
Abstract
Macrophage apoptosis is involved in atherosclerotic plaque development. The aim of this study was to evaluate the interrelationship between macrophage apoptosis and the endoplasmic reticulum (ER) stress in the tissue around the necrotic core (TANC) and in the periphery (P) of the same carotid plaques derived from patients undergoing carotid endarterectomy. Apoptosis was significantly higher in TANC than in P (p<0.001). mRNA and protein expression of the protein kinase-like ER kinase (Perk) and the nuclear erythroid-related factor 2 (Nrf2)-related survival genes was significantly higher in P than in TANC (p<0.01), while CCAAT/enhancer-binding protein homologous protein (Chop) and the apoptosis-related genes were higher in TANC than in P (p<0.01). The TANC extract was characterized by significantly higher concentrations of oxidized derivatives of polyunsaturated fatty acids (PUFAs) than the P extract (p<0.01). When THP-1 cells were incubated with P or TANC extracts there was a dose-dependent increase of Perk and Nrf2 or of Chop and of the apoptosis-related genes, respectively (p<0.01). Our observations lead to the hypothesis that ER stress induced by oxidized derivatives of PUFAs may promote macrophage apoptosis in TANC and favor the expansion of the necrotic core of the plaques, a major feature responsible for its disruption and acute luminal thrombosis. Antioxid. Redox Signal. 21, 850–858.
Introduction
M
The tissue around the necrotic core (TANC) of carotid plaques is characterized by an abnormal amount of apoptotic cells. This phenomenon may be related to a sustained endoplasmic reticulum (ER) stress since there is an abundance of the CCAAT/enhancer binding protein homologous protein (Chop) and apoptosis-related gene expression in TANC, while the protein kinase-like ER kinase (Perk) and survival genes prevail in periphery (P). This specular expression may be driven by different concentrations of oxidized derivatives of polyunsaturated fatty acids that, by binding to human thromboxane A2 (TP) and G2A receptors, may determine the direction toward cell apoptosis or survival. So, ER stress may promote macrophage apoptosis in TANC and favor the expansion of necrotic core, a feature responsible for its disruption and luminal thrombosis.
In ER-stressed macrophages, Chop has been demonstrated to induce ER oxidase 1α, which activates inositol triphosphate receptor-mediated release of calcium into the cytosol (8) and the subsequent activation of a calcium/calmodulin-dependent protein kinase II, which triggers apoptosis through different mechanisms (8).
Oxidized derivatives of arachidonic and linoleic acids have already been identified in human atherosclerotic plaques as markers of oxidative stress (7) and supposed to function as second hits to trigger apoptosis in macrophages exposed to low levels of ER stress (8). Some of these oxidized derivatives of arachidonic and linoleic acids such as 8-iso-prostaglandin-F2α (8-iso), the prototype of F2-isoprostanes, 9-hydroxyoctadecadienoic acid (9-HODE), and 15-hydroxyeicosatetraenoic acid (15-HETE) have also been shown to be mediators of important biological effects through their binding to the human thromboxane A2 (TP) receptor (1) and G2A receptor (6).
We have recently demonstrated that some oxidized derivatives of polyunsaturated fatty acids (PUFAs) contained in the tissue around the necrotic core (TANC) of human carotid plaques may have a role in the expansion of NC, by inducing defective efferocytosis (3). The aims of this study were to evaluate the interrelationship between macrophage apoptosis and the ER stress in TANC and in the periphery (P) of human carotid plaques and whether oxidized derivatives of PUFAs may have a role in promoting ER-induced macrophage apoptosis. Therefore, in TANC and in P of the same carotid plaques derived from patients undergoing carotid endarterectomy, we first quantitated apoptotic cells, the expression of Perk and Chop together with some genes correlated with apoptosis and Nrf2/ARE survival pathway, and the concentration of 8-iso, 9-HODE, and 15-HETE. Then, in the in vitro study, we evaluated the effect of TANC, P, and of normal artery (NA) extracts on the expression of Perk, Chop, and of apoptosis and Nrf2/ARE genes in macrophage-like THP-1 cells. Finally, we verified if 8-iso, 9-HODE, and 15-HETE increase the expression of Perk and Chop by binding to the TP and G2A receptors and the subsequent mobilization of intracellular Ca2+ (Ca2+i).
