
Abstract
Introduction
H
HDACi induce a number of direct anti-tumor effects, including growth arrest, differentiation, autophagy, and apoptosis (106, 108, 139). These effects are mediated through a number of mechanisms, including transcriptional changes, increased production of reactive oxygen species, altered signaling, and aberrant mitosis (15, 22, 76, 106). Among these, the most actively investigated is the alteration of gene expression (77). In this review, we will assess our current understanding of the key mechanisms by which HDACi regulate gene expression, and examine the spectrum and commonalities of HDACi-mediated transcriptional responses, both within and between tumor types.
Overview of HDACi-regulated gene expression
A number of microarray studies have established that HDACi treatment of tumor cell lines alters the expression of ∼0.5–20% of genes (25, 58, 107, 123, 135). Likely explanations for this disparity include the thresholds used to determine altered expression, drug concentrations used, and the time point of assessment, which would have an impact on the contribution of primary and secondary response genes. For example, a time course of HDACi treatment of acute T-cell leukemia cells demonstrated a 20-fold increase in the number of genes changed at 16 h compared with at 1 h post HDACi-treatment (123). In this review, we will focus, wherever possible, on the direct targets of HDACi, defined as those regulated rapidly and independently of new protein synthesis.
A consistent finding revealed by these studies is that approximately equal numbers of genes are up- and down-regulated after HDACi treatment (16, 107, 123). Notably, this ratio remains relatively constant at both early and late time points, indicating that direct and indirect HDACi-targets are similarly subjected to activation and repression.
Mechanisms of HDACi-mediated transcriptional regulation
Two major mechanisms by which HDACi modify gene expression have been investigated: the induction of histone acetylation and the induction of transcription factor acetylation (Fig. 1). We will focus on these mechanisms by highlighting evidence generated from gene-specific and whole transcriptome analyses. The ability of these mechanisms to explain HDACi-mediated transcriptional activation and repression will also be discussed.

Histone hyperacetylation
The hyperacetylation of lysine residues in core histone proteins is a consistent feature of HDACi treatment, first described in the late 1970s by Riggs et al. and Vidali et al. (130, 154). Histone acetylation is regulated by two opposing sets of enzymes: histone acetyltransferases (HATs), which add acetyl groups to lysine residues, and HDACs, which remove them. The eighteen HDACs that have been described in humans are classified into four classes based on their homology to the yeast HDACs Rpd3 (class 1), Hda1 (class II), or SIR2 (class 3). The fourth class comprises HDAC11, which has homology to both class I and II HDACs. The HDACi that will be discussed here (hydroxamic acids, carboxylic acids, benzamides, and cyclic tetrapeptides) are those which exclusively inhibit the zinc-dependent Class I (HDAC1, 2, 3, and 8) and class II HDACs (HDAC4, 5, 6, 7, 9, and 10), by binding the critical Zn2+ ion required for their enzymatic activity (14).
HDACs typically exist within large multi-subunit co-repressor complexes such as Sin3, NuRD, CoREST (HDACs 1 and 2), and NCoR/SMRT (HDAC3, 4, 5, and 7) (40, 76). These HDAC-containing complexes, along with HATs, are recruited to gene promoters via interactions with sequence-specific transcription factors, where they collectively establish the desired level of gene expression by regulating the acetylation status of lysine residues on surrounding histones. By inhibiting the activity of HDACs, HDACi shift the local equilibrium in favor of histone acetylation. Two general mechanisms by which histone lysine acetylation alters transcription can be envisioned: neutralization of the positive charge of lysines and subsequent establishment of a more permissive chromatin state, and enhanced recognition and binding by bromodomain (BRD)-containing epigenetic reader proteins.
Histone lysine acetylation and establishment of a permissive chromatin state
An association between histone acetylation and increased gene expression was first suggested by Allfrey almost 50 years ago (2, 3), and has since been validated in a number of studies using multiple approaches (60). Acetylation of lysine residues on histones has been suggested to induce a more permissive chromatin state (94). This likely facilitates the access of additional sequence-specific transcription factors to promoter regions, including transcriptional activators and repressors, as well as components of the basal transcriptional machinery. Consistent with a model in which a more open chromatin state facilitates the binding of both transcriptional activators and repressors, genome-wide ChIP-seq studies indicate that both HDACi-activated and repressed genes display increased promoter histone acetylation (68, 79).
Two ways in which histone acetylation induces a more open or permissive chromatin conformation have been proposed (Fig. 2). First, the acetylation of lysine neutralizes its positive charge. Acetylation of lysines on the N-terminal tails of core histones may, therefore, decrease their electrostatic interaction with negatively charged phosphate groups on the DNA backbone in adjacent nucleosomes, resulting in a more open higher-order chromatin structure (60, 155) (Fig. 2A). This charge neutralization model has been confirmed in vitro, where tetra-acetylation of the histone H4 tail significantly decreased its affinity for DNA (70). Second, a structural analysis of the nucleosome demonstrates that the positively charged histone H4 tail interacts with a highly negatively charged region on the H2A-H2B surface (101) (Fig. 2B). Acetylation of these lysines is likely to alleviate this interaction and facilitate the establishment of a more open chromatin state.

However, while the charge neutralization model has been widely implicated in the establishment of a more open chromatin conformation, there is limited direct in vivo evidence in support of this. One study suggesting this possibility was performed by Dion et al. in yeast (42). Here, the mutation of histone H4 lysine K5, K8, or K12 to arginine, which mimics the positively charged unacetylated lysine state, each induced a similar change in gene expression, which increased monotonically with the number of lysines mutated. However, while these findings are consistent with a model in which acetylation of these lysines incrementally increases the extent of chromatin relaxation due to progressive charge neutralization (42, 182), they are also consistent with a number of “reading mechanisms.” Specifically, decreasing the number of acetylated lysine residues on the histone tail may also progressively reduce the direct binding of BRD-containing proteins that regulate transcription (42). This alternative mechanism is discussed next.
Histone acetylation and recognition by BRD-containing proteins
Histone tail lysine acetylation serves as a key mark for the recognition and binding of BRD-containing proteins (12, 133). In humans, 42 BRD-containing proteins have been described. Several of these can impact transcription through mechanisms including modulation of chromatin-remodeling (BRM, BRG1) (50) and maintenance of higher-order chromatin structure (BRD4) (50, 164), provision of a scaffold for the recruitment of transcription factors (BRD1), transcriptional activation (BRPF1) and co-activation (P300, PCAF), transcriptional repression (BAZ2A, TIF1γ/TRIM33) (1, 187), initiation (TAF1), elongation (BRD4, TRIM33) (12, 41, 51, 133), gene bookmarking (BRD4, BRDT) (56, 185), and chromatin compaction (BRD4, BRDT) (52). Histone lysine hyperacetylation after HDACi treatment may alter the binding of BRD-containing proteins, which could either activate or repress transcription through these mechanisms (Fig. 3).
