn2+; (ii) not sensitive to temperature; and (iii) also found in Hv1-deficient mice.
Present knowledge on function and/or molecule expression of HV1 in different cells and tissues. Estimated expression levels are approximations and should be taken with caution.
ElPh, electrophysiology; IHC, immunohistochemistry; WB, western blot; qPCR, quantitative polymerase chain reaction; ISH, in situ hybridization; BCR, B-cell receptor signaling; NA, data is not available; ROS, reactive oxygen species; NOX, NADPH-dependent oxidase; PMA, phorbol-12-myristate-13-acetate; DC, dendritic cell; ICC, immunocytochemistry.
Innate Immune Cells
The innate immune system comprises a large number of different cell types. There are three types of granulocytes (neutrophils, eosinophils, and basophils), which are probably best studied with regard to H+ channels. The second subtype of innate immune cells is generally summarized as mononuclear phagocytes. Among those, monocytes (mobile precursors cells) and mature macrophages and dendritic cells (DCs) have been somewhat neglected as far as H+ channels are concerned, and most attention has been devoted to two tissue-specific mononuclear phagocytes, namely microglia (which we will discuss in the Central Nervous System section) and osteoclasts. Most of the innate immune cells are professional phagocytes and accumulate the phagocyte NOX within their phagosome as a way to kill bacteria. Given the key role of H+ channels for NOX2 function, it has been long anticipated that H+ channels would be present on phagosomes, and studies modeling the phagosomal potential, pH, and volume regulation predicted that voltage-gated proton channels should account for 95% of phagosome charge compensation in neutrophils (96). Biochemical evidence was provided for the presence of HV1 channels on the phagosomal membranes of mouse neutrophils and PLB-985 cell (107, 110), but direct functional evidence that HV1 channels contribute to the phagosomal pH and ROS homeostasis in neutrophils is still lacking. In DCs, intraphagosomal ROS production was decreased by HV1 ablation (120), but it is not clear whether the conditions are permissive for HV1 channel activation on phagosomes of mononuclear phagocytes, which produce much lower amounts of ROS. To complicate things further, the phagosomal pH regulation differs widely between the different phagocytic cell types. Macrophage phagosomes acidify rapidly (81), while neutrophil phagosomes remain neutral (66) and both a long-lasting alkalinization and a rapid acidification have been reported in DC phagosomes (120, 124). The function of H+ channels within phagosomes, thus, remains to be established and might depend on the specific type of phagocyte, as discussed in a recent report (El Chemaly et al., 2013 in press).
Neutrophil and eosinophil granulocytes
Expression
Neutrophil and eosinophil granulocytes are the cells that express the highest levels of HV1. Voltage-gated proton currents were detected using electrophysiological recordings in human and mouse neutrophils and eosinophils already 20 years ago (6, 32, 40, 109), and the underlying molecular entity was established in 2006 when human and mouse HV1 were cloned (114, 122). The expression of HVCN1 messenger ribonucleic acid (mRNA) and HV1 protein was shown in mouse leukocytes in 2009 (107, 115). Localization of HV1 on plasma and intracellular membranes of human neutrophils and eosinophils was shown in 2010 (110). The full-length protein can be detected in human granulocytes by western blot (WB) as a 30 kDa monomer or 70 kDa dimer (110). Immunocytochemistry data showed that HV1 partially colocalizes with NOX2 in the membrane of intracellular granules and in the plasma membrane (110). A recent study has demonstrated the presence of HV1 mRNA and protein in mouse eosinophils (154).
Function in neutrophils
HV1 proton channel function is probably best characterized in granulocytes and in particular in neutrophils. These cells represent the first line of defence against bacterial pathogens. They are capable of phagocytosis, which is accompanied by a release of large quantities of ROS by NOX2, a phenomenon termed “respiratory burst.” The role of HV1 in this process is considered as follows. During the respiratory burst, electrons from NADPH are transferred through the membrane, leading to depolarization, which inhibits the activity of NOX2 (39, 108). Moreover, H+ is accumulated in the cytoplasm, leading to acidification which will also inhibit NOX2 activity (92). HV1 is considered to sustain the activity of NOX2 by extruding the proton from the cytoplasm, therefore maintaining physiological membrane potential and re-establishing normal pH. These findings are corroborated by the analysis of neutrophils derived from HV1-deficient mice: The pH in the cytoplasm of HV1-deficient neutrophils on the activation of NOX2 with PMA is about one unit more acidic than in wild-type cells (6.46±0.17 vs. 7.18±0.15) (44) and 0.4 pH units more acidic (pHi 6.73±0.09 vs. 7.11±0.03) after the ingestion of opsonized zymosan particles (90), which confirms that HV1 is necessary for the extrusion of proton excess generated by NADPH oxidation. Moreover, HV1-deficient neutrophils are more depolarized than control cells (44), confirming that HV1 is needed to compensate charge changes induced by the electrogenic activity of NOX. Furthermore, due to the decreased driving force for Ca2+ on increased depolarization, Ca2+ influx is decreased in HV1-deficient neutrophils and Ca2+-dependent processes are, therefore, impaired. In particular, HV1-deficient neutrophils are characterized by increased spreading and reduced ability to migrate to the site of injury, probably due to the reduced Ca2+ influx and impaired actin depolymerization (44). An in vivo study of neutrophil migration in a model of peritonitis has confirmed that HV1-deficient neutrophils have reduced ability to migrate (154). However, phagocytosis in these cells is normal (115). Interestingly, chemotaxis and migration of HV1-deficient microglia, a phagocyte of the central nervous system (CNS) (see section Microglia), remain unaffected (146).
To which extent does HV1 deficiency impair ROS generation by NOX2? Initial studies measured ROS release into the extracellular space using either cytochrome C reduction (O2−) or Amplex Red fluorescence (H2O2). These studies clearly detected an attenuated NOX2-dependent release of ROS in neutrophils derived from HV1-deficient mice as compared with wild-type littermates (30%–75% reduction; (44, 107, 115). This effect is independent of the formation of functional NOX2 complex, as the expression levels of NOX2 and its subunits p47phox, p67phox, and p22phox are the same as in wild-type mice (107) and the electron current produced by the activity of NOX2 is preserved in HV1-deficient neutrophils (44, 90). In a simple model, this would suggest that the increased depolarization and cytosolic acidification due to HV1 deficiency leads to a decrease in NOX2 activity. However, a recent study has observed enhancement of a luminol chemiluminescence signal on the inhibition of HV1 and suggested that on HV1 inhibition a back diffusion of O2− into the cytoplasm occurs (28). This conclusion should be taken with caution. Indeed, luminol-enhanced chemiluminescence in neutrophils is entirely myeloperoxidase dependent (52), and the observed enhancement of the luminol signal on the inhibition of HV1 is probably due to ROS production in MPO-positive granules. Thus, further studies (e.g., using neutrophils from HV1-deficient mice or intracellular O2− probe hydroethidine) will be necessary to fully understand how HV1 enhances NOX2-dependent ROS release to the extracellular space.