Apoptotic cells and expression of Perk, Chop, and of the genes correlated with Nrf2/ARE pathway and apoptosis in NA, and in P and TANC of human carotid plaques
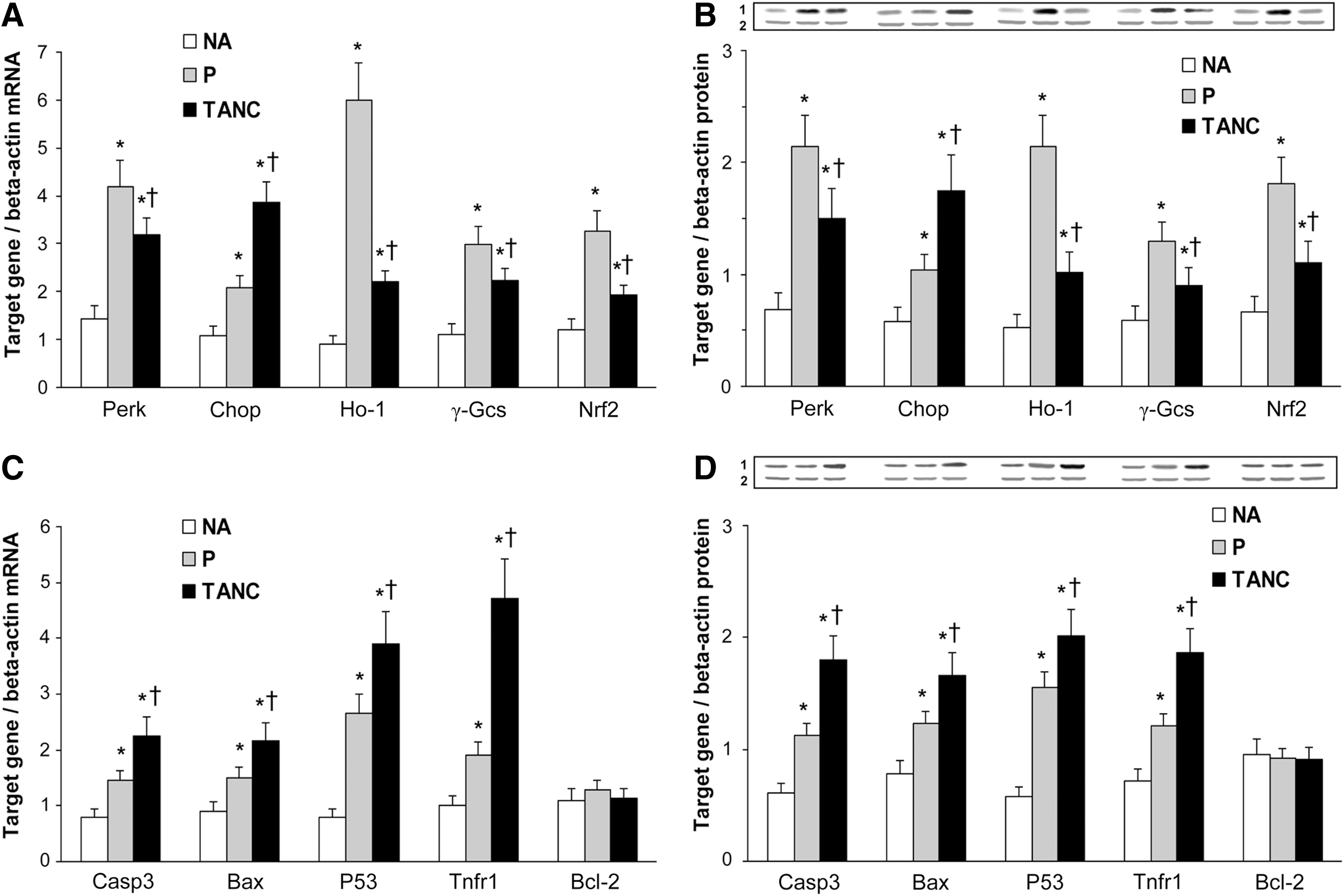
Figure 1 shows a representative section of segments derived from P (Fig. 1A) and TANC (Fig. 1B) of the carotid plaques. Positively macrophage apoptotic-stained cells resulted much higher in TANC than in P sections (p<0.001) (Fig. 1C). The mRNA and protein expression of Perk, Chop, and of survival genes (Nrf2, Ho-1, and γ-Gcs) resulted significantly higher in P and TANC of human carotid plaques than in NA (p<0.01) (Fig. 2A, B); the same genes were significantly more expressed in P than in TANC (p<0.01), while only Chop was higher in TANC than in P (p<0.01) (Fig. 2A, B). The apoptosis-related genes such as caspase (Casp)3, Bcl-2-associated X protein (Bax), tumor suppressor protein 53 (P53), tumor necrosis factor receptor 1 (Tnfr1) were found to be significantly more expressed in P and TANC than in NA (p<0.01), while B-cell leukemia/lymphoma-2 (Bcl-2) expression was similar in NA, P, and TANC (Fig. 2C, D). Casp3, Bax, P53, and Tnfr1 resulted significantly more expressed in TANC than in P (p<0.01) (Fig. 2C, D). These results are in line with