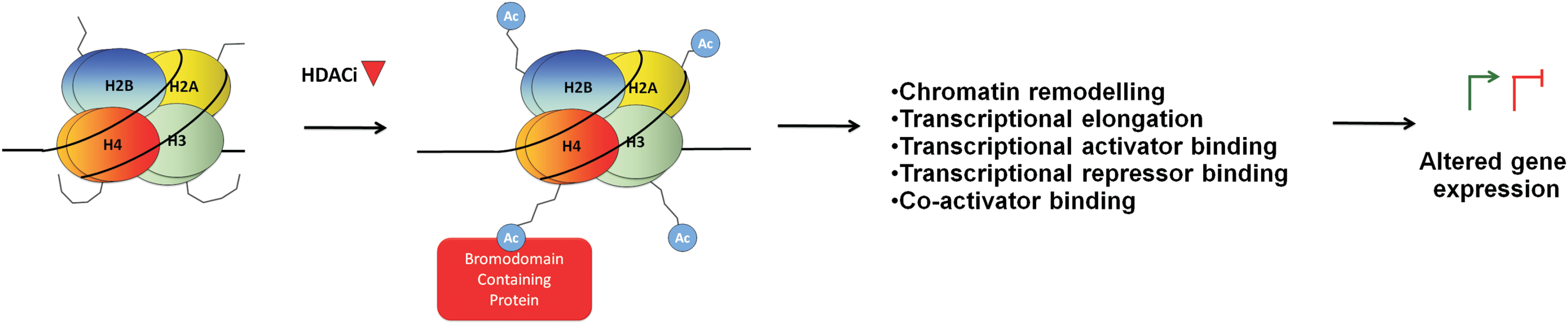
For example, with regard to transcriptional activation, BRD4 recognizes and binds to acetylated H4K5/8/12, which initiates transcriptional elongation (65, 142). BRD4 recognition and binding to acetylated H4K5 is also a key mechanism of gene bookmarking, by which the binding of BRD4 to gene promoters during interphase facilitates their rapid re-expression post mitosis (185). HDACi-induced histone hyperacetylation may, therefore, increase transcription through either of these mechanisms.
Conversely, histone acetylation may also facilitate transcriptional repression though enhanced binding of BRD-containing proteins. Specifically, the transcriptional repressor of TGFβ signaling, TIF1γ, binds sequentially acetylated lysines on the histone H3 tail (1). Binding to acetylated lysines increases the E3 ubiquitin ligase activity of TIF1γ and its ability to ubiquitinate its substrate SMAD4. This results in the disruption of SMAD4-containing transcriptional complexes and the repression of SMAD4 target genes (1). HDACi-induced histone hyperacetylation may, therefore, cause transcriptional repression by facilitating the recruitment of repressors such as TIF1γ. Furthermore, a paradoxical role for the B isoform of BRD4 in chromatin compaction after binding to acetylated histones has also recently been described (52). While the impact on transcription was not examined, increasing chromatin compaction would be expected to repress transcription.
Specific histone lysine residues acetylated by HDACi
Given the range of mechanisms by which histone hyperacetylation may modulate transcription, appreciation of the specific lysine residues that are acetylated by HDACi could provide insights into the corresponding alterations in gene expression.
Histone tail modifications
The nucleosome is composed of ∼147 bp of DNA wrapped around a histone octamer comprising two molecules each of histone H2A, H2B, H3, and H4. The four core histones contain numerous lysine residues that are amenable to acetylation, although acetylation events in the unstructured N-terminal histone tail domains have, by far, been the most extensively studied. Specificity among class I HDACs (HDACs 1, 2, 3, and 8) for deacetylating individual histone tail lysines has been investigated (75). For example, Johnson et al. reported that HDACs 1, 2, and 3 induced at least partial deacetylation of all lysines tested, including H2BK12, H2BK15, H2AK5, H3K14, H3K9/K18, H4K5, H4K8, and H4K12, although with varying efficiency. Furthermore, HDAC3 more robustly induced deacetylation of H2AK5 and H4K5 compared with HDAC1 (75).
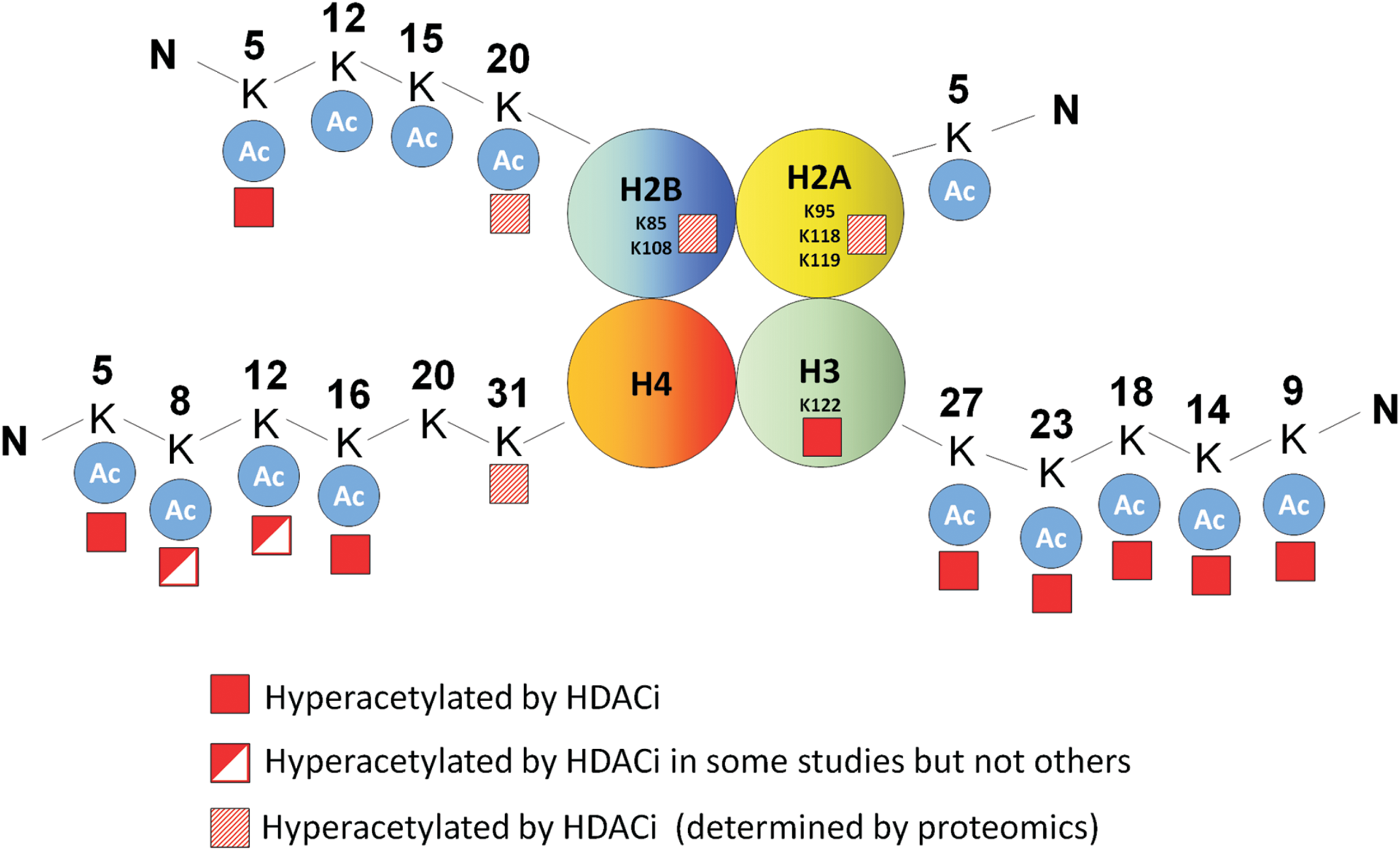
Consistent with their ability to simultaneously inhibit multiple class I HDACs (15), HDACi induce hyperacetylation of each of the four core histones (24, 177). Furthermore, these effects are evident within 30 min of HDACi treatment, consistent with the dynamic nature of histone acetylation changes, which typically have a half life of only a few minutes (182). An examination of acetylation changes induced by HDACi at each core histone using acid-urea gels indicates that all histones are hyperacetylated, with the most robust changes occurring on histone H4 (27, 39). This is consistent with recent proteomic profiling studies in which the most robust changes in acetylation after suberanilohydroxamic acid (SAHA) treatment occurred on histone H4 N-terminal lysines (33).