In terms of function, such reduction of ROS release by NOX2 in HV1-deficient neutrophils leads to a decrease in bacterial-killing capacity in vitro as shown by the analysis of survival of serum-opsonized Staphylococcus aureus (115). However, there was neither increased infection nor a statistically significant decrease in bacterial killing in vivo in mice inoculated i.p. with S. aureus or intranasally with Pseudomonas aeruginosa or Burkholderia cepacia (115). This indicates that even low activity of NOX2 is sufficient for innate immunity responses, at least in 2–3-month-old mice with pulmonary gavage of bacteria. It remains to be studied whether in aged mice or with other infection models an increased susceptibility of HV1-deficient mice can be unraveled.
Taken together, available data confirm that HV1 is required for optimal release of ROS by NOX2. However, the concept that HV1 deficiency inhibits NOX2 activity through enhanced plasma membrane depolarization and cytosolic acidification needs to be revisited. Indeed, sustained NOX2 activity, and a shift of ROS release to intracellular ROS accumulation (cytoplasm vs. myeloperoxidase-positive granules), appears a possible alternative.
Function in eosinophils
Eosinophils are nonphagocytic granulocytes that most likely participate in the killing of large pathogens, in particular parasites. Eosinophils are also involved in allergic reactions and, indeed, there is an up-regulation of HV1 expression in mouse lung eosinophils on an intranasal injection of the Aspergillus fumigatus extract (154). From a mechanistic point of view, HV1 in eosinophils is considered to contribute to NOX2-dependent ROS release. Similar as seen for neutrophils, eosinophils from HV1-deficient mice showed a ∼50% reduction in their capacity to produce O2− (lucigenin assay) and H2O2 (Amplex Red assay) (154). Further investigation demonstrated that contrary to neutrophils, HV1-deficient eosinophils had normal migration both in vitro and in vivo and normal Ca2+ responses. Interestingly, eosinophils from HV1-deficient mice seemed to be more susceptible to PMA-induced cell death as compared with wild-type eosinophils: They were unable to compensate for depolarization and cytosolic acidification caused by enhanced NOX2 activity.
Given the likely role of eosinophils in the killing of parasites, it will be important in the future to investigate parasitic infections in HV1-deficient mice. In addition, given the importance of eosinophils in allergic diseases, the importance of HV1 in allergy models warrants further attention.
Basophil granulocytes
Expression
H+ current can be detected in basophils (22, 100). This current can be activated by the PKC activator PMA, by chemotactic peptide N-formyl-methionyl-leucyl-phenylalanine (fMLF), and by IgE antibody mimicking allergen binding to IgE. Importantly, this current can be inhibited by HV1 inhibitor Zn2+ in a dose-dependent manner (1–100 μM) (100). However, to our knowledge, HV1 mRNA and protein have so far not been reported in basophils.
Function
Basophils are nonphagocytic granulocytes containing heparin, histamine, and other mediators of the immediate hypersensitivity response, which are released when IgE cross-links to the high-affinity Fc receptors on the cell surface. H+ channels are activated by agents that cause histamine release. HV1 inhibition by Zn2+ decreased histamine release caused by stimulation with anti-IgE stimulus and by PMA (85, 100). Zn2+ treatment also caused cytosolic acidification of basophil as shown by imaging of pH-sensitive fluorescent dye SNARF1. This suggests that HV1 activity is needed to extrude acid during basophil activation. Curiously, as compared with other granulocytes, basophils do not express NOX2 (26). Thus, H+ channel function in basophils appears to be independent of NOX2. However, the source of cytosolic acidification and the possible expression of other NOX enzymes in basophils have yet to be addressed.
Mast cells
Expression
Proton currents with typical characteristics of HV1 channel were detected in bone marrow-derived mast cells (75). No information is currently available on HV1 expression in mast cells at the level of mRNA or protein.
Function
Mast cells are tissue resident cells that are similar to basophils in many aspects. They play a critical role in type I allergic reactions. Antigen stimulation causes ROS production by mast cells, and NOX might be one of the sources of these ROS (62). However, the interaction of H+ channels with NOX enzymes in mast cells has not been studied so far. There is circumstantial evidence for the role of HV1 in mast cells: The putative HV1 inhibitor (−)-Epigallocatechin-3-gallate (EGCG) (see HV1 Pharmacology section) inhibited ROS generation in a mast cell line by interfering with the assembly of catalytically active NOX2 complex (105). Another study in human lung mast cells showed that histamine release was blocked by Zn2+ (84). Taken together, these data show that HV1 might participate in the regulation of histamine release. However, it is not yet fully understood whether this is an NOX-dependent or -independent HV1 function.
Monocytes/macrophages
H+ currents with characteristics of HV1 channels were recorded in mouse peritoneal macrophages (70); THP-1 monocyte cell line (34), and HV1 protein and current were detected in mouse macrophages (107). The function of HV1 in these cells is probably to sustain respiratory burst, but only a few experimental data are available (98).
Osteoclasts
Expression
A proton current with properties of HV1 channel was detected in rabbit and mouse osteoclasts (93, 106). No information on the expression of HV1 mRNA or protein in osteoclasts is currently available.
Function
Osteoclasts are cells of macrophage origin that are necessary for bone resorption. Osteoclasts release H+ ions to acidify the extracellular milieu and to dissolve the mineralized bone. Nordstrom et al. studied osteoclasts recovery from acid load and found that H+ current contributes to this process in a Zn2+-dependent manner (100 μM) (106). A number of studies indicate that Zn2+ can inhibit bone resorption in vitro (89, 148). Therefore, the H+ channel may provide H+ and promote bone resorption. Whether H+ channel function is linked to the activity of an NOX is not clear. It is known that osteoclasts express NOX1, NOX2, and NOX4, and are capable of oxidative burst. Therefore, H+ current in osteoclasts could play a similar role as in neutrophils, compensating for cytoplasm acidification on NOX activity. A study by Mori et al. shows that H+ currents in mouse osteoclasts were potentiated by PMA (93), a stimulus that promotes NOX activity, but a role for HV1 channels in sustaining ROS production by osteoclasts has not been established.