previous evidences that have indicated that UPR is chronically activated in cells of the atherosclerotic vascular wall and, in particular, in macrophages (8) and with the data of Myoishi et al. (5), who demonstrated that robust Chop expression and lesional apoptosis were more identified in advanced vulnerable plaques.

Quantitative analysis of the components of NA, P, and TANC extracts and their effect on the expression of Perk, Chop, and of the genes correlated with survival (Nrf2/ARE pathway) and apoptosis in THP-1 cells
The quantitative analysis of NA, P, and TANC extracts revealed that F2-isoprostanes (m/z 353.2), HETEs (m/z 319.2), and HODEs (m/z 295.3) were the major components of TANC and P extracts, their concentrations being significantly higher in TANC than in P (p<0.01) and NA (p<0.001) (Table 1); HETEs and HODEs also resulted higher in P than in NA (p<0.05) (Table 1).
Data are expressed as mean±SD (ng compound/mg tissue).
p from<0.05 to<0.001 versus NA; † p<0.01 versus P.
HETEs, total hydroxyeicosatetraenoic acids; HODEs, total hydroxyoctadecadienoic acids; NA, normal artery; P, periphery; TANC, tissue around the necrotic core.
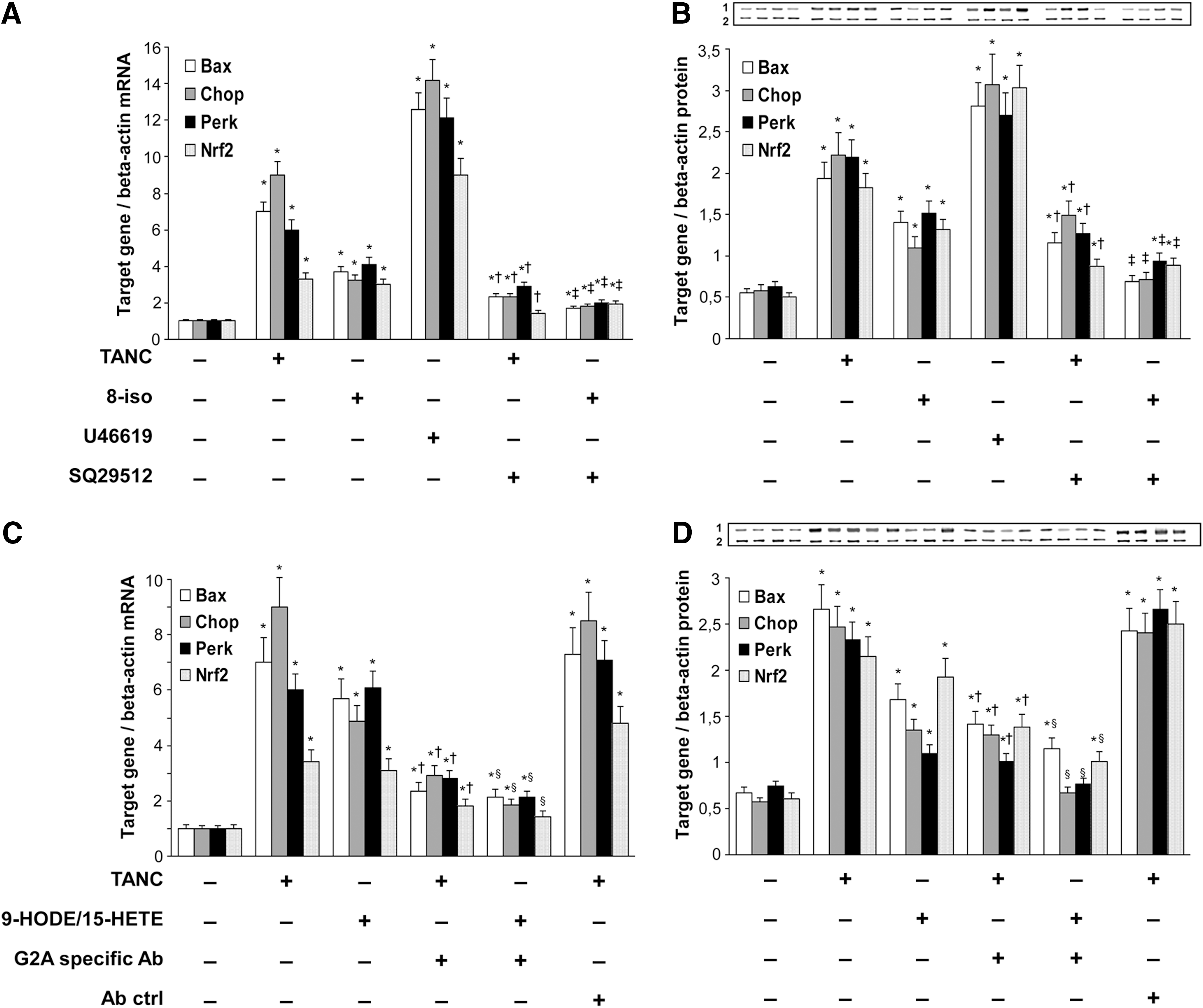
To understand if the oxidative derivatives of PUFAs present in the extracts were able to stimulate the expression of Perk and Chop and of the genes correlated with the Nrf2/ARE pathway and apoptosis, THP-1 cells were incubated with increasing amounts of NA, P, and TANC extracts. As shown in Figure 3, there was a dose-dependent increase of both Perk and Chop only after incubation with P (p<0.05) and TANC extracts (p<0.01). The rise, however, was much more evident for Perk when THP-1 cells were incubated with P, and for Chop when the cells were incubated with TANC (p<0.01). P also determined a dose-dependent increase of Nrf2 and Ho-1 (p<0.01), while TANC induced a less, but nondose-dependent increase (p<0.05). As for the genes correlated with apoptosis, both TANC and P dose dependently increased Bax and Tnfr1 expression (p from<0.001 to<0.01), but the rise was much higher by using TANC than P (p<0.001). The highest increase of Chop and of the genes correlated with apoptosis therefore was caused by TANC, while P determined the maximum increase of Perk and of the genes correlated with the Nrf2/ARE survival pathway. Even if preliminary, these results suggest that the concentrations of these oxidative derivatives may play a role in determining the direction toward apoptosis or survival of THP-1 cells.