Several studies have used specific antibodies to examine HDACi-induced acetylation of specific histone tail lysines. For example, on histone H2B, H2BK5 acetylation was observed in colon cancer cells treated with the HDAC1-specific inhibitor, MLRB-38489 (96), and neuroblastoma cells treated with a range of HDACi (95). For histone H3, HDACi induced hyperacetylation of H3K9 and H3K14 in porcine oocytes (48, 49, 148, 175, 188), and hyperacetylation of H3K18, H3K23, and H3K27 in colon cancer and neuroblastoma cells (95, 96). For histone H4, hyperacetylation of H4K5 and H4K16 has been observed in multiple cell lines (27, 46, 175, 188). In contrast, hyperacetylation of H4K8 and K4K12 was observed in some (19, 27, 47, 49, 95), but not in other studies (46, 188). These differences were not related to the HDACi used, and, instead, may be due to differences in the relative abundance of specific HATs and HDACs among cell types. It should also be noted that a limitation of some of these analyses is that they are an interrogation of histone acetylation changes on a genome-wide level. There is the possibility, therefore, that HDACi induce more subtle locus specific histone acetylation changes which are below the threshold of detection of these methods.
Histone core domain modifications
The histone modifications described earlier have focused on acetylation changes in the N-terminal histone tail. This has largely been due to methodological limitations favoring the analysis of the first 20–30 amino acids (111), and the subsequent development of antibodies that target these residues. Acetylation of lysine residues within the central portions of each histone, including the globular core domain required for histone–histone interactions (111), have also now been shown to have important functional consequences (72, 105, 150). For example, acetylation of H3K115 and H3K122, which are located in the DNA-histone interface of the nucleosome, reduces DNA–histone interactions and facilitates nucleosome repositioning, assembly, and disassembly (72, 105). Furthermore, a recent study generated a specific antibody to AcH3K122, and demonstrated a key role for this modification in transcriptional activation (150). Nucleosomes with AcH3K122 were globally enriched for other active histone marks, including H3K4me1 and H3K4me3 that are enriched at distal enhancers and active promoters, respectively. The authors further demonstrated that acetylation of H3K122 is mediated by p300/CBP and, importantly, observed increased acetylation of this mark in HDACi-treated MCF7 breast cancer cells (150).
Proteomic profiling of histone lysine acetylation
The advent of mass spectrometry is now enabling the investigation of all histone lysine modifications in an unbiased manner (111). Drogaris et al. used quantitative mass spectrometry to investigate the effect of HDACi on acetylation of lysine residues in the core histones in leukemia cells (43). Consistent with previous studies, the most significant changes observed were in histones H3 and H4, where acetylation was exclusively observed in the N-terminal tails (histone H3 K9, K14, K18, and K23 and histone H4 K5, K8, K12, and K16).
In a separate study, Choudhary et al. used mass spectrometry to comprehensively screen the acetylome of three cancer cell lines (MV4-11, Jurkat, and A431) and investigated the changes induced by the HDACi SAHA and MS-275 (33). These HDACi increased acetylation of ∼10% of all detectable acetylation sites in these cell lines, of which 28% were on histone proteins establishing histones as the primary targets of HDACi. This analysis confirmed the acetylation of several of the specific lysine tail modifications previously described, as well as highlighted some novel acetylation events in the tail regions of histone H2B (H2BK12 and H2BK20) and histone H4 (H4K31). Notably, this analysis also identified novel hyperacetylation events of lysine residues outside the histone tails of H2A and H2B (H2AK95, H2AK118, and H2BK108) (Fig. 4). Investigation of the functional significance of these modifications could provide novel insights into the mechanisms of HDACi-induced transcriptional changes.