Dendritic cells
Expression
The presence of HV1 proton currents and the expression of mRNA and protein were demonstrated in mouse bone marrow-derived DCs (120, 135).
Function
DCs are the cells of the immune system that are responsible for antigen presentation to T lymphocytes. They can become activated on stimulation with TLR4 ligands, such as LPS, and this process is accompanied by an increased production of ROS by NOX2. A recent study using whole-cell patch clamp demonstrated that cytoplasm acidification produced by the activity of NOX2 in mouse DCs is compensated by a proton extrusion via HV1 channel (135). This current depends on the maturation stimulus: It is increased on an acute stimulation with LPS and decreased after 24 h of incubation, paralleled with the production of ROS by NOX2. A second study showed that NOX2-mediated intraphagosomal ROS production controlled the level of proteolysis within phagosomes and was significantly compromised in HV1-deficient DC phagosomes or in the presence of the HV1 inhibitor ZnCl2 (50 μM; 31.18%±5.26% and 44.47%±9.44% reduction, respectively) (120). These results suggest that HV1 channels might mediate the antigen presentation process.
Adaptive Immune Cells
The most important cells of the adaptive immune system are B and T lymphocytes (CD4+ helper cells and CD8+ killer cells). B lymphocytes are responsible for antibody production; these cells express H+ channels as well as NOX2. T lymphocytes express low levels of H+ channels; however, the question whether they also express NOX2 remains controversial (48, 64).
B lymphocytes
Expression
Voltage-activated proton current with typical characteristics of HV1 current was detected in human peripheral blood B lymphocytes (126), and later studies have demonstrated the expression of HV1 protein in human and mouse B lymphocytes (16, 107).
Function
B lymphocytes are the cells of the immune system that are responsible for the production of antibodies. The maturation process is mediated by B-cell receptor (BCR), whose activation by antigen binding causes a sequence of downstream signaling events. This signaling is oxidation dependent, and NOX2 was identified as the major NOX responsible for BCR signaling (82). The role of HV1 in this process has been recently proposed (16). The authors found that HV1-deficient mice have normal B-cell development and steady-state serum immunoglobulin titers. However, NOX2-dependent ROS production in B lymphocytes from HV1-deficient mice was decreased by 60%–70%. The authors suggest that the decreased ROS production led to decreased downstream signaling from BCR, translating into impaired antibody production. Indeed, antigen-specific immunoglobulin titers after immunization with two different antigens (T lymphocyte-independent type 2 antigen NP-Ficoll or T lymphocyte-dependent antigen NP-KLH) were lower in HV1-deficient mice as compared with wild-type littermates; this appeared to be particularly the case for IgG2b. Taken together, these data suggest that HV1 is required for ROS production by NOX2 in mature B lymphocytes on antigen stimulation. However, the data still need to be reconciled with data from NOX2-deficient patient and mice, which suggest that in the absence of catalytically active NOX2 enzyme, there is increased antibody production (17, 116, 140). Furthermore, besides NOX2, B lymphocytes also express NOX5 and DUOX1 (9). The role of these NOX isoforms in B-lymphocyte function and the implications of HV1 in this process have yet to be addressed.
T lymphocytes
Expression
Voltage-activated proton currents were detected in human tonsillar T lymphocytes (68) and in human peripheral blood T lymphocytes (126). The expression of HVCN1 mRNA and HV1 protein by mouse T lymphocytes has been recently confirmed (123). The amplitude of proton current in T lymphocytes is 100 times smaller than in B lymphocytes. However, the current can be increased by about 13 times on stimulation with 50 ng/ml PMA for 24 h (126).
Function
T lymphocytes are cells of the specific immune system that recognize antigens presented by major histocompatibility complex I (MHCI) (CD8-positive killer T lymphocytes) or MHCII (CD4-positive helper T lymphocytes). A phenotype suggesting a role of HV1 in T lymphocytes biology was discovered by Sasaki et al. (123). HV1-deficient mice older than 6 months have splenomegaly, increased levels of anti-double-stranded DNA (dsDNA)antibody, a marker of systemic lupus erythematosus, and glomerular lesions similar to lupus nephritis. These mice had approximately 60% more of activated T lymphocytes (both CD4 and CD8). This phenotype was accentuated in response to infection with lymphocytic choriomeningitis virus (123). The authors further demonstrated that the late phase of O2− production by HV1-deficient T lymphocytes was significantly attenuated. These data suggest that in the absence of HV1, there is decreased ROS generation in lymphocytes and an increased number of activated T lymphocytes. This corroborates previous data, suggesting that low ROS generation may contribute to an autoimmune phenotype (125).
Central Nervous System
While H+ currents were first identified by electrophysiology (ElPh) in snail neurons (137), H+ channel expression in mammalian neurons is debated. ElPh recordings and immunoblotting suggest the presence of H+ channel in mammalian brain tissue (19, 42, 107, 146). However, microglia appears to be the major localization of H+ channels in the brain. No proton currents were detected in astrocytes (146), and no published information is available for oligodendrocytes.
Neurons
Expression
Although H+ current was first identified in snail neurons, no direct evidence of H+ current in mammalian neurons exists at present. Cheng et al. (19) reported a proton current in rat hippocampal neurons. However, this was probably not an HV1 current, as it was insensitive to Zn2+ and not temperature dependent. Moreover, in a later study, a similar current was detected in neurons from HV1-deficient mice (146).
Function
In snail neurons, H+ currents were proposed to compensate for the cytosolic acidification during depolarization (137). Functional data from mammalian neurons show that hippocampal basal synaptic transmission, plasticity, and N-methyl-D-aspartate (NMDA) receptor function are not altered in HV1-deficient mice as compared with wild-type animals (146). Learning and memory tests did not detect differences between HV1-deficient mice and wild-type mice under physiological conditions. The expression and function of HV1 in other neuronal subpopulations and/or in conditions of stress has yet to be established.
Microglia
Expression
The expression of HV1 in mouse and rat primary microglia and in microglia cultured from human fetal brain tissue has been detected by the means of ElPh, WB, and immunocytochemistry (42, 94, 107, 146).