Role of TP and G2A receptors on the expression of Perk, Nrf2, Bax, and Chop induced by TANC extract in THP-1 cells
To examine whether the fatty acid oxidative products present in plaque extract stimulated THP-1 cells by binding to TP or G2A receptors, we first incubated THP-1 cells with TANC extract, 8-iso, and TP receptor agonist U46619 and TP receptor antagonist SQ29512. Figure 4A and B shows that all these stimuli determined a significant increase of mRNA and protein expression of Bax, Chop, Perk, and Nrf2 (p from<0.01 to<0.001). The rise induced by TANC and 8-iso was significantly reduced by SQ29512, a specific TP antagonist (p<0.01). Then, we incubated THP-1 cells with TANC and with a mixture of 9-HODE and 15-HETE and looked at the effect of preincubation with the G2A-specific antibody. Figure 4C and D shows that the mRNA and protein expression of Bax, Chop, Perk, and Nrf2 expression was significantly induced by TANC and by a mixture of 9-HODE and 15-HETE (p from<0.01 to<0.001). The preincubation of the cells with an anti-G2A-specific antibody determined a significant decrease of the expression of Bax, Chop, Perk, and Nrf2 (p<0.01). TP receptors are membrane-bound, G-protein-coupled, seven-transmembrane receptors distributed widely in the cardiovascular systems. In addition to platelets and endothelial cells, TP receptors are also expressed in other cell types involved in atherothrombosis, such as smooth muscle cells (1). By binding to TP receptors, F2-isoprostanes have already been reported to promote endothelial cell activation, that is, adhesion molecule expression, vascular smooth muscle cell contraction, and platelet aggregation, thereby accelerating the progression of atherosclerotic lesions (1). The G2A receptor is a G-protein-coupled receptor that has been shown to function as a receptor for oxidized free fatty acids (6). HODEs and HETEs have recently been shown to increase the expression of Adam17 in THP-1 cells, an enzyme involved in defective efferocytosis (3). On the basis of these results, we are tempted to speculate that some oxidative derivatives of PUFAs present in TANC may increase the expression of Chop, Perk, and of the genes correlated with apoptosis by binding to TP and G2A receptors.
ACE, angiotensin-converting enzyme; AT, angiotensin.
Effect of TANC extract, 8-iso, 9-HODE, and 15-HETE on Ca2+i
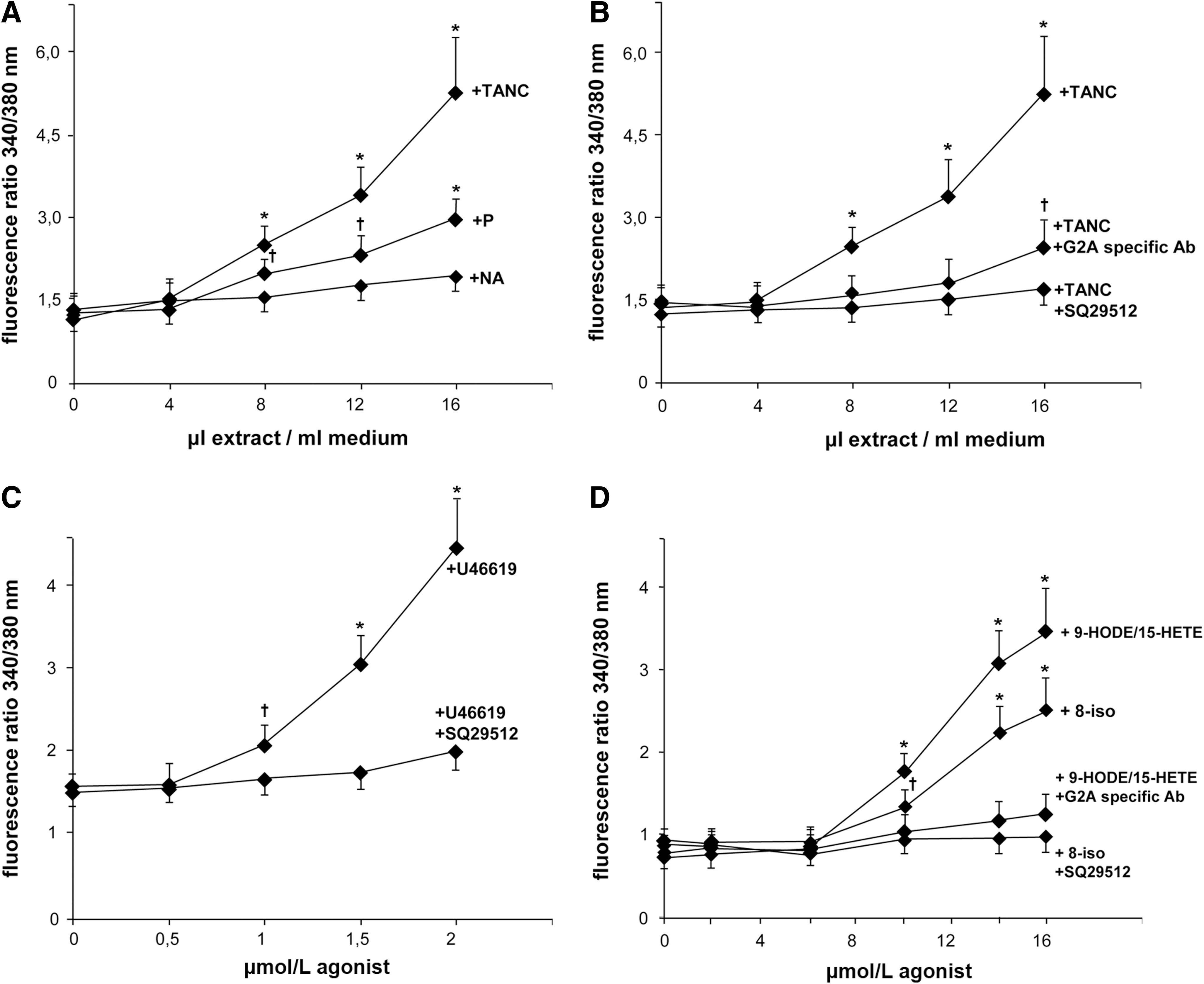