HDACi-induced histone acetylation and corresponding changes in gene expression
While several locus-specific studies have linked HDACi-induced gene expression changes with corresponding increases in promoter histone acetylation (64, 129, 160), ChIP-seq studies suggest that the majority of promoters hyper-acetylated by HDACi do not undergo corresponding gene expression changes. These findings indicate that promoter acetylation alone is not sufficient to alter transcription (46). For example, Hezroni et al. investigated the genome-wide effect of low-dose valproic acid (VPA) treatment on H3K9ac in mouse embryonic stem cells (68). While VPA increased H3K9ac at >10,000 loci, the expression of only ∼100 of these genes was altered (68). Similarly, Wang et al. reported that HDACi induced H3K9 and H4K16 hyperacetylation at >1000 gene promoters; however, <2% of these genes were altered in expression (165). This discrepancy was also observed in genome-wide ChIP-seq studies performed in the adult mouse hippocampus in vivo (99), and neonatal rat cardiomyocytes (79).
Several explanations for this disparity can be envisioned. First, while histone acetylation is expected to modulate transcription by facilitating transcription factor access to DNA, or by providing recognition sites for epigenetic reader proteins, if the abundance of these factors is limiting, histone acetylation alone is unlikely to be sufficient to modulate gene expression. Second, it is possible that specific combinations of histone modifications (e.g., H3K9Ac and H3K4me3) are required for efficient recognition and binding by epigenetic reader proteins, in order to affect transcriptional change (128). For instance, the transcriptional repressor TIF1γ, preferentially binds to histone H3 that is unmodified at H3K4 and sequentially acetylated at H3K18 and H3K23 (1). Histone hyperacetylation of specific residues alone, therefore, may not be sufficient to induce corresponding transcriptional changes.
Nevertheless, these findings also illustrate that acetylation of histones only partially explains the gene expression changes induced by HDACi, and indicates that other mechanisms also likely play a role. One such mechanism is the acetylation of non-histone proteins, particularly transcription factors.
Hyperacetylation of transcription factors
A second mechanism by which HDACi regulate gene expression is through the direct acetylation of transcription factors. Several transcription factors that are modified by acetylation have been described, including p53 (61), multiple STAT (189), and specificity protein (Sp) (157) family members, GATA1 (18), E2F1 (110), HMG (116), ERα (83), MYC (122), MYB (149), TCF (161), NF-κB p65 (26), and components of the basal transcriptional machinery (TFIIEβ and TFIIF) (74). Depending on the transcription factor, and in some cases the specific lysine residue modified, acetylation can modulate transcription factor binding to DNA (61), recognition by BRD-containing epigenetic readers (190), homo and heterodimerization (89, 179), and subcellular localization (62). Acetylation can also impact other post-translational modifications of the same residue, such as ubiquitination, and potentially methylation and sumoylation. For instance, acetylation of c-myc (153) and SMAD7 (59) can protect them from ubiquitination and proteasomal degradation. These pleiotropic effects of acetylation on transcription factor function may contribute, at least in part, to the bidirectional changes in gene expression induced by HDACi. Next, we review several of the major transcription factors regulated via HDACi-induced acetylation.
P53
P53 was the first non-histone protein shown to be post-translationally modified by acetylation (61). Several p53 lysine residues subject to acetylation have now been identified and include residues within the DNA binding domain (K120, K164, and K292), tetramerization domain (K305, K320), and C-terminal domain (K351, K357, K370, K373, K381, K382, and K386) (21). While studies in mouse models have demonstrated that modifications of these lysines are not essential for p53-induced cell cycle arrest and apoptosis (90), several in vitro studies have linked p53 acetylation with increased DNA binding and target gene expression (21, 61), and induction of apoptosis. For example, acetylation of p53 K120 mediated by the MYST family HAT TIP60/MOF is required for p53-dependent expression of the pro-apoptotic genes BAX and PUMA, and mutation of this residue to arginine abrogates p53-induced apoptosis (146).
Two models have been proposed for how acetylation of p53 may enhance its DNA binding and transcriptional activation of target genes. The first allosteric model suggests that the C-terminus of p53 negatively regulates the DNA binding domain of the molecule, which is relieved upon acetylation-mediated neutralization of the positive charge on C-terminal lysines (61). Second, the acetylation of p53 has been proposed to facilitate the recruitment of transcriptional co-activators such as HATs, which enhance transcription by inducing local histone hyperacetylation (10, 186).
The mechanism of p53 deacetylation has also been investigated, with both class I (HDAC1/NuRD complex) (103) and class III HDACs (SIRT1) (102) shown to play a role. The role of HDAC1 in p53 deacetylation suggested that p53 may be subjected to hyperacetylation after HDACi treatment, which was subsequently demonstrated in several studies (24, 186). These include hepatoma cells where SAHA induced acetylation of p53 on lysines K320, K373, and K382 (24), and lung cancer cells where the HDACi depsipeptide also induced acetylation of K373 and K382 (186). In a subsequent study, Zhao et al. demonstrated that depsipeptide-mediated induction of p21 was dependent on Sp1/Sp3 binding sites within the p21 promoter as well as p53 binding sites. Notably, using ChIP analysis, the authors demonstrated that p53 Ac-K373/382 bound preferentially to the p21 promoter after depsipeptide treatment. Furthermore, overexpression of WT p53 in the p53 null H1299 cell line enhanced depsipeptide induction of p21, whereas p53 with lysine to arginine mutations at these two acetylation sites did not (186). Finally, Terui et al. demonstrated that re-expression of p53 in p53-mutant gastric cancer cells enhanced HDACi-induced apoptosis, which correlated with hyperacetylation of p53 on known deacetylation sites for HDAC1, K320, K373, and K382 (147). Among these, acetylation of K320 and K373 appeared most important, as cells overexpressing p53 with lysine to arginine mutations at these sites were not sensitized to HDACi induction of the pro-apoptotic p53 target genes PIG3 and NOXA, or apoptosis (147).
Sp1
The Sp family of transcription factors comprises eight members (Sp1–8), with each located in close proximity of an HOX gene cluster (157). Extensive post-translational modifications, including phosphorylation, acetylation, glycosylation, and sumoylation of several Sp family members, have been reported (157). Sp1 can be acetylated at a single lysine residue (K703), which is likely mediated by the HAT p300 (144, 157). When acetylated, Sp1 has been suggested to act as a transcriptional repressor as a mutation of K703 to alanine, increased the ability of Sp1 to induce promoter activity of its target gene, 12(S)-lipoxygenase (157). Sp1 also interacts with HDACs 1 and 2, suggesting that these HDACs may potentially function in Sp1 deacetylation (143).