Function
Microglia are the resident macrophage-like cells in the CNS. They become activated in a number of acute and chronic brain disorders, including brain injury, ischemia, and neurodegeneration of various origins (13). As in other macrophages, the expression and function of HV1 is tightly correlated with the expression and function of NOX2. HV1 appears to support the activity of NOX2 in microglia by extruding the excess of proton accumulated in the cytoplasm due to the electron transport through the NOX. Similar to observations in neutrophils (44, 115), PMA-stimulated ROS production in microglial cells detected by the oxidation of hydroethidine was decreased in microglia from HV1-deficient mice (146). However, chemotaxis and migration of microglia remained unaffected (146).
Other Cells
In this review, we discuss preferentially cell types (i) that express high levels of H+ channels and (ii) where a putative function for these H+ channels has been proposed and experimentally evaluated. However, interestingly, many other tissues and cells also express H+ channels (30), although at relatively low levels. Indeed, it is very difficult to find H+ channel-negative cells for heterologous expression of H+ channels (usually the green monkey kidney cell lines COS-7 is used for this purpose). However, for most of the cell types and tissues that express low levels of H+ channels, a specific function has not been defined. It is possible that such functions will be identified over the next years. It also cannot be excluded that such low levels of H+ channels are a safety mechanism that protects cells from extreme acidification under stress conditions. Besides immune cells and CNS cells, there are three cell types in which high activity and a physiological function for H+ channels have been documented: airway epithelia, spermatozoa, and cardiac fibroblasts.
Airway epithelia
Expression
Alveolar epithelial cells from rat were the first mammalian tissue where voltage-gated proton currents were identified by patch clamp (29). Further studies have also demonstrated the presence of H+ currents in primary human tracheal epithelial cells (46) and of HVCN1 mRNA in the nasal mucosa cell line JME (63). To our knowledge, no data on HV1 protein expression in airway epithelia have been published.
Function
Recently, H+ channel activity was shown to contribute to the regulation of acidity of airway surface liquid (ASL) in primary tracheal epithelium and in nasal mucosa cell line JME/CF15 (46, 63, 128). Iovannisci et al. demonstrated that H+ current in human nasal epithelium cell line was completely abolished by 10 μM ZnCl2 and by RNAi-mediated decrease of HV1 expression. The proton current was sensitive to mucosal pH: Acid secretion was increased with an increase of mucosal pH above 7. Interestingly, these authors identified a naturally occurring mutation (M91T), resulting in a channel with slightly different properties: Higher pH (∼0.5 points) or stronger depolarization (∼20mV) were needed to induced acid secretion by M91T HV1 as compared with wild-type channel (as already discussed in the section SNPs). Another study addressed the possibility of HV1 cooperation with an NOX in lung epithelia (128). They found that in tracheal epithelium, DUOX1 and DUOX2 mRNAs are highly expressed. The authors propose that DUOX activity in this tissue generates the driving force for H+ channel activity needed to maintain the acidification of ASL. Thus, in airway epithelia, the role of HV1 might not be simply to get rid of an excess of H+ produced by DUOX activity, but also to provide an additional protective mechanism through acidification of the ASL.
Spermatozoa
Expression
A recent development of a method to perform patch-clamp recordings on human sperm enabled the measurement of H+ current in these cells. This current was sensitive to known blockers of HV1, Zn2+ (IC50 222±36 nM), and hanatoxin. The current was also potentiated by oleic acid (100 μM) and AA (30 μM), known activators of HV1 (78). HV1 expression was also confirmed by in situ hybridization, WB, and immunocytochemistry, showing high levels of HV1 expression in the principal piece of the sperm flagellum. As opposed to human sperm, there was no H+ channel activity in mouse sperm.
Function
The cytoplasm of quiescent spermatozoa is acidic (pH<6.5), and its alkalinization is required for sperm motility, chemotaxis, and acrosome reaction. A seminal paper from Lishko et al. suggests that HV1 activity is necessary for sperm activation: Functional assays showed that capacitation of spermatozoa occurred along with an increase in the amplitude of voltage-gated proton current. Interestingly, seminal fluid has an unusually high concentration of HV1 blocker Zn2+ (2.2±1.1 mM), which might be a physiological mechanism of keeping the spermatozoa in the quiescent state. ROS production is also necessary for sperm maturation (27). One of the possible sources of ROS in spermatozoa is NOX5, a member of the NOX family that is highly expressed in human testis (4, 8, 99). The abundant expression of both HV1 and NOX5 in human testis raises the question as to whether HV1 activity in the testis is linked to electrogenic activity of an NOX. The relationship between HV1 and NOX5 was addressed in a recent study (99). Using coimmunoprecipitation, they showed that NOX5 interacted with HV1 in sperm cells, suggesting that these two molecules work as a functional unit. Furthermore, Zn2+ (1 mM) or RNAi-mediated inhibition of HV1 in K562 leukemia cells overexpressing NOX5 resulted in a substantial decrease in O2− production, supporting this hypothesis. Zn2+ treatment (1 mM) also significantly inhibited O2− production in human spermatozoa stimulated by PMA, ionomycin, or H2O2. The authors also found a significant correlation between the percent of motile sperm and basal levels of O2− production.
Another important factor in sperm activation is Ca2+ current mediated by pH-dependent flagellar Ca2+ channel, cation channels of sperm (CatSper) channel, which is inhibited by acidic intracellular pH (79), and can be activated by progesterone (129). Interestingly, spermatozoa from a CatSper2-deficient patient not only retained fully functional proton channel HV1, but also its current density was up-regulated. This may indicate a possible linked developmental regulation between HV1 and CatSper channels in human spermatozoa.
It should be noted that sperm activation in mice (and probably also in rats) must have some fundamental differences with the situation in humans. Not only do they lack HV1 expression in sperm cells, but also these animals entirely lack NOX5 on a genomic level. In addition, progesterone activates CatSper only in humans and not in mice.