We found that TANC extract dose dependently increased Ca2+i in THP-1 cells. As shown in Figure 5A, TANC extract evoked a dose-dependent Ca2+i mobilization in THP-1 cells (p<0.01) that was significantly higher than that induced by P and NA extracts (p<0.01). The Ca2+i mobilization was significantly reduced when THP-1 cells were incubated with the TP antagonist SQ29512 or with the anti-G2A-specific antibody (p<0.01) (Fig. 5B). We also observed a dose-dependent Ca2+i mobilization after incubation of THP-1 cells with increasing concentrations of the TP agonist U46619, 8-iso, and the mixture of 9-HODE and 15-HETE (Fig. 5C, D) (p<0.01). Again, the TP antagonist SQ29512 and the anti-G2A-specific antibody reduced Ca2+i mobilization (p<0.01). The fact that both the TP and G2A receptors, once activated by different ligands, are known to cause a rapid increase of Ca2+i (1, 6) and the demonstration that specific antagonists of the receptors counteract this phenomenon as well as the expression of Perk and Chop, may preliminarily indicate that the oxidized derivatives of PUFAs contained in TANC may promote apoptosis also through these receptors.
Notes
Materials and methods
All the investigations conform to the Declaration of Helsinki. The study was approved by the Ethics Committee of the University of Verona and all participants provided written consent.
Collection of human specimens
We collected 107 carotid plaque specimens from 117 asymptomatic consecutive patients undergoing carotid endarterectomy according to the present guidelines. After removal from the carotid artery, the plaques were placed in chelex-100-treated phosphate-buffered saline (PBS) containing EDTA (0.75 mg/ml), BHT (0.02 mg/ml), and RNA stabilization reagent (RNALater; Qiagen, Hilden, Germany). The plaques were examined macroscopically to individualize the site of the maximum plaque thickening and then transversely dissected into two segments. One of the two segments was further dissected in three specimens for histology, immunohistochemistry, and protein expression; the other segment was used to prepare TANC and P extracts. In brief, after removing NC, the segment close to NC was used to obtain TANC extract, while P was derived from the periphery of the same human carotid plaques. The segments were blotted dry and immediately stored at −80°C. NA extracts were collected from 10 patients undergoing splenectomy.