Indeed, Sp1 has been shown to become hyperacetylated after HDACi treatment. Huang et al. demonstrated hyperacetylation of Sp1 by the HDACi trichostatin A (TSA) in pancreatic cancer cells (71), while Waby et al. used a novel anti acetyl-Sp1 antibody to demonstrate robust Sp1 acetylation in the nucleus of HCT116 cells treated with multiple HDACi (158). Waby et al. also demonstrated that acetylated Sp1 had reduced binding affinity for the p21 and Bax promoters, which were subsequently induced in expression (158). Swingler et al. also demonstrated Sp1 acetylation after HDACi treatment of HeLa cells. This was correlated with transcriptional activation of its target gene, MMP28 (145); however, whether this was mediated by increased or decreased Sp1 binding to the MMP28 promoter was not investigated. Additional studies across multiple loci are, therefore, required to determine the consequence of Sp1 acetylation on its transcriptional activity.
Sp3
The Suske lab was the first to demonstrate acetylation of Sp3 and identified lysine 551, located within the Sp3 inhibitory domain, as the residue modified (20). Sp3 was subsequently shown to associate with class I HDACs in T5 breast cancer cells (143), suggesting that they may play a role in its deacetylation. Similar to Sp1, Sp3 acetylation was suggested to reduce Sp3 transcriptional activity, as a mutation of K551 enhanced Sp1/Sp3 reporter activity (20). Notably, however, this same residue was subsequently shown to be subject to sumoylation, complicating the interpretation of whether this lysine confers loss of Sp3 transcriptional activity due to acetylation or sumoylation (134).
Several studies have demonstrated Sp3 hyperacetylation in response to HDACi treatment (5, 167). In contrast to the original studies (20), Ammanamanchi et al. suggested that HDACi-induced acetylation of Sp3 converts it from a repressor into an activator (5). This was based on the finding that overexpression of unmodified Sp3 repressed the expression of its target gene, TGFβRII; whereas co-transfection with p300 or co-treatment with an HDACi, both of which induced Sp3 acetylation, induced TGFβRII expression (5). However, these studies did not eliminate the possibility that p300 or HDACi treatment may induce TGFβRII expression through an Sp3-indpendent mechanism. Therefore, as for Sp1, additional studies are required to establish the consequences of Sp3 acetylation on its transcriptional activity.
STAT1
Acetylation of several members of the STAT family (STAT1, STAT2, STAT3, STAT5b, and STAT6) has been described (189), of which STAT1, STAT3, and STAT5b are hyperacetylated in response to HDACi (89, 163). In melanoma cell lines, HDACi selectively increased total STAT1 levels in HDACi sensitive lines. STAT1 also became hyperacetylated after HDACi treatment, due to the dissociation of HDAC1 and HDAC3, and increased interactions with CBP. Hyperacetylated STAT1 bound more strongly to NF-κB p65, inducing its cytoplasmic sequestration. This resulted in decreased expression of the pro-survival NF-κB target genes Bcl-XL and survivin, and the induction of apoptosis (89).
STAT3
Two groups identified lysine 685 in the C-terminus of STAT3 to be subject to p300-mediated acetylation after cytokine treatment in prostate and hepatoma cell lines (163, 179). Yuan et al. demonstrated that STAT3 acetylation could be reversed by overexpression of class I HDACs, particularly HDAC3, and enhanced by TSA treatment (179). Acetylation of STAT3 promoted its dimerization, cytokine-induced DNA binding, and transcription of its target genes in prostate cancer cells (179). Conversely, in diffuse large B-cell lymphoma, the HDACi panobinostat induced STAT3 acetylation at K685. This induced a parallel decrease in STAT3 phosphorylation, its cytoplasmic accumulation, and decreased expression of its pro-survival target gene, MCL1 (62). STAT3 acetylation, therefore, has differing transcriptional outcomes depending on the cell type.
E2F-1
The E2F family of transcription factors (E2F1–7) plays a central role in cell cycle progression by coordinating the expression of multiple genes required for S-phase entry. However, E2F1, in particular, plays a dual role, also being required for DNA damage-induced apoptosis through transcriptional activation of p73, APAF1, caspase 7, and p14ARF, which stabilizes p53 (124). The Kouzarides lab first demonstrated that E2F1 is acetylated by PCAF and p300, at lysines 117, 120, and 125. These residues lie adjacent to the DNA binding domain, and are deacetylated by HDAC1 (109). Acetylation of E2F1 increased its DNA binding capacity and ability to activate transcription through stabilization of this inherently unstable protein. Acetylation of E2F1 was subsequently shown to be increased after DNA damage, and to be recruited to the P73 promoter (73, 124). With regard to HDACi treatment, Ozaki et al. demonstrated increased E2F1 acetylation after TSA treatment of HeLa cells by immunoprecipitating E2F1 and immunoblotting for acetylated lysine, and demonstrated that E2F1 acetylation correlated with increased expression of p73 (120).
NF-κB p65
NF-κB plays a key role in regulating inflammatory and anti-apoptotic responses. The most abundant NF-κB complex, which comprises the p50 and p65 (RelA) subunits, is typically sequestered in the cytoplasm by the inhibitory protein, IκB. In response to stimuli such as TNF, IκB becomes phosphorylated and targeted for proteasomal degradation. This liberates NF-κB to translocate to the nucleus and drives gene expression by binding to NF-κB regulatory elements. In 2001, Warner Greene's lab demonstrated that p65/RelA is acetylated by p300 and deacetylated by HDAC3 on lysine residues K218, K221, and K310 (26). Consistent with the role of HDAC3 in deacetylation, the treatment of 293T cells with TSA induced hyperacetylation of p65. Importantly, p65 acetylation inhibited its interaction with IκB, and was proposed to serve as a switch that controls the duration of NF-κB transcriptional responses (26).
A number of other studies have also demonstrated the acetylation of NF-κB by HDACi, and linked this to increased transcriptional activity. For example, Dai et al. reported that the treatment of leukemia cells with SAHA and MS-275 induced hyperacetylation of p65, its accumulation in the nucleus, and NF-κB transcriptional activation (36). Activation of NF-κB also elicited a cytoprotective effect on HDACi-induced apoptosis, as its inhibition enhanced HDACi-induced apoptosis. However, a direct role for NF-κB acetylation in this process was not demonstrated (36). Likewise, Liu et al. demonstrated acetylation of NF-κB in response to SAHA treatment in non small cell lung cancer cell lines and identified K310 as the key residue involved. Utilizing an acetyl-p65-K310 antibody, they further demonstrated that SAHA treatment increased the recruitment of acetylated p65 to the promoter of the pro-survival gene cIAP2, which corresponded with increased promoter activity (98).