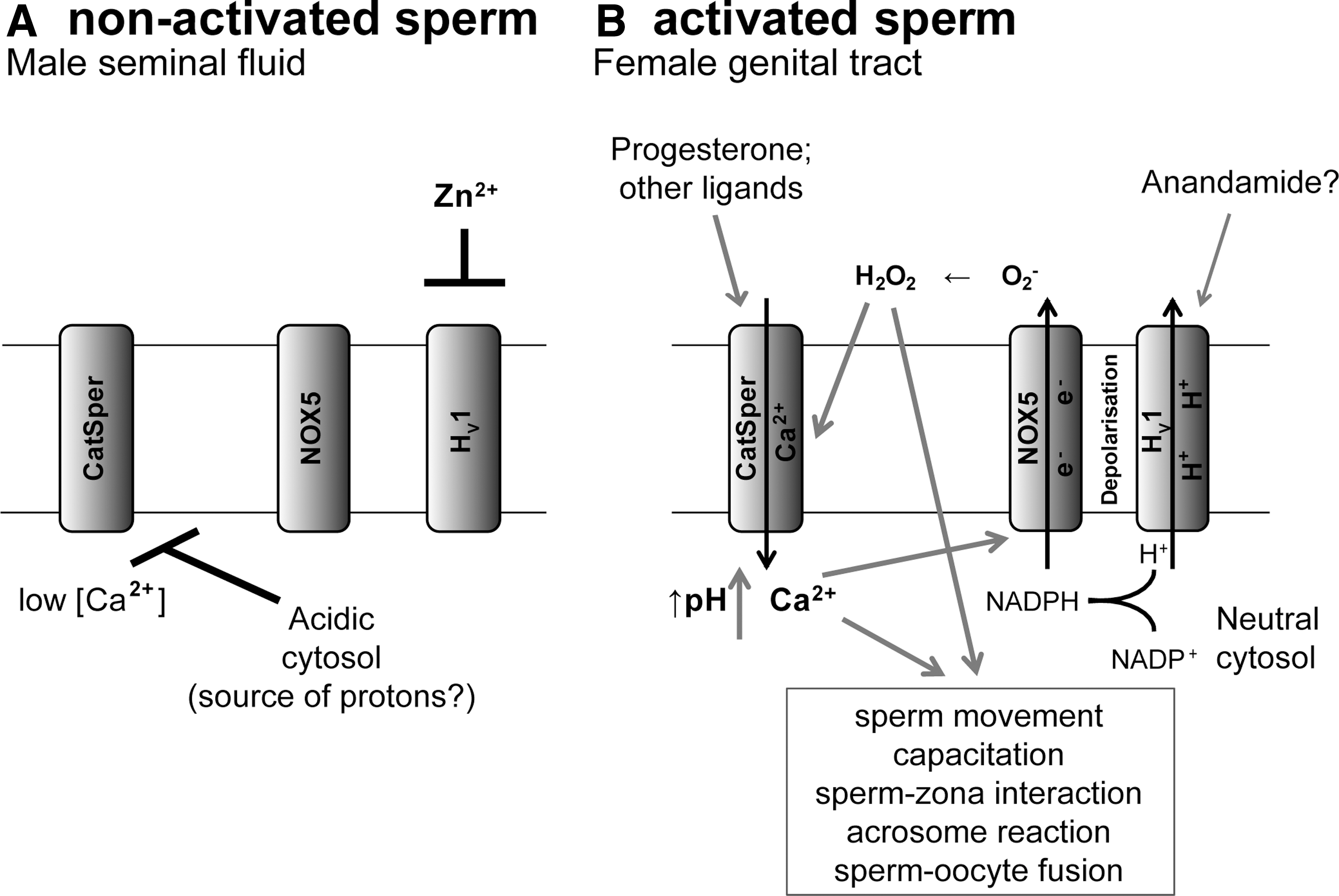
Figure 4 summarizes the role and potential interaction of three key players in human sperm activation: HV1, the Ca2+-activated NOX5, and the sperm-specific Ca2+ channel CatSper. These proteins regulate three intracellular signaling systems with crucial importance for sperm activation: intracellular pH, redox potential, and cytosolic free Ca2+ concentration. Although details of their dynamic interaction still need to be elucidated, the following working hypothesis can be formulated. High Zn2+ concentrations in the male reproductive tract block spermatocyte H+ channels and lead to acidic sperm cytoplasm. It should be noted, however, that while the total Zn2+ concentrations in the male reproductive tract are indeed very high, little is known about the free Zn2+ concentration, which is the determinant for HV1 inhibition. The fact that H+ channels are involved in cytosolic pH maintenance in resting spermatocytes suggests that these cells might have high basal H+ production and/or lack efficient other H+ extrusion mechanisms. On arrival of sperm cells in the female reproductive tract, H+ channels will be activated because of low Zn2+ concentration and possibly also through the endocannaboid Anandamide (78). It should be noted, however, that it is not clear as to what extent the Anandamide concentrations in the female genital tract are sufficiently high to activate Hv1. Simultaneously, CatSper is activated through progesterone, as well as through the HV1-dependent cytosolic pH neutralization. CatSper-dependent Ca2+ influx activates the NOX5, which, due to its electrogenicity and cytosolic H+ generation, also requires HV1 for optimal function. NOX5 further enhances the activity of HV1 through depolarization and the activity of CatSper through H2O2. The conversion of O2 − into H2O2 consumes H+ ions. This has two important functional consequences for the system: (i) HV1 provides a driving force for dismutation of O2− to the signaling molecule H2O2, and (ii) H+ ion consumption through O2− dismutation supports HV1 function by preventing excessive H+ ion accumulation at the extracellular site of the plasma membrane, which would inhibit HV1. Thus, in a synergistic cooperation, the three sperm proteins abolish the inhibitory cytosolic acidification, increase the oxidative environment, and elevate cytosolic-free Ca2+ concentration. pH-, redox-, and Ca2+-signaling then collaborate and activate sperm effector functions, such as sperm movement, capacitation, sperm-zona pellucida interaction, acrosome reaction, and sperm-oocyte fusion.
Cardiac fibroblasts
Expression
H+ currents with characteristics of HV1 current were detected in human cardiac fibroblast by patch clamp (43). No information on mRNA and protein expression is available.
Function
Cardiac fibroblasts are needed for structural support and play an important role in cardiac tissue remodeling on ischemic condition. El Chemaly et al. propose that H+ current in these cells is important to regulate membrane potential in pathological conditions when other conductances are reduced and cytosolic pH changes in response to physiological activity and during ischemia (43). The predominant NOX isoform in cardiac fibroblasts appears to be NOX4 (24). At this point, it is not clear whether HV1 might contribute to NOX4 function.
HV1 Pharmacology
Nowadays, only a few compounds that are capable of inhibiting proton currents have been described. Three types of molecules obviously have an inhibitory effect on voltage-gated proton channels: polyvalent cations, a tarantula toxin, and guanidine derivatives (Table 3). In addition, there are several compounds that block H+ currents; however, it is not clear whether they are direct channel inhibitors. All of these molecules often have multiple targets, and their potency of proton channel inhibition is quite low. Thus, none of the hitherto described HV1 inhibitors is specific and can be considered a drug candidate. However, these compounds can be used as pharmacological tools for a better understanding of HV1 and its physiological function. These compounds also might pave the way to clinically useful HV1 inhibitors in the future and we will, therefore, review the available data in this section. However, in the long run, specific HV1 targeting compounds will be needed and they will not necessarily be derived from existing molecular scaffolds. Thus, large high-throughput screens will be required. Availability of a crystal structure of HV1 should also enable a more rational, structure-based approach to the development of HV1-active drugs.