Histology, immunohistochemistry, and apoptosis detection
Segments for histology and immunohistochemistry were fixed in paraformaldehyde 4% for 2–4 h and embedded in paraffin as previously described (3). Sections of each segment were stained with hematoxylin–eosin for histological analysis to evaluate NC size, fibrous cap thickness and rupture, and presence of calcification. The carotid plaques were classified histologically as previously described (3). In this study, only carotid plaques with a fibrous cap atheroma or a thin fibrous cap atheroma, markedly infiltrated by macrophages and an underlying NC, were taken into consideration (n=41). Details of the main characteristics of the patients undergoing endarterectomy and of the plaques considered are described in Table 1. Immunostaining of CD68-positive cells was performed as previously described (3). Slices were incubated with 2% normal serum, 0.3% Triton X-100, 1% bovine serum albumin for 20 min, and then with the primary antibody for 1 h at room temperature (PG-M1; Dako, Aachen, Germany). The sections were then incubated with a complementary secondary antibody for 1 h and then with avidin–biotin for 30 min. Diaminobenzidine was used as chromogen and sections were counterstained with hematoxylin.
Detection of apoptotic cells in paraffin-embedded sections of each segment (P and TANC) was performed with the ApopTag Peroxidase In situ Oligo Ligation (ISOL) detection kit (Chemicon International, Inc., Darmstadt, Germany) following the manufacturer's instruction. Briefly, the tissue sections were deparaffinized with proteinase K. The endogenous peroxidase activity was quenched with 3% H2O2 and incubated with an equilibration buffer. Working strength terminal deoxynucleotidyl transferase was then applied for 1 h at 37°C. The slices were then stained with an anti-digoxigenin peroxidase conjugate for 30 min at room temperature. After washing, the peroxidase substrate diaminobenzidine was added. The reaction was stopped when the color developed. Slices were extensively washed with H2O and mounted with a glass coverslip in an Aqua-Mount medium (Lerner Laboratories, New Haven, CT). Apoptosis was expressed as a percentage of the total number of nuclei.
Preparation of NA, P, and TANC extracts
NA, P, and TANC extracts were prepared as previously described (1). Briefly, the segments, still frozen, were immediately placed in the chelex-100-treated PBS buffer containing EDTA (0.75 mg/ml) and BHT (0.02 mg/ml). The samples were homogenized using an Ultra-Turrax T8 blade homogenizer (IKA Labortechnik, Staufen, Germany) for 5 min and the fine tissue powders derived from TANC and P of all plaques and from all normal spleen arteries were put together. The powder was extracted as previously described (3). Since no hydrolysis of cholesterol ester and triglycerides was performed, only the free forms of oxidative derivatives of PUFAs were extracted (3). 8-iso, 9-HODE, 15-HETE, and their deuterated internal standards were purchased from Cayman Chemical Company (Ann Arbor, MI).
Evaluation of components of NA, P, and TANC extracts
Mass spectrometry analysis of NA, P, and TANC extracts was performed on an ion trap mass spectrometer (model 1100; Agilent Technologies, Waldbronn, Germany) by reverse-phase HPLC using an Alltech Vydac C18 column (100×0.3 mm, 3 μm ODS) (Deerfield, IL, USA) equipped with a guard column at 37°C; the sample was eluted in the isocratic mode using methanol/ammonium acetate (10 nM) 95:5 v:v at a flow rate of 25 μl/min as previously described (3). Analytical quantification of F2-isoprostanes, HODEs, and HETEs was obtained by relating the peak areas with 8-iso, 9-HODE, and 15-HETE and with areas of the deuterated internal standards (Cayman Chemical Company).
Cell cultures
The macrophage-like THP-1 cell line was cultured and differentiated as previously described (3). THP-1 cells were incubated with NA, P, and TANC extracts to evaluate the expression of Perk, Chop, and apoptosis genes. In some experiments, THP-1 cells were preincubated with SQ29512 (a TP receptor antagonist) (10 μM) and with U46619 (a TP receptor agonist) (1 μM) (Sigma-Aldrich, Milan, Italy), G2A receptor-specific antibody (2 μg/ml) (SC-9692; Santa Cruz, Heidelberg, Germany), or normal goat IgG (2 μg/ml) for 1 h, and incubated with TANC extract, 8-iso (10 μM), and a mixture of 9-HODE and 15-HETE (10 μM). Early apoptosis and cell viability were determined using the AnnexinV-FITC Kit (Bender MedSystems GmbH, Vienna, Austria) and 7-amino-actinomycin D (7-AAD) (BD Biosciences, Buccinasco, Italy) by flow cytometry. Endotoxin contamination of cell cultures was routinely excluded with the chromogenic Limulus amebocyte lysate assay (Sigma-Aldrich).