Conversely, Kiernan et al. identified K122 and K123 to be acetylated on p65. Furthermore, they demonstrated that acetylation of these lysines facilitated its removal from DNA by IκB, and subsequent export into the cytoplasm, thus serving as a mechanism of inactivating NF-κB-mediated transcription (82). Notably, however, the authors did not demonstrate that TSA treatment alone was able to induce hyperacetylation of these residues. Nevertheless, NF-κB likely represents a factor whose transcriptional activity may be enhanced or reduced depending on the specific lysine subjected to acetylation.
Other acetylated transcription factors
As described earlier, to address the effect of HDACi treatment on protein lysine acetylation on a global scale, Matthias Mann's lab used quantitative mass spectrometry to assess acetylation changes in response to HDACi treatment in three cancer cell lines (33). A number of novel acetylated proteins were identified, including SMARCC1, a component of the SWI/SNF chromatin remodeling complex; HMGA1, which plays a role in nucleosome phasing; HMGB1, which is involved in DNA bending; and NONO, which plays a role in DNA unwinding and DNA repair. Perhaps most notable was the acetylation of MED6, a component of the mediator co-activator complex that plays a central role in RNA pol II-dependent transcription (6). The functional consequences of acetylation of these proteins warrant detailed investigation and could provide new perspectives into the mechanisms of HDACi-regulated gene expression.
Collectively, these findings illustrate that a number of transcription factors are hyperacetylated after HDACi treatment. Depending on the transcription factor, this can result in increased or reduced transcriptional activity, although in some cases the same transcription factor may be differentially activated by acetylation depending on the cell type or the specific lysine residue that is modified. Furthermore, acetylation can modulate the subcellular localization, dimerization capability, and ability to interact with other transcription factors, which could collectively explain the myriad transcriptional changes induced by HDACi (Fig. 5). Finally, the spectrum of acetylation-regulated transcription factors that are similarly or differentially expressed between tumor and normal cells, and between different cell types, could provide insights into the cell-type-specific changes in gene expression induced by these agents.

Examination of HDACi-induced gene expression changes across cell types
Having discussed the primary mechanisms by which HDACi regulate gene expression, it is of interest to examine how this impacts HDACi-mediated gene expression changes in tumor and normal cells, as well as both within and between different tumor types.
Comparison of HDACi-induced gene expression changes in normal and tumor cells
HDACi preferentially inhibit proliferation and induce apoptosis in tumor cells. This has been demonstrated in multiple cell culture studies where HDACi induce apoptosis in tumor cell lines at concentrations that do not affect normal cell viability (8, 126, 151). HDACi also inhibit the growth of xenografts with no discernable toxicity to animals (121). Finally, the clinical use of HDACi is now well established and these drugs are generally well tolerated (45, 81). However, the mechanistic basis for the tumor selectivity of these agents is not well understood, and the extent to which this is due to the differential regulation of gene expression has not been extensively examined.
As in tumor cells, HDACi induces histone hyperacetylation in several normal cell types, including peripheral blood mononuclear cells (63), fibroblasts (58), neuronal cells (17), and hepatocytes (121), suggesting that these agents are likely to also impact gene expression in normal cells. Demonstrating this is indeed the case, Chiba et al. identified 143 up-regulated and 155 repressed genes in primary hepatocytes treated with TSA (29). However, in contrast, the corresponding transcriptional response induced in hepatoma cells was vastly different with only one gene (PRDX1) similarly altered in expression. The basis for this difference remains unknown but may provide an explanation for the resistance of normal hepatocytes to HDACi-induced apoptosis (121).
HDACi-regulated genes were also recently compared in normal and transformed human foreskin fibroblasts (16). Remarkably, and in contrast to the findings of Chiba et al., 89% of the genes altered by SAHA in transformed fibroblasts were similarly altered in normal fibroblasts. However, it should be taken into consideration that these cell lines were derived from the same origin, and both cell types are rapidly proliferating. Notably, despite the overall similarity, a sub-set of genes selectively regulated by SAHA in transformed cells were identified, which were enriched for transcripts involved in the regulation of apoptosis. This finding provides a possible explanation for the selectivity of HDACi to induce apoptosis in transformed cells (16).
In summary, these studies suggest that the differential response of tumor and normal cells to HDACi may be linked to the differential transcriptional responses induced; however, this needs to be directly validated and the mechanistic basis should be further investigated.
HDACi-induced gene expression changes within a tumor type
The overlap in HDACi-regulated genes in a specific tumor cell type was compared using two colon cancer cell lines by LaBonte et al. (92). Of the 2448 genes altered in expression by SAHA in HT29 cells, 860 (35%) were also changed in HCT116 cells (92). To expand on these findings using our own microarray data, we examined the overlap in gene expression changes in SW403 and HCT116 colon cancer cells treated with sodium-butyrate for 24 h. Of the 155 genes induced or repressed two-fold or greater in SW403 cells, 99 (64%) were similarly changed in HCT116 (Fig. 6). In contrast, <1% of probes were changed in the opposite orientation (not shown). Similarly, Chiba et al. used microarrays to compare HDACi-regulated genes across six hepatoma cell lines. While the overall extent of overlap was not reported, consistent induction and repression of 57 and 119 genes were observed, respectively (29). Finally, our own laboratory performed a microarray study to compare the gene expression changes induced by HDACi in five sensitive and five resistant colon cancer cell lines (171). This analysis identified 48 genes that were preferentially induced and 44 genes which were preferentially repressed in sensitive cell lines. Among the selectively induced subset were seven immediate-early response genes, FOS, JUN, ATF3, EGR1, EGR3, ARC, and NR4A1. This study, therefore, identified a transcriptional signature that was consistently induced by HDACi in multiple colon cancer cell lines (171), and furthermore a reproducible transcriptional response associated with HDACi-induced apoptosis. Collectively, these studies demonstrate that there is a considerable overlap in the transcriptional response induced by HDACi within a tumor type.