Only for the first three compounds (Zn2+, hanatoxin, 2GBI) convincing evidence for direct HV1 inhibtion exist. The mechanism of action of the other compounds might be indirect.
A range of IC50 from 200 nM to 100 μM has been reported depending on the cell system and the buffer solution.
Three-dimensional structure of Hanatoxin solved using NMR spectroscopy was taken from
2GBI, 2-guanidinobenzimidazole; DEPC, diethylpyrocarbonate; BCECF, 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein; NA, data is not available.
Zn2+ and polyvalent cations
Zn2+ and other polyvalent cations have been used for many years as proton current inhibitors for in vitro experiments. Half-maximal Zn2+ concentrations for H+ current inhibitions were generally in the range of 1 μM. It should be noted that this refers to free Zn2+ concentrations, and because of Zn2+ binding by proteins, metal chelators, and possibly other compounds, higher total Zn2+ concentrations were used depending on the buffer solutions (44). Before the identification of HV1 and availability of HV1-deficient mice, inhibition by Zn2+ was the gold standard to identify putative H+ currents. Of note, though, HV1 channels from some species have been shown to be insensitive to Zn2+ inhibition, probably due to the lack of Zn2+ binding histidines at key locations (130, 136). However, since Zn2+ ions are implicated in many other physiological processes, the usefulness of Zn2+ as a specific H+ channel blocker is limited. Molecular mechanisms of Hv1 inhibition by Zn2+ have been extensively reviewed previously (30, 31, 35). In brief, Zn2+ appears to compete with H+ for binding to the external surface of the HV1 channel. This changes the membrane potential perceived by the channel, and, therefore, stronger voltage needs to be applied to elicit the proton current.
Hanatoxin
Voltage sensors are known to be inhibited by venom toxins. Studies based on chimeras from different voltage sensors identified a modular unit composed of S3 and S4 helices, named paddle motif, which is highly conserved between different voltage-sensing proteins. The binding of hanatoxin, a toxin from venom of tarantula Grammostola spatulata (1) to the paddle motive, inhibits ion fluxes through various voltage-dependent ion channels. HV1 channels also possess the paddle motif and were blocked by hanatoxin, which shifted the activation to more positive voltages, consistent with its predicted site of action (1). Interestingly, a mutation in the third TM segment of HV1 (D185A; see Fig. 1) abolished this inhibitory effect. This work shows that the use of hanatoxin can be instrumental for further understanding the structure of HV1 and could contribute to the development of new specific inhibitors of this ion channel.
Guanidine derivatives
Many inhibitors of ion channels are open channel blockers, including quaternary ammonium ions and cationic derivatives of local anesthetics. These compounds usually bind to the inside pore domain of an open channel and block the ion current. A recent study screened guanidine derivatives with different steric features in human HV1 channels expressed in Xenopus oocytes. This work identified several molecules that are capable of blocking the depolarization-induced H+ current (59). The authors performed structure-activity analysis and found that even small differences in structure significantly affected the affinity. The most potent compounds were 2-guanidinobenzimidazole (2GBI) and 1-(1,3-benzothiazol-2-yl)guanidine, and their inhibitory effect depended on the presence of 2 guanidine groups conjugated on a five-membered aromatic ring. The inhibition by 2GBI had an IC50 of 38 μM and was reversible. Further experiments with 2GBI allowed to obtain information on the binding site and to shed light on the location of the pore of HV1: (i) The inhibitory effect of 2GBI was only observed on intracellular application; (ii) site-directed mutagenesis demonstrated that amino acid F150 in the S2 helix of HV1 is important for the blocking by 2GBI (Fig. 1). The results of this study confirm that guanidine derivatives represent an interesting pharmacological tool to investigate the molecular mechanism of HV1 inhibition. The discovery of this interaction is a promising basis for a further analysis of structure-activity relationship for HV1 and might provide a molecular backbone for the development of more potent inhibitors. Interestingly, guanidine analogues showed neuroprotective potential in an in vitro model of ischemia (10), and it was recently shown that HV1-deficient mice are protected in the model of stroke (146). None of the neuroprotective guanidine analogs has been tested for efficacy of HV1 inhibition, but it will be of interest to see whether these compounds act—at least in part—through HV1. In order to be of interest as for drug development, it will be necessary to develop cell-permeant guanidine analogs.
H+ current inhibitors with potentially indirect effects
Diethylpyrocarbonate (DEPC) is a histidine-modifying reagent that has been used as a biochemical tool to understand the role of histidines in the function of proteins. It was shown to inhibit NOX-dependent H+ current in human eosinophils and O2− production on human neutrophils (5, 83). Indeed, histidines are known to be important for proton binding by HV1: There is a competition between H+ and Zn2+ binding at His140 and His193. However, since the inhibitory effects of DEPC were only observed with an activated NOX and the effect was not found in eosinophils from CGD patients, it is more likely that DEPC acts through the modification of hemes in NOX2.
Three recent papers report the identification of compounds with inhibitory activity on H+ currents: tricyclic antidepressants (imipramine, amitryptiline, and desipramine), selective serotonin reuptake ihibitor fluoxetine, the morphine-derivative dextromethorphan (DM), and a tea catechin flavonoid EGCG. All of these compounds were found to inhibit H+ current in BV2 mouse microglial cells (67, 131, 132). The mechanism of H+ current inhibition by imipramine and DM was ascribed to the fact that they form weak organic bases, a mechanism of H+ channel inhibition previously discussed (30). With regard to EGCG, the authors argue that its inhibition of the H+ channel is pH independent. After a detailed analysis of the known properties of EGCG, they hypothesize that this compound might interfere with the activity of the H+ channel by modifying the structure of the lipid bilayer. Antidepressants, DM, and EGCG have been previously reported to have neuroprotective properties in multiple models of neurodegeneration and neuroinflammation (54, 80, 119, 147, 153). However, these drugs have multiple targets, including NOX2 (18, 153), and currently, there is no evidence that the neuroprotective effect is due to H+ channel inhibition.