RNA isolation and quantitative real-time PCR
Fresh, frozen tissue samples were cut and transferred into the rotor-stator homogenizer with the QIAzol Lysis buffer and magnetic beads to obtain tissue lysates. Total RNA isolation was performed with the RNeasy plus Universal Mini Kit on QiaCube instrument (Qiagen) according to the manufacturers' instructions. The RNA concentration and quality were evaluated with the RNA 6000 Nano LabChip kit by using the Bioanalyzer 2100 microcapillary electrophoresis system (Agilent Technologies, Inc., Santa Clara, CA). Reverse transcription was performed using the IScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). The relative expression levels of genes of Perk, Nrf2, Ho-1, γ-Gcs, Chop, Casp3, Bax, P53, Tnfr1, and Bcl-2 were performed in triplicate using the QuantiTect Primer Assay and QuantiTect SYBR Green PCR Kit (Qiagen) on the MyiQ Thermal Cycler (Bio-Rad). QuantiTect Hs-ACTB Assay (Qiagen) was used as normalizer.
Western blotting analysis
Western blotting analysis was performed as previously described (3). The following primary antibodies against the indicated proteins were used: Perk (ab65142; Abcam, Cambridge, United Kingdom), Nrf2 (sc-13032; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), Ho-1 (sc-10789; Santa Cruz Biotechnology, Inc.), γ-Gcs (ab41463; Abcam), Chop (ab11419; Abcam), Casp3 (AAS-103; Stressgen Bioreagents Corp., Victoria, BC, Canada), Bax (AAS-040; Stressgen Bioreagents Corp.), P53 (ab26; Abcam), Tnfr1 (ab19139; Abcam), Bcl-2 (AAS-070; Stressgen Bioreagents Corp.), and β-actin (sc-130656; Santa Cruz Biotechnology, Inc.). As secondary antibodies, appropriate horseradish peroxidase-conjugated antibodies were used and the bands visualized according to a standard chemiluminescence protocol (Immobilon Western; Millipore, Billerica, MA). Protein expression was quantified using the VersaDoc Imaging System (Bio-Rad).
Calcium measurement
Ca2+i measurement in THP-1 cells was made by monitoring the intensity of Fura2 (Molecular Probes, Eugene, OR) fluorescence. THP-1 cells were washed twice in PBS, resuspended in the modified Ca++/Mg++-free Hank's buffered salt solution containing 10 mM HEPES, pH 7.6, and 0.1% bovine serum albumin (HBSS buffer) at 107 cells/ml and incubated in the dark with 5 μM Fura2/AM for 45 min at 37°C. Subsequently, the cells were collected by centrifugation (600 g, 5 min), washed once in an equal volume of HBSS buffer, resuspended in HBSS buffer at 107 cell/ml, and kept at room temperature in the dark until use. Fura2 fluorescence was recorded in THP-1 cells at 37°C with gentle stirring using a Shimadzu RF-5000 spectrofluorometer at excitation wavelengths of 340 and 380 nm and emission wavelength of 510 nm. The rapid, transient rise and fall in Ca2+ levels in response to ligand stimulation were interpreted as receptor-mediated Ca2+i mobilization. The calibration of the signal was performed in each sample by adding 0.2 Triton X-100 to obtain the maximal fluorescence (Fmax) and adding 1 mM EGTA to obtain the minimal fluorescence (Fmin).
Statistical analysis
The data are presented as mean±SD. Statistical analysis was performed by two-tailed unpaired Student's t-test and by one- or two-way analysis of variance for repeated measures followed by the post hoc Tukey test for multiple comparisons using the SYSTAT program and statistical software manual (SYSTAT, Inc., Evanston, IL) for Macintosh. Statistical significance was inferred at p-values<0.05.
Footnotes
Acknowledgments
The authors thank the past and present members of our laboratory for their contribution to various aspects of histology and immunohistochemistry. Especially, the authors thank Stefania Manfro for the excellent histological preparations.