Comparison of HDACi-induced gene expression changes between tumor types
With regard to the similarity in HDACi-regulated genes between tumor types, Monks et al. identified a core subset of HDACi regulated genes commonly regulated across heterogeneous cancer cell lines. The authors initially identified 238 genes altered by HDACi in colon cancer cells (113). When 25 of these genes, including several involved in cell cycle progression (CCNA2, CCNB1, and TYMS), were examined for altered expression in seven additional cell lines derived from various other tumor types, the majority were similarly regulated (113).
Likewise, Glaser et al. used microarray profiling to compare HDACi-induced gene expression changes in breast and bladder carcinoma cell lines, and identified 13 commonly regulated genes (58). These included p21, Hep27, histone 2B, and metallothionein 1X, which were commonly up-regulated, and TYMS and CTP synthase, which were commonly repressed. However, this represented only 1–2% of the total genes regulated by HDACi in these cell lines, indicating that the vast majority of HDACi-induced changes are cell-type specific. These findings indicate that although there is extensive variability in the transcriptional changes induced by HDACi between different cancer cell lines, there are a core subset of HDACi target genes which are commonly regulated across cell types. A partial list of these genes that are likely directly regulated by HDACi (altered in expression within 6–12 h), in at least two distinct cell types, is provided in Table 1.
Commonly regulated HDACi-target genes
The strongest evidence that HDACi induce at least some similarities in gene expression, irrespective of cell type, comes from studies of the CDKN1A gene. CDKN1A (p21WAF1/CIP1) is a cyclin-dependent kinase inhibitor that is induced by HDACi in a protein synthesis-independent manner, establishing it as a direct transcriptional target (7). Consistent with regulation by HDACs, siRNA-mediated knockdown of individual class I and II HDACs increases p21 expression in multiple cell types (137). CDKN1A induction in response to HDACi treatment has been observed in mouse (152) and human cells, as well as in cell lines derived from multiple tumor types, including colorectal (7), breast (66), prostate (91), lung (125), melanoma (85), CTCL (183), multiple myeloma (112), and a range of leukemia cell lines (132, 156), establishing it as a universally induced HDACi target gene.
In addition to being a p53 target gene, the proximal p21 promoter is GC rich and contains 6 Sp1/Sp3 binding sites. Promoter deletion studies identified these sites as essential for HDACi-mediated induction (138), and a model has emerged in which Sp1 or Sp3, or both, recruit HATs and HDACs to the p21 promoter, which compete to establish a dynamically regulated basal level of p21 expression (9, 39, 173). Treatment with HDACi alters this equilibrium in favor of HATs, leading to local histone hyperacetylation and p21 transcriptional activation (39).
Several pieces of evidence support this model. First, both Sp1 and Sp3 have been shown to interact with class I HDACs as well as the class II HDAC, HDAC4, and re-ChIP experiments have demonstrated that the recruitment of HDACs to the p21 promoter is Sp1/Sp3-dependent (170). Second, a p300 dominant negative mutant was able to attenuate both TSA and Sp1-induced p21 expression (173). Finally, the pharmacological inhibitor of Sp1/Sp3, mithramycin, and siRNA-mediated knockdown of Sp1 attenuated HDACi-mediated p21 induction (170).
Sp1/Sp3 transcription factors in HDACi-regulated gene expression
In addition to CDKN1A, Sp1 and or Sp3 have been reported to be critical for HDACi-mediated regulation of a number of other targets. These include genes involved in cell cycle arrest such as p57 (34) and INK4D (178), apoptosis induction such as Bak (30), DR5 (86) and TRAIL (174), and a range of other genes involved in a variety of cellular processes—NHE3 (4), Galectin 1 (100), IGFBP3 (159), MMP11(11), 5-LO (136), CYP46A1 (119), ENaC (180), eNOS (53), DLC-1 (84), NECL1 (54), ADAMTS1 (32), and EC-SOD (181).
In our own study in which we identified 48 genes consistently induced by HDACi in sensitive colon cancer cell lines, we demonstrated a key role for Sp1 and Sp3 in inducing expression of the majority of these targets. First, the promoters of these genes were found to have a significantly higher GC content and to be enriched for Sp1/Sp3 binding sites when compared with a control set of un-induced genes. This finding is similar to that reported by Moore et al., who also observed an enrichment of Sp1 binding sites in genes induced by TSA in pancreatic cancer cells (115). Furthermore, we observed that co-treatment with the Sp1/Sp3 inhibitor mithramycin, or dual knockdown of Sp1 and Sp3, markedly attenuated induction of many of these genes, which notably also attenuated HDACi-induced apoptosis (171).
Interestingly, although less extensively investigated, HDACi also mediates transcriptional repression of specific genes in an Sp1/Sp3-dependent manner. This was clearly illustrated for the anti-apoptotic gene bcl-2, which is directly repressed by HDACi in lymphoma cell lines (44). In this study, Duan et al. (44) demonstrated that bcl-2 repression by TSA was accompanied by dissociation of Sp1 from the bcl-2 promoter, and that deletion of the Sp1 binding site attenuated HDACi-mediated repression. Similarly, sodium-butyrate treatment caused direct repression of the c-Src oncogene in hepatocellular carcinoma cells, most likely in an Sp1/Sp3-dependent manner (88). This was suggested by the finding that the magnitude of repression of c-Src promoter activity was similar to that mediated by the deletion of two Sp1/Sp3 sites (88). On a broader scale, our microarray analysis of genes altered by HDACi in colon cancer cell lines demonstrated that as observed for induced genes (171), the promoters of HDACi-repressed genes also had an overrepresentation of Sp1/Sp3 binding sites compared with the control gene set (Mariadason et al., unpublished).
Sp1 and Sp3 are abundantly and ubiquitously expressed transcription factors (97, 169). HDACi-mediated induction of Sp1/Sp3-regulated genes such as p21 across multiple cell types is, therefore, consistent with the expression profile of these transcription factors. However, GC-rich Sp1/Sp3-regulated genes are also among the most robustly regulated by HDACi (29, 57, 171), indicating that these promoters are exquisitely sensitive to HDACi. The mechanistic basis for this intriguing observation has not been clearly elucidated, although a number of possible reasons can be envisioned.