Pharmacological modulation of HV1 is not necessarily limited to inhibition. Indeed, as discussed next, in some disease situations, such as autoimmune disease and male infertility, HV1 activation might be considered a relevant pharmacological approach. Currently, no drug-like molecules are known to activate HV1. However, proton currents are pharmacologically enhanced by unsaturated long-chain fatty acids such as AA and others (32, 69, 78, 127). It appears that these fatty acid effects are due to a direct pharmacological activation of HV1. A better understanding of the involved binding sites might lead to the discovery of drug-like HV1 activators. In general, ion channel activators are more difficult to identify than ion channel inhibitors, as binding to the channel pore usually produces inhibition. However, it is likely that once large-scale screenings are performed, compounds which activate HV1 by shifting the voltage- or pH dependency of channel activation could be identified.
Potential Therapeutic Indications for HV1 Modulators
At present, there are no clinically used drugs that target HV1. However, there are several reasons to think that HV1 might become an interesting drug target in the future (Fig. 5): (i) HV1 is involved in the regulation of several relevant physiological and pathophysiological functions; however, there is no severe spontaneous pathology associated with HV1 deficiency in mice. (ii) Globally speaking, it is well established that ion channels are valuable drug targets: They often regulate critical elements of cellular signaling and since they are typically localized on the cell surface, they are readily accessible for small binding. (iii) Pathways dependent on the phagocyte NOX2 could be pharmacologically modulated without a direct NOX2 inhibition. Interestingly, when looking at knock-out mice, it appears that HV1 deficiency is more advantageous than NOX2 deficiency. ROS production in neutrophils is a crucial host defense mechanism, and a complete block of ROS generation would not be desirable. Neutrophils from HV1 knock-out mice have a residual ROS release of approximately 30%. This reduction does not lead to a decreased bacterial host defense in vivo, as assessed by infection models with S. aureus, P. aeruginosa, or B. cepacia (115). This confirms other observations that a relatively low residual activity of NOX is sufficient for innate immune responses. (iv) Not all HV1 functions are directly related to NOX activity. Thus, new interesting therapeutic indications are emerging that could not necessarily be targeted with NOX-active drugs. Examples for these are histamine release from basophils and H+ extrusion in cancer cells (see section Cancer). (v) In addition to development of HV1 inhibitor, HV1 activators might be of interest for certain types of pathologies. The most obvious indication at this point might be male infertility and possibly also autoimmune disease (see section Autoimmune disorders). However, the risk of on-target side effects of HV1 activators cannot be predicted based on currently available information.

Inflammatory diseases
No NOX2-independent function has been attributed to HV1 in neutrophils and eosinophils. Thus, the impact of HV1 inhibition in these cell types would be essentially a mild to moderate NOX2 inhibition, to the best of our knowledge without immunosuppression. The inhibition of neutrophil ROS generation through targeting HV1 is obviously of interest in a variety of inflammatory pathologies, including septic shock, and other infectious diseases with an overshooting inflammatory component (e.g., bacterial meningitis, pulmonary infections, and hepatitis), ischemia/reperfusion injury, and stroke (microglia ROS generation probably is also important in this pathology, see section CNS diseases). The inhibition of HV1 by bivalent cations should be carefully evaluated in prosthetics surgery: Cobalt ions released from metal prosthesis can promote bacterial infections in patients with metal-on-metal total hip arthroplasty (25). It should be noted that this list is similar to possible indications of inhibitors of the phagocyte NOX (76), and future research will need to determine which of both is the more promising therapeutic avenue.
Allergic diseases
Eosinophils, mast cells, and basophils
Histamine release accompanying allergic reactions is a redox-dependent process. In addition, eosinophils are the cells with the highest levels of NOX2-dependent ROS release, which probably also contributes to allergic reactions. HV1 has been shown to sustain NOX2 activity in mast cells and in eosinophils and to participate in acid extrusion by basophils. Therefore, HV1 inhibition can be potentially used to reduce histamine secretion from several cell types and excessive ROS release from eosinophils, which may become an interesting concept for the treatment of allergies.
Autoimmune disorders
Aging HV1-deficient mice spontaneously develop mild autoimmunity [(123); see section T lymphocytes]. Along these lines, a study on gene expression in blood of patients with autoimmune inflammatory bowel disease detected an inverse correlation between the expression levels of HV1 and the activity of Crohn's disease, with the expression of HV1 being higher in the remission than in an active phase of disease (53). The mechanisms underlying the enhanced autoimmunity in the absence of HV1 are not understood. Indeed, B-cell receptor signaling was decreased in HV1-deficient mice (16). Thus, further studies to understand mechanisms will be necessary. However, one might speculate that HV1 activation could be a useful therapeutic approach in certain types of autoimmune diseases. It should be noted that there is also an increased autoimmune phenotype in NOX2-deficient humans and mice. As shown in Table 4, at this point it is not clear whether there is a common denominator of the NOX2- and HV1-associated autoimmunity.
Enhanced autoimmunity is observed with HV1 deficiency and with NOX2 deficiency. No systematic comparison of the two autoimmune phenotypes has been performed. This table summarizes available evidence from separate studies.
NA, data is not available; CGD, chronic granulomatous disease; dsDNA, double-stranded deoxyribonucleic acid.
B-cell-derived tumors
Certain lymphoid tumors are characterized by hyper-reactive B-lymphocyte receptor signaling. Therefore, HV1 inhibition has been suggested as a therapeutic approach for this type of tumor (16).
Osteoporosis
Given the role of HV1-dependent H+ secretion in acid-dependent calcium phosphate mobilization by osteoclasts, HV1 inhibition might be an attractive concept for the treatment of osteoporosis. The underlying mechanism might be simply H+ secretion; however, in the case of osteoclasts, there is also an interesting link to NOX enzymes. NOX-dependent ROS generation has been implicated in osteoclast activity and, hence, in bone resorption (93, 121, 149, 150). However, while no bone alterations have been associated with NOX1 and NOX2 deficiency (9), NOX4-deficient mice appear to have increased bone density (Goettsch et al., 2013, in press). Thus, it is possible that H+ channels, in addition to their impact on extracellular acidity, may contribute to bone resorption through supporting NOX4 activity in osteoclasts. Thus, HV1 inhibitors might target osteoporosis through the blocking of two osteolytic principles, namely acid secretion and ROS generation. However, no bone-related phenotype has been detected in HV1-deficient mice so far.
CNS diseases
There are at least two mechanisms dealing with how HV1 could be implicated in CNS diseases. HV1 might modulate neuronal pH homeostasis. Indeed, it has been suggested that H+ channels might contribute to proton extrusion from metabolically active neurons (87). This HV1 function would be neuroprotective rather than contributing to disease. However, inappropriate HV1 function (e.g., dysregulated phosphorylation) or SNPs altering HV1 activation properties might perturb normal neural pH homeostasis and, therefore, possibly also contribute to disease. To date, there is no experimental evidence for these scenarios and—as pointed out earlier—there is presently no convincing evidence for functional expression of HV1 in mammalian neurons. However, HV1 is abundantly expressed in microglia, where it might contribute to CNS disease through supporting NOX function. Indeed, oxidative stress, at least in part due to ROS generation by NOX, is a major contributor to the development of CNS diseases. A recent study by Wu et al. demonstrated that HV1-deficient mice are protected in the pMCAO model of ischemic stroke (146). The infarct size in these mice was smaller, the neurological score was less severe as compared with the control mice, and molecular markers of cell death indicated the neuroprotective effect of HV1 deficiency. Authors explain this effect by the reduced production of ROS by microglia. In the absence of HV1, NOX2 causes excess depolarization of the membrane, which inhibits electron transfer by the oxidase, therefore abolishing ROS production. The authors also addressed the role of HV1 in excitotoxicity. They observed that NMDA-induced neuronal death in primary cultures of neurons and microglia was not rescued by the absence of HV1. This indicates that the beneficial effect of HV1 deficiency in the model of stroke is dependent on its regulation of microglial activity rather than its contribution to excitotoxicity. These data suggest that HV1 inhibition could be beneficial for the treatment of stroke. By extrapolation, on might notice that HV1 inhibition could also be of interest for the treatment of other neurodegenerative processes accompanied by excessive production of ROS by microglia, such as, for example, stroke, Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis.
Pulmonary disease
No data on pulmonary disease models in HV1-deficient mice are currently available, and no spontaneous airway phenotype has been detected in HV1-deficient mice so far. However, based on the two documented physiological functions of HV1 in airway epithelia, acidification of the ASL and facilitation of ROS generation by DUOX NOX (see section Airway epithelia), one may speculate on a possible role of HV1 inhibitors in lung diseases. Maintaining the pH of ASL at a mildly acidic pH is important for normal physiology, and dysregulation might contribute to disease. In particular, overshooting acidification might be involved in the development of airway inflammation, as well as chronic obstructive lung disease, asthma, and cystic fibrosis [reviewed in Fischer and Widdicombe (45)]. The facilitation of ROS generation by DUOX NOX similarly might contribute to airway inflammation. Thus, HV1 inhibitors might be of interest for the treatment of inflammatory airway diseases.
Male fertility
HV1 is highly expressed in the testis, and its activity is crucial for sperm motility, chemotaxis, and acrosome reaction (78, 99) and therefore for male fertility. Interestingly, low levels of HV1 expression are correlated with male infertility (111) and, more specifically, patients with teratozoospermia (i.e., abnormal sperm morphology) have reduced expression of HV1 (Geo Profiles, NCBI ID: 38039902; ID: 38148335; ID: 38065002). Altogether, these findings suggest that HV1 activation in human spermatozoa could be an interesting concept for male infertility. Conversely, HV1 inhibition could be considered a tool for contraception.
Cancer
Otto Warburg described unique metabolic properties of cancer cells, namely a mostly nonoxidative breakdown of glucose (145). He also hypothesized that unique metabolic state leads to increased acidity in cancers. This should, indeed, make cancer cells particularly dependent on efficient proton extrusion pathways, in particular proton channels. Indeed, several recent publications point toward a privileged role of HV1 in cancer cells (142 –144). For example, expression of HV1 protein was associated with an invasive phenotype in breast cancer and in colorectal cancer (142 –144): HV1 expression was increased on the surface of highly metastatic cells and correlated with tumor progression. Associations between HV1 levels and tumor size, tumor classification, lymph node status, and clinical stage were detected. Tumor cells are highly metabolically active, resulting in low pH inside the cell. Voltage-gated proton channel expression is probably up-regulated in tumor cells in response to intracellular acidosis in order to regulate the pH. In fact, the inhibition of HV1 expression by short-interfering ribonucleic acid (siRNA) in highly metastatic MDA-MB-231 cells reduced intracellular pH measured using BCECF pH-sensitive fluorescent probe and decreased the invasion and migration potential of these cells. Moreover, HV1 silencing in grafted cells reduces tumor growth in nude mice injected sub-cutaneously with MDA-MB-231 cells. On the other hand, the expression of HV1 in tumors might be linked to the role of NOX4 in breast cancer (152). Taken together, HV1 inhibition might become an interesting concept to limit invasiveness of breast cancer, and possible other types of tumors. There is also an interest in HV1 as a general biomarker for tumors. Indeed, several patents (WO 2010110346 A1, EP 1961825 A1, US 20110190157; EP 2253721 A1; EP 2115170 A2; US 20110217297 A1) suggest that HV1 could be useful as a biomarker for leukemias and solid tumors.
Outlook
Will H+ channel active drugs be used in the future? If yes, for which disease indications and will there be inhibitors or rather activators? Generally speaking, ion channels tend to be good drug targets, and H+ channels might not be an exception to this rule. In addition, given the benign phenotype of the HV1-deficient mice, no major or target side effects are expected for HV1 inhibitors. Therefore, the possible use of H+ channel active drugs relies mostly on their potential indications. At this point, the following diseases appear most likely to be amenable to an H+ channel-based treatment. In our opinion, the strongest candidates at this point are as follows: (i) HV1 inhibitors as anti-inflammatory drugs; (ii) HV1 inhibitors for ischemic stroke; (iii) HV1 activators for certain forms of infertility; and (iv) HV1 inhibitors for the treatment of invasive breast cancer and possibly other types of tumors.
Thus, taken together, research on HV1 modulators and their potential indications is only at a preliminary stage. With this limitation in mind, we believe that it will be possible to develop HV1-active and -selective compounds and that such compounds will find their place in the clinical medicine of the future.
Footnotes
Acknowledgments
The authors thank Vincent Jaquet, Hedi Peterson, Julien Cachat, and Christine Deffert for helpful discussions and Freddy Heitz, Antoine El Chemaly, and Vincent Jaquet for careful editing of this article. T.S. received funding from the European Community's Framework program under grant agreement Neurinox (Health-F2-2011-278611).