One possibility is that these promoters comprise a specific chromatin architecture which predisposes them to HDACi-mediated regulation. In this regard, ChIP-seq studies have now defined a major class of primary response genes characterized by GC-rich Sp1-bound promoters which are associated with highly inducible genes, including FOS, NR4A1, EGR1, and EGR3 (65, 127). In the basal state, these promoters comprise a unique chromatin conformation that includes high levels of the positive histone modifications H3K9/K14ac and H3K4me3, unstable nucleosome occupancy, and a lack of SWI/SNF nucleosome remodeling complexes. These promoters also harbor preassembled RNA Pol-II, which is paused at the proximal promoter (65, 127), indicating their existence in a poised state. Importantly, Sp1 is critical for the maintenance of this poised state, as its knockdown results in a loss of RNA-Pol-II from these promoters (65). These genes are particularly sensitive to induction by stimuli such as serum, which induces their expression through facilitating transcriptional elongation. This process is triggered by acetylation of K5, K8, and K12 on histone H4, which are marks recognized by the epigenetic reader, BRD4. BRD4, in turn, recruits pTEFb, which induces transcriptional elongation by phosphorylating RNA Pol-II (65) and by antagonizing the negative elongation factors NELF and DSIF.
Several lines of evidence suggest that these same promoters are also amenable to regulation by HDACi. First, a number of the genes that harbor these promoters, including FOS, EGR1, EGR3, and NR4A1, are rapidly and highly induced by HDACi in colon cancer cells in an Sp1/Sp3-dependent manner (171). Second, independent ChIP-Seq analyses have demonstrated that promoters with similar features are the most prone to hyper-acetylation after HDACi treatment (68). Finally, HDACi treatment can induce the specific histone modifications (H4K5/8/12Ac) that attract BRD4 to mediate transcriptional elongation. Collectively, a model can be envisioned by which the inhibition of Sp1 or Sp3-associated HDACs at these promoters induces specific histone acetylation marks that are recognized by epigenetic readers such as BRD4. This, in turn, recruits factors such as pTEFb, which activate Pol-II to drive expression through transcriptional elongation (Fig. 7).

However, the proposed model also needs to accommodate the finding that GC rich promoters are also capable of being repressed by HDACi. Interestingly, while we propose that increased transcriptional elongation may play a role in HDACi-mediated up-regulation of gene expression, there is also evidence that HDACi conversely represses genes by attenuating this process. This was first described by Heruth et al., who reported that HDACi-mediated repression of c-Myc in colon cancer cells was mediated by a block in c-Myc transcriptional elongation (67). This finding was confirmed by Wilson et al., who utilized FISH probes targeting the 5′-and 3′-ends of the c-Myc transcript to demonstrate that HDACi treatment induced an increase in transcription initiation but a concomitant decrease in transcriptional elongation (172). Using a similar approach, the Augenlicht group also subsequently demonstrated that the inhibition of transcriptional elongation is a key mechanism by which HDACi inhibits cyclin D1 expression (104).
Two studies have examined the frequency of HDACi-mediated blockade of transcriptional elongation on a genome-wide scale. Most recently, Kim et al. used global run-on sequencing (Gro-seq) to identify genes repressed by HDACi in this manner in breast cancer cells. This revealed that highly expressed and high copy number loci, such as Erbb2, were particularly prone to inhibition of transcriptional elongation by HDACi (87). Second, Daroqui and Augenlicht designed microarrays that enabled interrogation of the relative level of expression of the 5′ and 3′ ends of mRNA transcripts. Using these arrays, they identified 367 genes (2% of analysed loci) with transcripts that were preferentially down-regulated at the 3′ relative to the 5′ end in colon cancer cells treated with HDACi, including c-Myc and cyclin D1 (38). A further analysis of these 367 transcripts using a custom-designed microarray with probes which tiled across these genes confirmed that 42 of these genes displayed significantly lower expression at the 3′ compared with the 5′ end, and were, thus, potentially paused on HDACi treatment (38). Analysis of these 42 gene promoters reveals that 68% harbor CpG islands, indicating that GC-rich promoters are particularly prone to pausing in response to HDACi treatment.
These findings suggest that the HDACi-mediated induction and repression of genes harboring GC-rich promoters may be due to enhanced and attenuated transcriptional elongation, respectively. One possibility is that these contrasting outcomes are related to the relative contributions made by the highly related Sp1 and Sp3 transcription factors, which bind Sp1/Sp3 binding sites with an equal affinity. Furthermore, as discussed earlier, both of these factors are themselves subject to HDACi-mediated acetylation, which may selectively occur at some promoters depending on the specific HATs and HDACs with which they are locally associated. Notably, acetylation of Sp1 in response to HDACi treatment was associated with its dissociation from the bcl-2 promoter and bcl-2 repression (44). One possibility, therefore, is that by dissociating from promoter regions after its acetylation, Sp1 is no longer able to mediate transcriptional elongation, which, consequently, results in repression through transcriptional pausing (Fig. 8). Sp1 and Sp3 may also work in combination or in competition at specific loci to elicit a net effect. For example, in colon cancer cells, attenuation of HDACi-mediated induction of EGR1, EGR3, and GADD45 expression was best achieved by combinatorial knockdown of Sp1 and Sp3, demonstrating that both transcription factors are required for the induction of these genes (171).

Detailed genome-wide studies of promoter occupancy of these factors in the basal and acetylated state, and the impact of HDACi treatment on their binding profiles will be required to address these mechanisms. Furthermore, these findings would need to be integrated with corresponding gene expression changes, histone modifications, and patterns of BRD4 and Pol-II binding to establish a comprehensive model with the scope to understand both the induction and repression of gene expression induced by HDACi.
Summary and Conclusions
HDACis are an emerging class of anti-cancer therapeutics with activity in several hematological cancers and potential activity in some solid tumors when used in combination. These drugs elicit extensive transcriptional changes in tumor cells, with a subset of genes, particularly those regulated in an Sp1/Sp3-dependent manner, reproducibly regulated across a broad range of cell types. HDACi also induce and repress an approximately similar number of genes. Here, two models of HDACi-regulated gene expression are reviewed—histone acetylation and transcription factor acetylation—with consideration given to how these events may explain both transcriptional induction and repression. How these mechanisms may intersect at GC-rich promoters is also considered.
It is hoped that through a detailed understanding of the specific mechanisms by which HDACi modulates gene expression, the clinical development of these agents as anti-cancer therapeutics can be further enhanced. In particular, this information may facilitate the identification of tumors that are likely amenable to treatment with HDACi and enable the design of rational drug combinations, which can further enhance the clinical utility of these agents.
Footnotes
Acknowledgments
The authors thank Amardeep Dhillon and Hoanh Tran for a critical reading of this article. J.M.M. is the recipient of an Australian Research Council Future Fellowship (FT0992234), and J.W.T.T. is the recipient of an Australian Postgraduate Award PhD scholarship.